
Molecular Dynamics Simulation Study of the Self-Assembly of Phenylalanine Peptide Nanotubes

 ,
,
Abstract
:1. Introduction
2. Model Details and Methodology for Numerical MD Experiments
2.1. Main Software Platform Used
2.2. Model Details and Force Technique Assembly
3. Results of MD Analysis and Discussion of Results
4. Calculation of the Chirality of Phenylalanine Helical Nanotubes from the Dipole Moment Data of Their Constituent Phenylalanine Molecules
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brandon, C.J.; Martin, B.P.; McGee, K.J.; Stewart, J.J.P.; Braun-Sand, S.B. An approach to creating a more realistic working model from a protein data bank entry. J. Mol. Model. 2015, 21, 3. [Google Scholar] [CrossRef]
- Orsi, M. Molecular simulation of self-assembly. In Self-Assembling Biomaterials. Molecular Design, Characterization and Application in Biology and Medicine, 1st ed.; Azevedo, H.S., da Silva, R.M.P., Eds.; Woodhead Publishing Series in Biomaterials; Elsevier: Amsterdam, The Netherlands, 2018; pp. 305–318. [Google Scholar]
- Hospital, A.; Goñi, J.R.; Orozco, M.; Gelpi, J. Molecular dynamics simulations: Advances and applications. Adv. Appl. Bioinform. Chem. 2015, 8, 37–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Gunsteren, W.F.; Berendsen, H.J.C. Computer Simulation of Molecular Dynamics: Methodology, Applications, and Perspectives in Chemistry. Angew. Chem. Int. Ed. Engl. 1990, 29, 9921023. [Google Scholar] [CrossRef]
- Karplus, M.; Petsko, G.A. Molecular dynamics simulations in biology. Nature 1990, 347, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Kamberaj, H. Molecular Dynamics Simulations in Statistical Physics: Theory and Applications; Springer: Cham, Switzerland, 2020. [Google Scholar]
- Hollingsworth, S.A.; Dror, R.O. Molecular dynamics simulation for all. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [Green Version]
- Pachahara, S.K.; Subbalakshmi, C.; Nagaraj, R. Formation of nanostructures by peptides. Curr. Protein Pept. Sci. 2017, 18, 920–938. [Google Scholar] [CrossRef]
- Mendes, A.C.; Baran, E.T.; Reis, R.L.; Azevedo, H.S. Self-assembly in nature: Using the principles of nature to create complex nanobiomaterials. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2013, 5, 582–612. [Google Scholar] [CrossRef]
- Lee, O.S.; Stupp, S.I.; Schatz, G.C. Atomistic molecular dynamics simulations of peptide amphiphile self-assembly into cylindrical nanofibers. J. Am. Chem. Soc. 2011, 133, 3677–3683. [Google Scholar] [CrossRef]
- Lehninger, A.L. Biochemistry—The Molecular Basis of Cell Structure and Function; Worth Publishers, Inc.: New York, NY, USA, 1972. [Google Scholar]
- Bystrov, V. Computer Simulation Nanostructures: Bioferroelectric Amino Acids. Bioferroelectricity: Peptide Nanotubes and Thymine Nucleobase; LAP LAMBERT Academic Publishing: Riga, Latvia, 2020; ISBN 978-620-2-91926-5. [Google Scholar]
- Levin, A.; Hakala, T.A.; Schnaider, L.; Bernardes, G.J.L.; Gazit, E.; Knowles, T.P.J. Biomimetic peptide self-assembly for functional materials. Nat. Rev. Chem. 2020, 4, 615–634. [Google Scholar] [CrossRef]
- Holmstedt, B.; Frank, H.; Testa, B. (Eds.) Chirality and Biological Activity; Liss: New York, NY, USA, 1990. [Google Scholar]
- Tverdislov, V.A. Chirality as a primary switch of hierarchical levels in molecular biological systems. Biophysics 2013, 58, 128–132. [Google Scholar] [CrossRef]
- Tverdislov, V.A.; Malyshko, E.V. On regularities in the spontaneous formation of structural hierarchies in chiral systems of nonliving and living matter. Phys.-Uspekhi 2019, 62, 354–363. [Google Scholar] [CrossRef]
- Bystrov, V.S.; Zelenovskiy, P.S.; Nuraeva, A.S.; Kopyl, S.; Zhulyabina, O.A.; Tverdislov, V.A. Chiral peculiar properties of self-organization of diphenylalanine peptide nanotubes: Modeling of structure and properties. Math. Biol. Bioinform. 2019, 14, 94–124. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Mao, K.; Chen, S.; Zhu, H. Chirality Effects in Peptide Assembly Structures. Front. Bioeng. Biotechnol. 2021, 9, 703004. [Google Scholar] [CrossRef]
- Lemak, A.S.; Balabaev, N.K. A Comparison Between Collisional Dynamics and Brownian Dynamics. Mol. Simul. 1995, 15, 223–231. [Google Scholar] [CrossRef]
- Lemak, A.S.; Balabaev, N.K. Molecular dynamics simulation of a polymer chain in solution by collisional dynamics method. J. Comput. Chem. 1996, 17, 1685–1695. [Google Scholar] [CrossRef]
- Likhachev, I.; Bystrov, V. Assembly of a phenylalanine nanotube with a molecular dynamic manipulator. Math. Biol. Bioinform. 2021, 16, 244–255. [Google Scholar] [CrossRef]
- Ghadiri, M.R.; Granja, J.; Milligan, R.A.; McRee, D.E.; Khazanovich, N. Self assembling organic nanotubes based on cyclic peptide architecture. Nature 1993, 366, 324–327. [Google Scholar] [CrossRef]
- Ghadiri, M.R.; Kobayashi, K.; Granja, J.R.; Chadha, R.K.; McRee, D.E. The structural and thermodynamic basis for the formation of self-assembled peptide nanotubes. Angew. Chem. Int. Ed. Engl. 1995, 34, 93–95. [Google Scholar] [CrossRef]
- Görbitz, C.H. Nanotube formation by hydrophobic dipeptides. Chem. Eur. J. 2001, 7, 5153–5159. [Google Scholar] [CrossRef]
- Görbitz, C.H. The structure of nanotubes formed by diphenylalanine, the core recognition motif of Alzheimer’s b-amyloid polypeptide. Chem. Commun. 2006, 1, 2332–2334. [Google Scholar] [CrossRef]
- Görbitz, C.H. Hydrophobic dipeptides: The final piece in the puzzle. Acta Cryst. 2018, B74, 311–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reches, M.; Gazit, E. Controlled patterning of aligned self-assembled peptide nanotubes. Nat. Nanotechnol. 2006, 1, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Sedman, V.L.; Adler-Abramovich, L.; Allen, S.; Gazit, E.; Tendler, S.J.B. Direct observation of the release of phenylalanine from diphenilalanine nanotubes. J. Am. Chem. Soc. 2006, 128, 6903–6908. [Google Scholar] [CrossRef]
- Scanlon, S.; Aggeli, A. Self-assembling peptide nanotubes. Nano Today 2008, 3, 22–30. [Google Scholar] [CrossRef]
- Shklovsky, J.; Beker, P.; Amdursky, N.; Gazit, E.; Rosenman, G. Bioinspired peptide nanotubes: Deposition technology and physical properties. Mater. Sci. Eng. B. 2010, 169, 62–66. [Google Scholar] [CrossRef]
- Adler-Abramovich, L.; Aronov, D.; Beker, P.; Yevnin, M.; Stempler, S.; Buzhansky, L.; Rosenman, G.; Gazit, E. Self-assembled arrays of peptide nanotubes by vapour deposition. Nat. Nanotechnol. 2009, 4, 849–854. [Google Scholar] [CrossRef] [PubMed]
- Adler-Abramovich, L.; Gazit, E. The physical properties of supramolecular peptide assemblies: From building block association to technological applications. Chem. Soc. Rev. 2014, 43, 6881–6893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kol, N.; Adler-Abramovich, L.; Barlam, D.; Shneck, R.Z.; Gazit, E.; Rousso, I. Self-assembled peptide nanotubes are uniquely rigid bioinspired supramolecular structures. Nano Lett. 2005, 5, 1343–1346. [Google Scholar] [CrossRef]
- Zelenovskiy, P.; Kornev, I.; Vasilev, S.; Kholkin, A. On the origin of the great rigidity of self-assembled diphenylalanine nanotubes. Phys. Chem. Chem. Phys. 2016, 18, 29681–29685. [Google Scholar] [CrossRef] [Green Version]
- Adler-Abramovich, L.; Reches, M.; Sedman, V.; Allen, S.; Tendler, S.J.B.; Gazit, E. Thermal and chemical stability of diphenylalanine peptide nanotubes: Implications for nanotechnological applications. Langmuir 2006, 22, 1313–1320. [Google Scholar] [CrossRef]
- Akdim, B.; Pachter, R.; Naik, R.R. Self-assembled peptide nanotubes as electronic materials: An evaluation from first-principles calculations. Appl. Phys. Lett. 2015, 106, 183707. [Google Scholar] [CrossRef]
- Amdursky, N.; Molotskii, M.; Aronov, D.; Adler-Abramovich, L.; Gazit, E.; Rosenman, G. Blue luminescence based on quantum confinement at peptide nanotubes. Nano Lett. 2009, 9, 3111–3115. [Google Scholar] [CrossRef]
- Handelman, A.; Lavrov, S.; Kudryavtsev, A.; Khatchatouriants, A.; Rosenberg, Y.; Mishina, E.; Rosenman, G. Nonlinear Optical Bioinspired Peptide Nanostructures. Adv. Opt. Mater. 2013, 1, 875–884. [Google Scholar] [CrossRef]
- Nikitin, T.; Kopyl, S.; Shur, V.Y.; Kopelevich, Y.V.; Kholkin, A.L. Low-temperature photoluminescence in self-assembled diphenylalanine microtubes. Phys. Lett. A 2016, 380, 1658–1662. [Google Scholar] [CrossRef]
- Bystrov, V.S. Photo-Ferroelectricity in di-phenylalanine peptide nanotube. Comput. Condens. Matter 2018, 14, 94–100. [Google Scholar] [CrossRef]
- Esin, A.; Baturin, I.; Nikitin, T.; Vasilev, S.; Salehli, F.; Shur, V.Y.; Kholkin, A.L. Pyroelectric effect and polarization instability in self-assembled diphenylalanine microtubes. Appl. Phys. Lett. 2016, 109, 142902. [Google Scholar] [CrossRef]
- Kholkin, A.; Amdursky, N.; Bdikin, I.; Gazit, E.; Rosenman, G. Strong Piezoelectricity in Bioinspired Peptide Nanotubes. ACS Nano 2010, 4, 610–614. [Google Scholar] [CrossRef]
- Nguyen, V.; Zhu, R.; Jenkins, K.; Yang, R. Self-assembly of diphenylalanine peptide with controlled polarization for power generation. Nat. Commun. 2016, 7, 13566. [Google Scholar] [CrossRef] [Green Version]
- Bdikin, I.; Bystrov, V.S.; Delgadillo, I.; Gracio, J.; Kopyl, S.; Wojtas, M.; Mishina, E.; Sigov, A.; Kholkin, A.L. Polarization switching and patterning in self-assembled peptide tubular structures. J. Appl. Phys. 2012, 111, 074104. [Google Scholar] [CrossRef] [Green Version]
- Bystrov, V.S. Computer Simulation Nanostructures: Bioferroelectric Peptide Nanotubes. Bioferroelectricity: Peptide Nanotubes; LAP Lambert Academic Publishing: Saarbruecken, Germay, 2016. [Google Scholar]
- Bystrov, V.S.; Zelenovskiy, P.S.; Nuraeva, A.S.; Kopyl, S.; Zhulyabina, O.A.; Tverdislov, V.A. Molecular modeling and computational study of the chiral-dependent structures and properties of the self-assembling diphenylalanine peptide nanotubes. J. Mol. Model. 2019, 25, 199. [Google Scholar] [CrossRef]
- Zelenovskiy, P.S.; Nuraeva, A.S.; Kopyl, S.; Arkhipov, S.G.; Vasilev, S.G.; Bystrov, V.S.; Gruzdev, D.A.; Waliszek, M.; Svitlyk, V.; Shur, V.Y.; et al. Chirality-Dependent Growth of Self-Assembled Diphenylalanine Microtubes. Cryst. Growth Des. 2019, 19, 6414–6421. [Google Scholar] [CrossRef]
- Nuraeva, A.; Vasilev, S.; Vasileva, D.; Zelenovskiy, P.; Chezganov, D.; Esin, A.; Kholkin, A.L.; Kopyl, S.; Romanyuk, K.; Shur, V.Y. Evaporation-Driven Crystallization of Diphenylalanine Microtubes for Microelectronic Applications. Cryst. Growth Des. 2016, 16, 1472–1479. [Google Scholar] [CrossRef]
- Bystrov, V.S.; Coutinho, J.; Zelenovskiy, P.; Nuraeva, A.; Kopyl, S.; Zhulyabina, O.; Tverdislov, V. Structures and properties of the self-assembling diphenylalanine peptide nanotubes containing water molecules: Modeling and data analysis. Nanomaterials 2020, 10, 1999. [Google Scholar] [CrossRef] [PubMed]
- Bystrov, V.S.; Coutinho, J.; Zhulyabina, J.A.; Kopyl, S.A.; Zelenovskiy, P.S.; Nuraeva, A.S.; Tverdislov, V.A.; Filippov, S.V.; Kholkin, A.L.; Shur, V.Y. Modelling and physical properties of diphenylalanine peptide nanotubes containing water molecules. Ferroelectrics 2021, 574, 78–91. [Google Scholar] [CrossRef]
- German, H.W.; Uyaver, S.; Hansmann, U.H.E. Self-Assembly of Phenylalanine-Based Molecules. J. Phys. Chem. A 2015, 119, 1609–1615. [Google Scholar] [CrossRef]
- Adler-Abramovich, L.; Vaks, L.; Carny, O.; Trudler, D.; Magno, A.; Caflisch, A.; Frenkel, D.; Gazit, E. Phenylalanine assembly into toxic fibrils suggests amyloid etiology in phenylketonuria. Nat. Chem. Biol. 2012, 8, 701–706. [Google Scholar] [CrossRef]
- Bystrov, V.; Sidorova, A.; Lutsenko, A.; Shpigun, D.; Malyshko, E.; Nuraeva, A.; Zelenovskiy, P.; Kopyl, S.; Kholkin, A. Modeling of Self-Assembled Peptide Nanotubes and Determination of Their Chirality Sign Based on Dipole Moment Calculations. Nanomaterials 2021, 11, 2415. [Google Scholar] [CrossRef]
- Sidorova, A.; Bystrov, V.; Lutsenko, A.; Shpigun, D.; Belova, E.; Likhachev, I. Quantitative Assessment of Chirality of Protein Secondary Structures and Phenylalanine Peptide Nanotubes. Nanomaterials 2021, 11, 3299. [Google Scholar] [CrossRef]
- Likhachev, I.V.; Balabaev, N.K.; Galzitskaya, O.V. Elastic and Non-elastic Properties of Cadherin Ectodomain: Comparison with Mechanical System. In Advances in Computer Science for Engineering and Education II. ICCSEEA 2019; Hu, Z., Petoukhov, S., Dychka, I., He, M., Eds.; Advances in Intelligent Systems and Computing; Springer: Berlin/Heidelberg, Germany, 2020; pp. 555–566. [Google Scholar] [CrossRef]
- Glyakina, A.V.; Likhachev, I.V.; Balabaev, N.K.; Galzitskaya, O.V. Comparative mechanical unfolding studies of spectrin domains R15, R16 and R17. J. Struct. Biol. 2018, 201, 162–170. [Google Scholar] [CrossRef]
- Likhachev, I.V.; Balabaev, N.K. Trajectory analyzer of molecular dynamics. Mat. Biol. Bioinform. 2007, 2, 120–129. [Google Scholar] [CrossRef]
- Likhachev, I.V.; Balabaev, N.K.; Galzitskaya, O.V. Available Instruments for Analyzing Molecular Dynamics Trajectories. Open Biochem. J. 2016, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Cieplak, P.; Kollman, P.A. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000, 21, 1049–1074. [Google Scholar] [CrossRef]
- HyperChem 8. Tools for Molecular Modeling; Professional Edition for Windows AC Release 8.0 USB (on CD); Hypercube. Inc.: Gainesville, FL, USA, 2011.
- Stewart, J.J.P. Optimization of Parameters for Semiempirical Methods. I. Method. J. Comput. Chem. 1989, 10, 209–220. [Google Scholar] [CrossRef] [Green Version]
- Stewart, J.J.P. Optimization of Parameters for Semiempirical Methods. II. Applications. J. Comput. Chem. 1989, 10, 221–264. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. [Google Scholar] [CrossRef] [Green Version]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods VI: More modifications to the NDDO approximations and re-optimization of parameters. J. Mol. Model. 2013, 1, 1–32. [Google Scholar] [CrossRef] [Green Version]
- Stewart, J.J.P. MOPAC: A semiempirical molecular orbital program. J. Comput. Aided Mol. Des. 1990, 4, 1–105. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Stewart Computational Chemistry. MOPAC2016. Available online: http://openmopac.net/MOPAC2016.html (accessed on 30 January 2022).
- Stewart, J.J.P. Accuracy of PM7 and PM6-D3H4. Available online: http://openmopac.net/PM7_and_PM6-D3H4_accuracy/Accuracy%20of%20PM7%20and%20PM6-D3H4.html (accessed on 30 January 2022).
- Rocha, G.B.; Freire, R.O.; Simas, A.M.; Stewart, J.J.P. RM1: A reparameterization of AM1 for H, C, N, O, P, S, F, Cl, Br, and I. J. Comput. Chem. 2006, 27, 1101–1111. [Google Scholar] [CrossRef]
- Szabo, A.; Ostlund, N. Modern Quantum Chemistry; Macmillan: New York, NY, USA, 1985. [Google Scholar]
- Clark, T.A. Handbook of Computational Chemistry; John Wiley and Sons: New York, NY, USA, 1985. [Google Scholar]
- Ramachandran, K.I.; Gopakumar, D.; Namboori, K. Semiempirical Methods. In Computational Chemistry and Molecular Modeling; Springer: Berlin/Heidelberg, Germany, 2008. [Google Scholar] [CrossRef]
- Van der Kamp, M.W. Semiempirical Quantum Mechanical Methods. In Encyclopedia of Biophysics; Roberts, G.C.K., Ed.; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar] [CrossRef]
- Pople, J.A.; Beveridge, D.L. Approximate Molecular Orbital Theory; McGraw-Hill: New York, NY, USA, 1970. [Google Scholar]
- Sidorova, A.E.; Lutsenko, A.O.; Shpigun, D.K.; Malyshko, E.V.; Tverdislov, V.A. Methods for Determining the Chirality sign of the Helical and Superhelical Protein Structures. Biophysics 2021, 66, 421. [Google Scholar] [CrossRef]
- Sidorova, A.E.; Malyshko, E.V.; Lutsenko, A.O.; Shpigun, D.K.; Bagrova, O.E. Protein Helical Structures: Defining Handedness and Localization Features. Symmetry 2021, 13, 879. [Google Scholar] [CrossRef]
- Lu, H.; Schulten, K. Steered molecular dynamics simulations of force-induced protein domain unfolding. Proteins Struct. Funct. Bioinform. 1999, 35, 453–463. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [Green Version]
- Tian, F.; Li, G.; Zheng, B.; Liu, Y.; Shi, S.; Deng, Y.; Zheng, P. Verification of sortase for protein conjugation by singlemolecule force spectroscopy and molecular dynamics simulations. Chem. Commun. 2020, 56, 3943–3946. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Tong, B.; Sun, L.; Shi, S.; Zheng, B.; Wang, Z.; Dong, X.; Zheng, P. N501Y mutation of spike protein in SARS-CoV-2 strengthens its binding to receptor ACE2. Elife Sci. 2021, 10, e69091. [Google Scholar] [CrossRef]
- Glyakina, A.V.; Likhachev, I.V.; Balabaev, N.K.; Galzitskaya, O.V. Right- and left-handed three-helix proteins. II. Similarity and differences in mechanical unfolding of proteins. Proteins 2014, 82, 90–102. [Google Scholar] [CrossRef]
- Glyakina, A.V.; Likhachev, I.V.; Balabaev, N.K.; Galzitskaya, O.V. Mechanical stability analysis of the protein L immunoglobulin-binding domain by full alan;ne screening using molecular dynamics simulations. Biotechnol. J. 2015, 10, 386–394. [Google Scholar] [CrossRef]










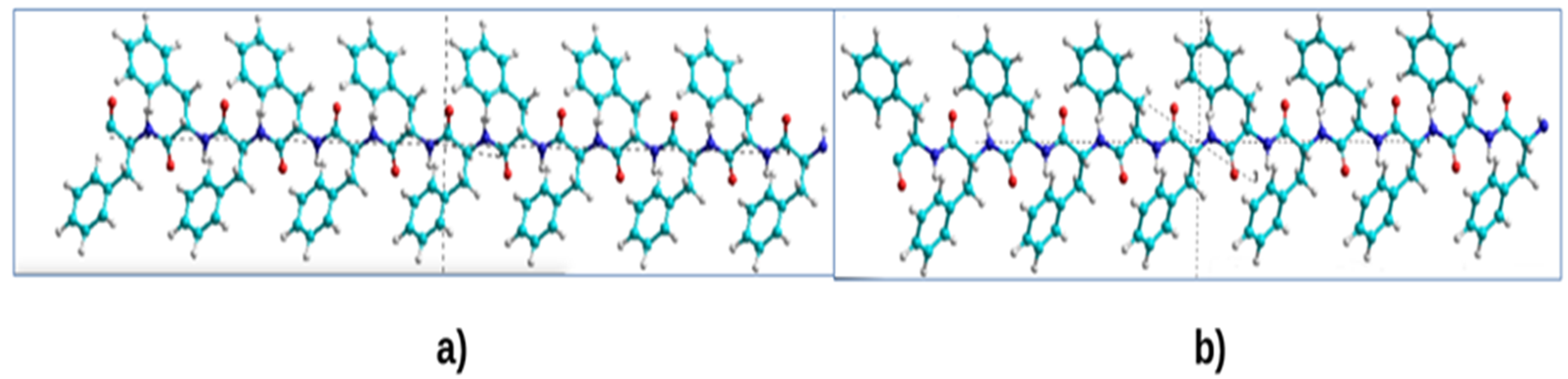{kind=link}
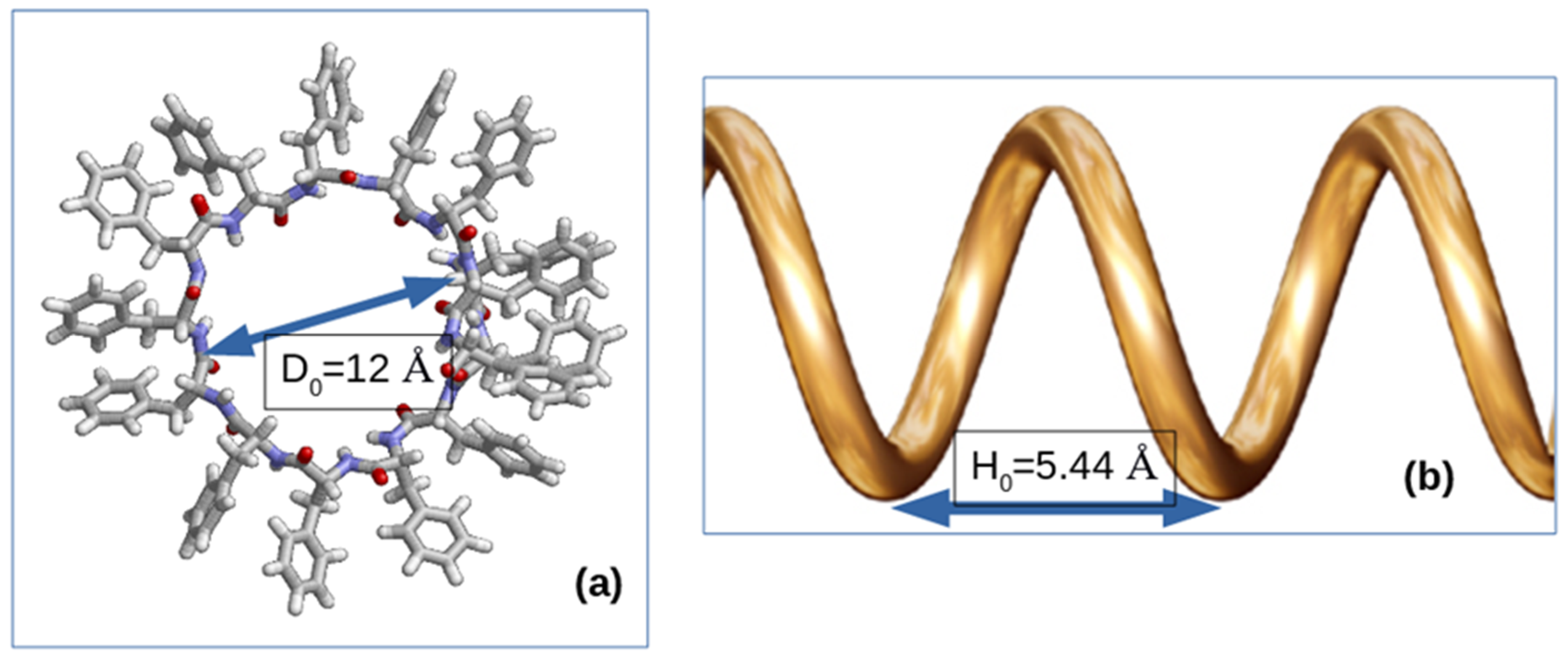{kind=link}
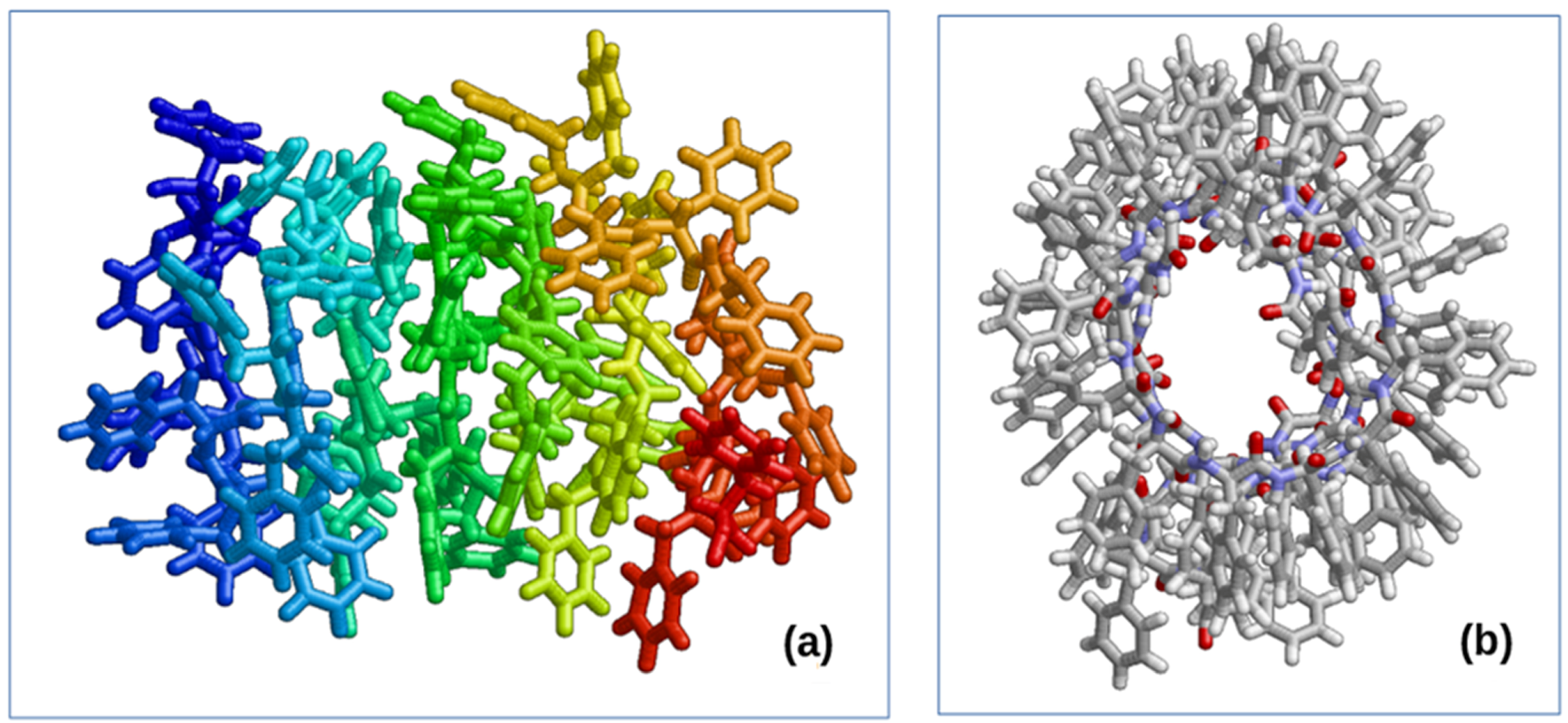{kind=link}
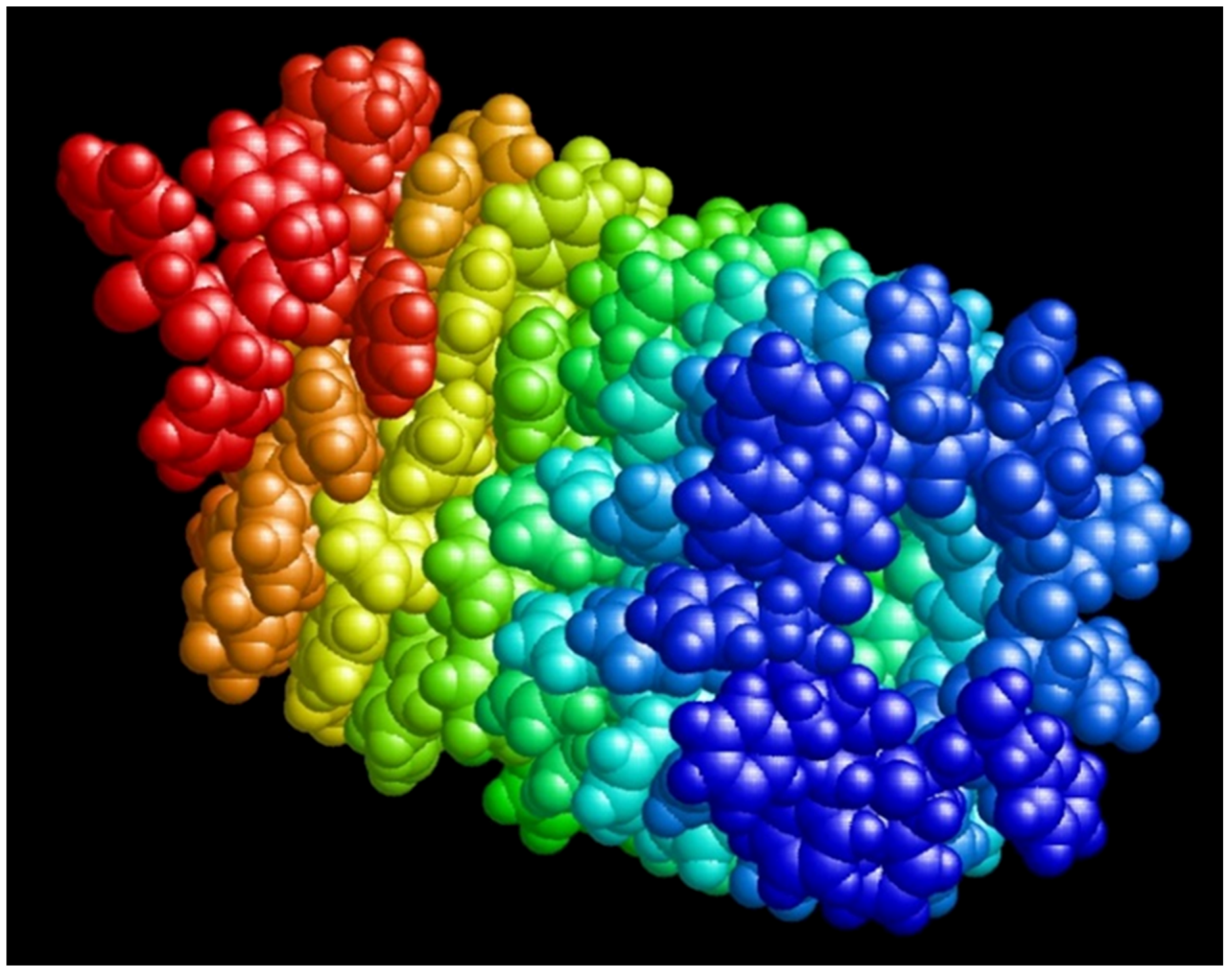{kind=link}
{kind=link}
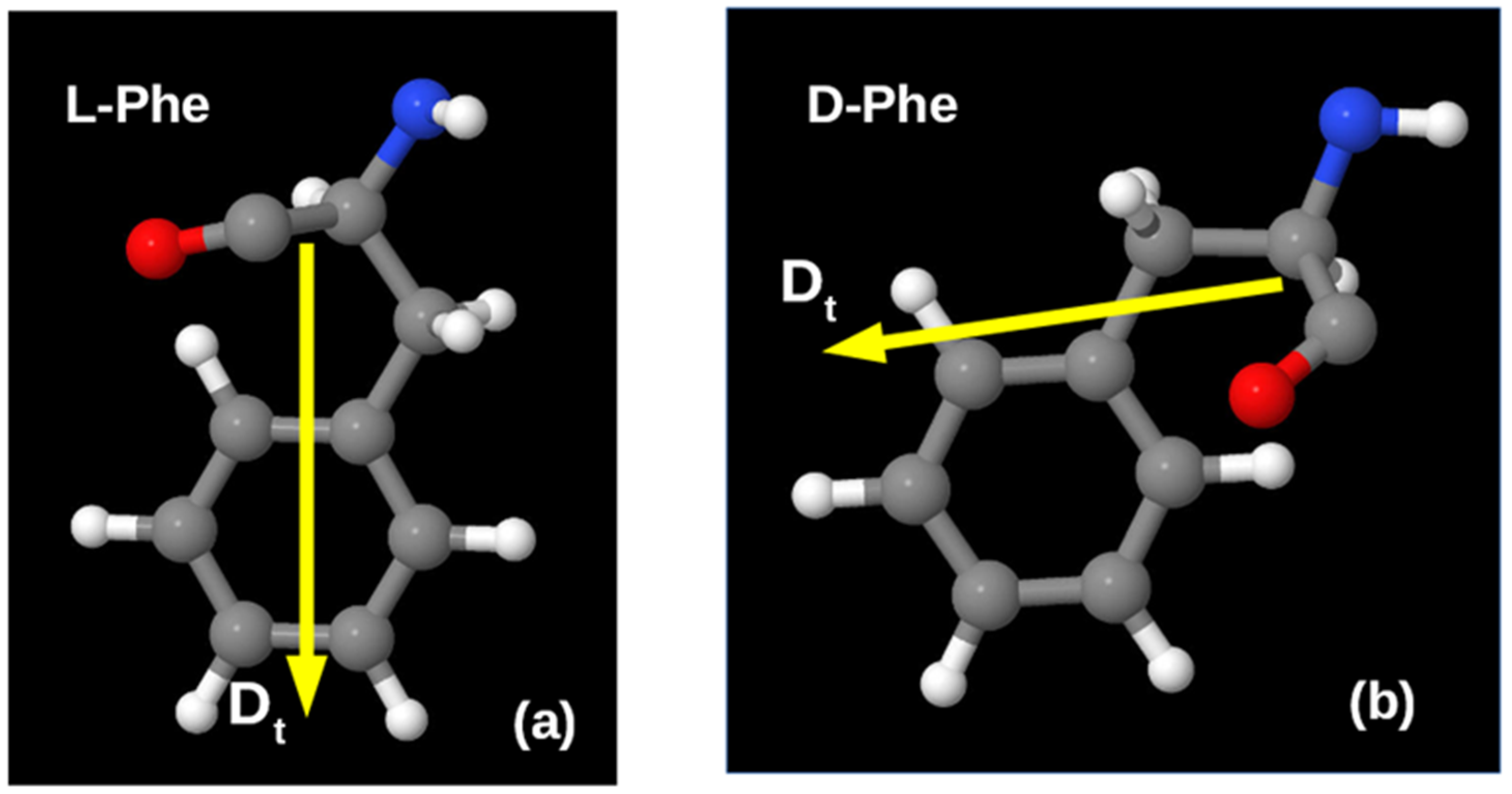{kind=link}
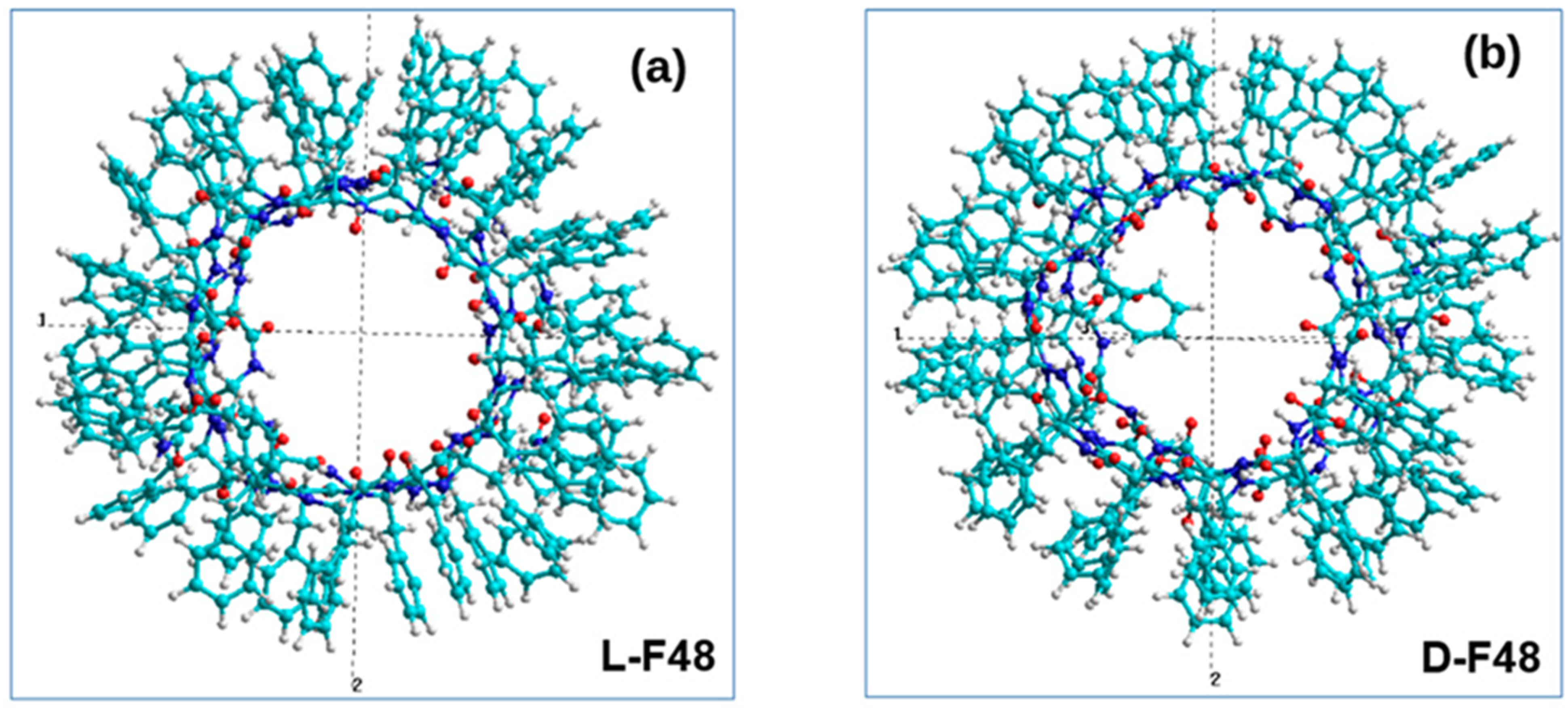{kind=link}
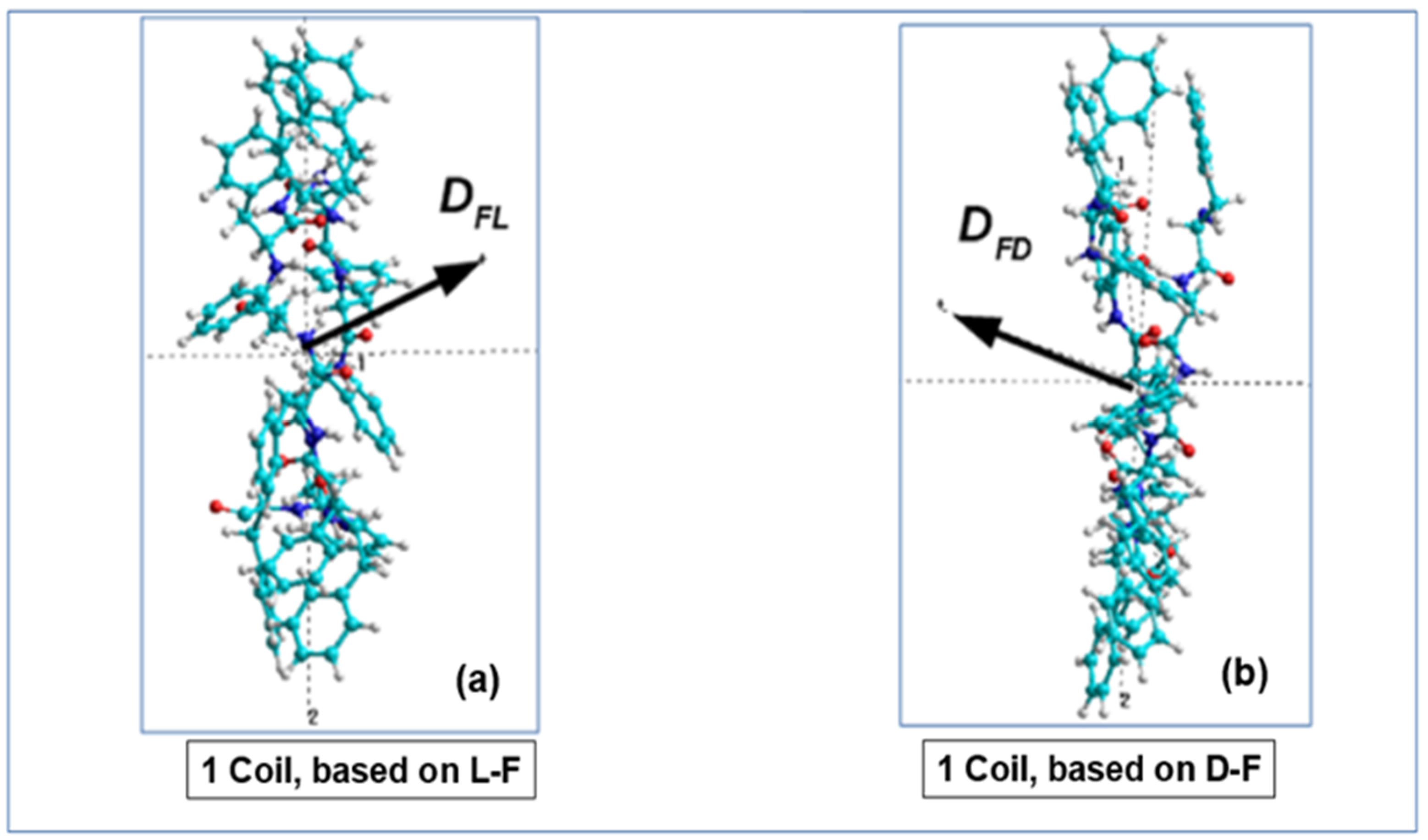{kind=link}
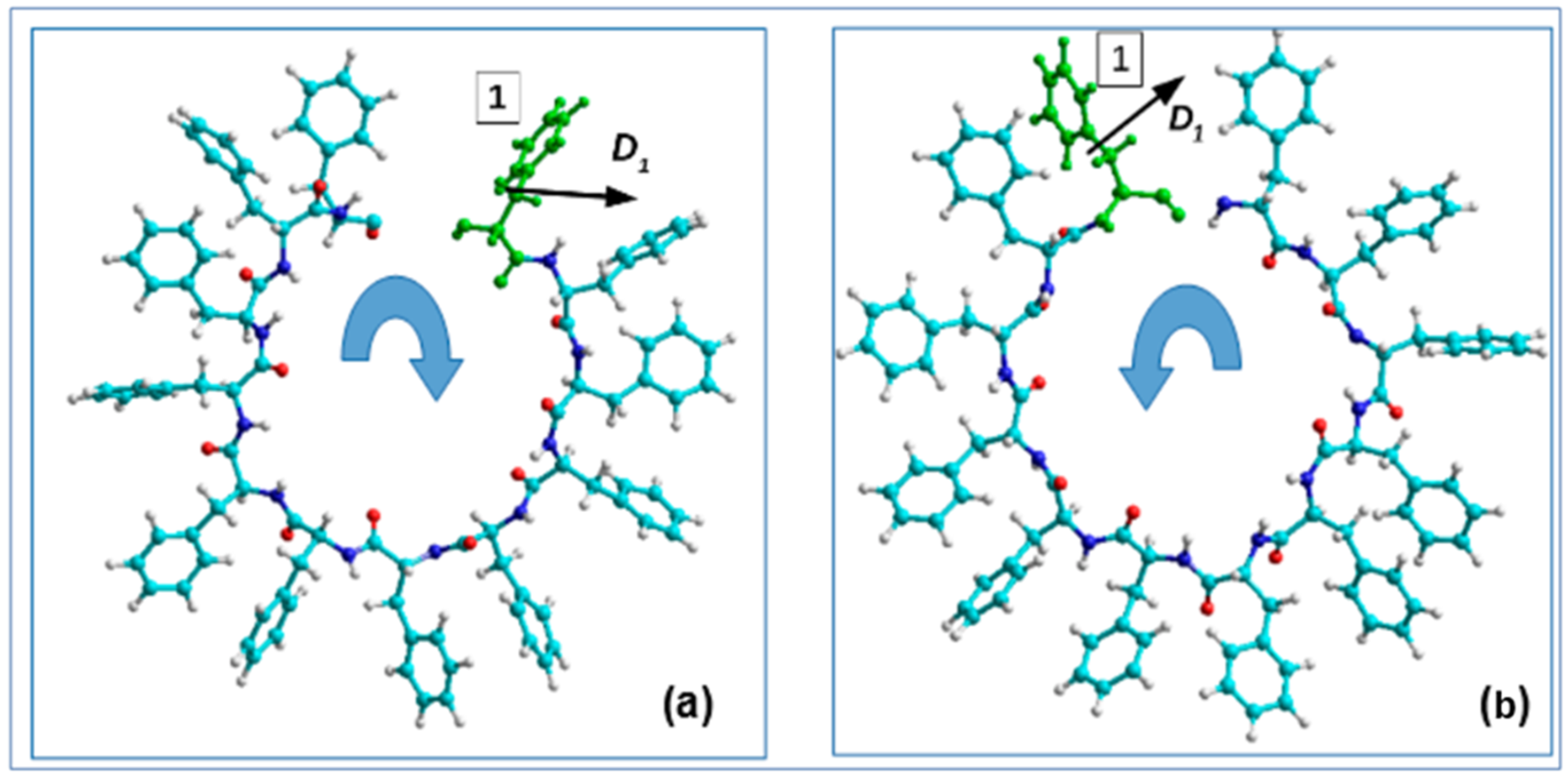{kind=link}
| Implementation Number | D-Monomer | L-Monomer | ||
|---|---|---|---|---|
| Number of Left Helices | Number of Right Helices | Number of Left Helices | Number of Right Helices | |
| 1 | 0.5 | 0.5 | 1 | |
| 2 | 0.5 | 0.5 | 1 | |
| 3 | 1 | 1 | ||
| 4 | 1 | 1 | ||
| 5 | 1 | 0.2 | 0.8 | |
| 6 | 1 | 0.5 | 0.5 | |
| 7 | 1 | 1 | ||
| 8 | 0.5 | 0.5 | 1 | |
| 9 | 1 | 1 | ||
| 10 | 1 | 0.2 | 0.8 | |
| 11 | 0.2 | 0.8 | 1 | |
| 12 | 1 | 1 | ||
| 13 | 1 | 1 | ||
| 14 | 1 | 1 | ||
| 15 | 1 | 1 | ||
| 16 | 1 | 1 | ||
| 17 | 1 | 1 | ||
| 18 | 1 | 1 | ||
| 19 | 1 | 1 | ||
| 20 | 1 | 1 | ||
| 21 | 0.5 | 0.5 | 0.2 | 0.8 |
| 22 | 0.5 | 0.5 | 1 | |
| 23 | 1 | 1 | ||
| 24 | 1 | 1 | ||
| 25 | 1 | 1 | ||
| 26 | 1 | 1 | ||
| 27 | 1 | 0.1 | 0.9 | |
| 28 | 0.5 | 0.5 | 1 | |
| 29 | 1 | 1 | ||
| 30 | 0.5 | 0.5 | 1 | |
| 31 | 0.5 | 0.5 | 1 | |
| 32 | 1 | 0.2 | 0.8 | |
| Total | 26.2 | 5.8 | 3.4 | 28.6 |
| # | Method | L-F | D-F | ||
|---|---|---|---|---|---|
| SP/1SCF | Opt | SP/1SC | Opt | ||
| 1 | PM3 | 1.978 | 1.981 | 1.831 | 1.512 |
| 2 | PM7 | 3.167 | 2.684 | 2.756 | 3.323 |
| 3 | PM6-D3H4 | 2.973 | 2.578 | 2.608 | 2.374 |
| 4 | RM1 | 2.470 | 2.172 | 2.252 | 4.123 |
| Initial Chirality Type of F Molecule | L-F | D-F |
|---|---|---|
| Calculating method | RM1 RHF | RM1 RHF |
| Computed data ctotal, debye3 | 20.266 | −19.647 |
| Normalized data cnorm | 1.219 | −0.674 |
| Chirality PNT sign | Positive | Negative |
| Chirality PNT symbol | D | L |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bystrov, V.; Likhachev, I.; Sidorova, A.; Filippov, S.; Lutsenko, A.; Shpigun, D.; Belova, E. Molecular Dynamics Simulation Study of the Self-Assembly of Phenylalanine Peptide Nanotubes. Nanomaterials 2022, 12, 861. https://doi.org/10.3390/nano12050861
Bystrov V, Likhachev I, Sidorova A, Filippov S, Lutsenko A, Shpigun D, Belova E. Molecular Dynamics Simulation Study of the Self-Assembly of Phenylalanine Peptide Nanotubes. Nanomaterials. 2022; 12(5):861. https://doi.org/10.3390/nano12050861
Chicago/Turabian StyleBystrov, Vladimir, Ilya Likhachev, Alla Sidorova, Sergey Filippov, Aleksey Lutsenko, Denis Shpigun, and Ekaterina Belova. 2022. "Molecular Dynamics Simulation Study of the Self-Assembly of Phenylalanine Peptide Nanotubes" Nanomaterials 12, no. 5: 861. https://doi.org/10.3390/nano12050861