
Safety Profile of Rapamycin Perfluorocarbon Nanoparticles for Preventing Cisplatin-Induced Kidney Injury

 ,
,
Abstract
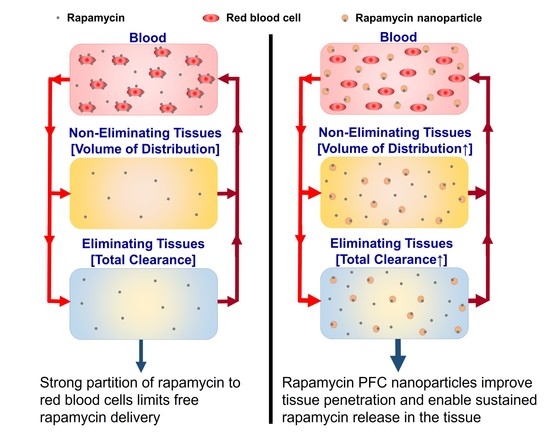
:
1. Introduction
2. Materials and Methods
2.1. Rapamycin Perfluorocarbon (PFC) Formulation and Characterization
2.2. Western Blot
2.3. Cell Viability Evaluation
2.4. Animal Procedures
2.4.1. Evaluation of Therapeutics in Mitigating Cisplatin Induce Acute Kidney Injury
2.4.2. Single Intravenous Administration of Unformulated Free Rapamycin and Rapamycin-Loaded Nanoparticles for Pharmacokinetic Analysis
2.4.3. Effects of Free Rapamycin or Rapamycin Nanoparticles in Immune Responses
2.4.4. Blood Test
2.4.5. Blood Urea Nitrogen (BUN) Test
2.5. LC/MS/MS Assay of Rapamycin in Nanoparticles, Blood, and Tissue
2.5.1. Sample Processing
2.5.2. Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS)
2.5.3. Calibration and Quantification
2.6. Pharmacokinetic Analysis
2.7. Transthoracic Echocardiography and Data Analyses
2.8. Splenocytes Isolation and Stimulation
2.9. Cytokine Assays and Cell Proliferation
2.10. Flow Cytometry
2.11. Anti-Nanoparticle Antibody Formation
2.12. Statistics
3. Results
3.1. Physical Characterization of Rapamycine Perfluorocarbon (PFC) Nanoparticles
3.2. Rapamycin PFC Nanoparticles Provide Therapeutic Benefits in Cisplatin-Induced Acute Kidney Injury
3.3. Neither Free Rapamycin Nor Rapamycin PFC Nanoparticles Affect Cell Viability
3.4. Differential Kinetics and Distribution of Free Rapamycin and Rapamycin PFC Nanoparticles
3.5. Safety Profile after One Dose of Administration
3.6. Effects of Chronic Treatment with Rapamycin PFC Nanoparticles on Immune Responses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Miller, K.D.; Siegel, R.L.; Lin, C.C.; Mariotto, A.B.; Kramer, J.L.; Rowland, J.H.; Stein, K.D.; Alteri, R.; Jemal, A. Cancer treatment and survivorship statistics, 2016. CA Cancer J. Clin. 2016, 66, 271–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NCI. Cancer Statistics; NCI: Rockville, MD, USA, 2019. [Google Scholar]
- Hull, M.C.; Morris, C.G.; Pepine, C.J.; Mendenhall, N.P. Valvular dysfunction and carotid, subclavian, and coronary artery disease in survivors of hodgkin lymphoma treated with radiation therapy. JAMA 2003, 290, 2831–2837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oeffinger, K.C.; Mertens, A.C.; Sklar, C.A.; Kawashima, T.; Hudson, M.M.; Meadows, A.T.; Friedman, D.L.; Marina, N.; Hobbie, W.; Kadan-Lottick, N.S.; et al. Chronic health conditions in adult survivors of childhood cancer. N. Engl. J. Med. 2006, 355, 1572–1582. [Google Scholar] [CrossRef]
- Moslehi, J. The cardiovascular perils of cancer survivorship. N. Engl. J. Med. 2013, 368, 1055–1056. [Google Scholar] [CrossRef] [Green Version]
- Denlinger, C.S.; Sanft, T.; Baker, K.S.; Broderick, G.; Demark-Wahnefried, W.; Friedman, D.L.; Goldman, M.; Hudson, M.; Khakpour, N.; King, A.; et al. Survivorship, Version 2.2018, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2018, 16, 1216–1247. [Google Scholar] [CrossRef]
- Patane, S. Cardiotoxicity: Cisplatin and long-term cancer survivors. Int. J. Cardiol. 2014, 175, 201–202. [Google Scholar] [CrossRef]
- Al-Mamgani, A.; de Ridder, M.; Navran, A.; Klop, W.M.; de Boer, J.P.; Tesselaar, M.E. The impact of cumulative dose of cisplatin on outcome of patients with head and neck squamous cell carcinoma. Eur. Arch. Otorhinolaryngol. 2017, 274, 3757–3765. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, P.A. Anthracycline cardiotoxicity: An update on mechanisms, monitoring and prevention. Heart 2018, 104, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, B.; Vancamp, L.; Krigas, T. Inhibition of Cell Division in Escherichia Coli by Electrolysis Products from a Platinum Electrode. Nature 1965, 205, 698–699. [Google Scholar] [CrossRef]
- NCI. The “Accidental” Cure—Platinum-Based Treatment for Cancer: The Discovery of Cisplatin; NCI: Rockville, MD, USA, 2014. [Google Scholar]
- Hardaker, W.T., Jr.; Stone, R.A.; McCoy, R. Platinum nephrotoxicity. Cancer 1974, 34, 1030–1032. [Google Scholar] [CrossRef]
- Ward, J.M.; Young, D.M.; Fauvie, K.A.; Wolpert, M.K.; Davis, R.; Guarino, A.M. Comparative nephrotoxicity of platinum cancer chemotherapeutic agents. Cancer Treat. Rep. 1976, 60, 1675–1678. [Google Scholar]
- Gonzales-Vitale, J.C.; Hayes, D.M.; Cvitkovic, E.; Sternberg, S.S. The renal pathology in clinical trials of cis-platinum (II) diamminedichloride. Cancer 1977, 39, 1362–1371. [Google Scholar] [CrossRef]
- Lebwohl, D.; Canetta, R. Clinical development of platinum complexes in cancer therapy: An historical perspective and an update. Eur. J. Cancer 1998, 34, 1522–1534. [Google Scholar] [CrossRef]
- Lippman, A.J.; Helson, C.; Helson, L.; Krakoff, I.H. Clinical trials of cis-diamminedichloroplatinum (NSC-119875). Cancer Chemother. Rep. 1973, 57, 191–200. [Google Scholar]
- Chawla, L.S.; Eggers, P.W.; Star, R.A.; Kimmel, P.L. Acute kidney injury and chronic kidney disease as interconnected syndromes. N. Engl. J. Med. 2014, 371, 58–66. [Google Scholar] [CrossRef] [Green Version]
- Wu, V.C.; Wu, C.H.; Huang, T.M.; Wang, C.Y.; Lai, C.F.; Shiao, C.C.; Chang, C.H.; Lin, S.L.; Chen, Y.Y.; Chen, Y.M.; et al. Long-term risk of coronary events after AKI. J. Am. Soc. Nephrol. 2014, 25, 595–605. [Google Scholar] [CrossRef] [Green Version]
- Bucaloiu, I.D.; Kirchner, H.L.; Norfolk, E.R.; Hartle, J.E., 2nd; Perkins, R.M. Increased risk of death and de novo chronic kidney disease following reversible acute kidney injury. Kidney Int. 2012, 81, 477–485. [Google Scholar] [CrossRef] [Green Version]
- Skinner, R.; Parry, A.; Price, L.; Cole, M.; Craft, A.W.; Pearson, A.D. Persistent nephrotoxicity during 10-year follow-up after cisplatin or carboplatin treatment in childhood: Relevance of age and dose as risk factors. Eur. J. Cancer 2009, 45, 3213–3219. [Google Scholar] [CrossRef]
- Sehgal, S.N.; Baker, H.; Vezina, C. Rapamycin (AY-22,989), a new antifungal antibiotic. II. Fermentation, isolation and characterization. J. Antibiot. 1975, 28, 727–732. [Google Scholar] [CrossRef] [Green Version]
- Vezina, C.; Kudelski, A.; Sehgal, S.N. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J. Antibiot. 1975, 28, 721–726. [Google Scholar] [CrossRef]
- Pritchard, D.I. Sourcing a chemical succession for cyclosporin from parasites and human pathogens. Drug Discov. Today 2005, 10, 688–691. [Google Scholar] [CrossRef]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Y.; Wu, J.; Yang, R.; Zhang, L.; Ma, Z. Rapamycin-induced autophagy plays a pro-survival role by enhancing up-regulation of intracellular ferritin expression in acute lymphoblastic leukemia. Exp. Oncol. 2020, 42, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Ravikumar, B.; Floto, R.A.; Rubinsztein, D.C. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ. 2009, 16, 46–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dan, H.C.; Cooper, M.J.; Cogswell, P.C.; Duncan, J.A.; Ting, J.P.; Baldwin, A.S. Akt-dependent regulation of NF-{kappa}B is controlled by mTOR and Raptor in association with IKK. Genes Dev. 2008, 22, 1490–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozkok, A.; Ravichandran, K.; Wang, Q.; Ljubanovic, D.; Edelstein, C.L. NF-kappaB transcriptional inhibition ameliorates cisplatin-induced acute kidney injury (AKI). Toxicol. Lett. 2016, 240, 105–113. [Google Scholar] [CrossRef]
- Shen, W.; Jia, N.; Miao, J.; Chen, S.; Zhou, S.; Meng, P.; Zhou, X.; Tang, L.; Zhou, L. Penicilliumin B Protects against Cisplatin-Induced Renal Tubular Cell Apoptosis through Activation of AMPK-Induced Autophagy and Mitochondrial Biogenesis. Kidney Dis. 2021, 7, 278–292. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.J.; Chung, J.; Fiorentino, D.F.; Flanagan, W.M.; Blenis, J.; Crabtree, G.R. Rapamycin selectively inhibits interleukin-2 activation of p70 S6 kinase. Nature 1992, 358, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Li, F.; Mahato, R.I. Poly(ethylene glycol)-block-poly(2-methyl-2-benzoxycarbonyl-propylene carbonate) micelles for rapamycin delivery: In vitro characterization and biodistribution. J. Pharm. Sci. 2011, 100, 2418–2429. [Google Scholar] [CrossRef] [Green Version]
- Yanez, J.A.; Forrest, M.L.; Ohgami, Y.; Kwon, G.S.; Davies, N.M. Pharmacometrics and delivery of novel nanoformulated PEG-b-poly(epsilon-caprolactone) micelles of rapamycin. Cancer Chemother. Pharmacol. 2008, 61, 133–144. [Google Scholar] [CrossRef] [Green Version]
- Zeng, S.; Xiong, M.P. Trilayer micelles for combination delivery of rapamycin and siRNA targeting Y-box binding protein-1 (siYB-1). Biomaterials 2013, 34, 6882–6892. [Google Scholar] [CrossRef] [Green Version]
- Haeri, A.; Sadeghian, S.; Rabbani, S.; Anvari, M.S.; Ghassemi, S.; Radfar, F.; Dadashzadeh, S. Effective attenuation of vascular restenosis following local delivery of chitosan decorated sirolimus liposomes. Carbohydr. Polym. 2017, 157, 1461–1469. [Google Scholar] [CrossRef]
- Haeri, A.; Sadeghian, S.; Rabbani, S.; Anvari, M.S.; Boroumand, M.A.; Dadashzadeh, S. Use of remote film loading methodology to entrap sirolimus into liposomes: Preparation, characterization and in vivo efficacy for treatment of restenosis. Int. J. Pharm. 2011, 414, 16–27. [Google Scholar] [CrossRef]
- Elsaid, N.; Somavarapu, S.; Jackson, T.L. Cholesterol-poly(ethylene) glycol nanocarriers for the transscleral delivery of sirolimus. Exp. Eye Res. 2014, 121, 121–129. [Google Scholar] [CrossRef]
- Cyrus, T.; Zhang, H.; Allen, J.S.; Williams, T.A.; Hu, G.; Caruthers, S.D.; Wickline, S.A.; Lanza, G.M. Intramural delivery of rapamycin with alphavbeta3-targeted paramagnetic nanoparticles inhibits stenosis after balloon injury. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 820–826. [Google Scholar] [CrossRef] [Green Version]
- Bibee, K.P.; Cheng, Y.J.; Ching, J.K.; Marsh, J.N.; Li, A.J.; Keeling, R.M.; Connolly, A.M.; Golumbek, P.T.; Myerson, J.W.; Hu, G.; et al. Rapamycin nanoparticles target defective autophagy in muscular dystrophy to enhance both strength and cardiac function. FASEB J. 2014, 28, 2047–2061. [Google Scholar] [CrossRef] [Green Version]
- Winter, P.M.; Neubauer, A.M.; Caruthers, S.D.; Harris, T.D.; Robertson, J.D.; Williams, T.A.; Schmieder, A.H.; Hu, G.; Allen, J.S.; Lacy, E.K.; et al. Endothelial alpha(v)beta3 integrin-targeted fumagillin nanoparticles inhibit angiogenesis in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2103–2109. [Google Scholar] [CrossRef] [Green Version]
- Halade, G.V.; Kain, V.; Ingle, K.A. Heart functional and structural compendium of cardiosplenic and cardiorenal networks in acute and chronic heart failure pathology. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H255–H267. [Google Scholar] [CrossRef]
- Winter, P.M.; Cai, K.; Caruthers, S.D.; Wickline, S.A.; Lanza, G.M. Emerging nanomedicine opportunities with perfluorocarbon nanoparticles. Expert Rev. Med. Devices 2007, 4, 137–145. [Google Scholar] [CrossRef]
- Danaei, M.; Dehghankhold, M.; Ataei, S.; Hasanzadeh Davarani, F.; Javanmard, R.; Dokhani, A.; Khorasani, S.; Mozafari, M.R. Impact of Particle Size and Polydispersity Index on the Clinical Applications of Lipidic Nanocarrier Systems. Pharmaceutics 2018, 10, 57. [Google Scholar] [CrossRef] [Green Version]
- Vargas, I.; Stephenson, D.J.; Baldwin, M.; Gaut, J.P.; Chalfant, C.E.; Pan, H.; Wickline, S.A. Sustained local inhibition of thrombin preserves renal microarchitecture and function after onset of acute kidney injury. Nanomedicine 2021, 38, 102449. [Google Scholar] [CrossRef] [PubMed]
- Djebli, N.; Rousseau, A.; Hoizey, G.; Rerolle, J.P.; Toupance, O.; Le Meur, Y.; Marquet, P. Sirolimus population pharmacokinetic/pharmacogenetic analysis and bayesian modelling in kidney transplant recipients. Clin. Pharm. 2006, 45, 1135–1148. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Cohen, E.E.; House, L.K.; Ramirez, J.; Zhang, W.; Ratain, M.J.; Bies, R.R. Nonlinear population pharmacokinetics of sirolimus in patients with advanced cancer. CPT Pharmacomet. Syst. Pharmacol. 2012, 1, e17. [Google Scholar] [CrossRef]
- Shin, H.J.; Jo, M.J.; Jin, I.S.; Park, C.W.; Kim, J.S.; Shin, D.H. Optimization and Pharmacokinetic Evaluation of Synergistic Fenbendazole and Rapamycin Co-Encapsulated in Methoxy Poly(Ethylene Glycol)-b-Poly(Caprolactone) Polymeric Micelles. Int. J. Nanomed. 2021, 16, 4873–4889. [Google Scholar] [CrossRef] [PubMed]
- Ferron, G.M.; Mishina, E.V.; Zimmerman, J.J.; Jusko, W.J. Population pharmacokinetics of sirolimus in kidney transplant patients. Clin. Pharmacol. Ther. 1997, 61, 416–428. [Google Scholar] [CrossRef]
- Emoto, C.; Fukuda, T.; Cox, S.; Christians, U.; Vinks, A.A. Development of a Physiologically-Based Pharmacokinetic Model for Sirolimus: Predicting Bioavailability Based on Intestinal CYP3A Content. CPT Pharmacomet. Syst. Pharmacol. 2013, 2, e59. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, J.J.; Kahan, B.D. Pharmacokinetics of sirolimus in stable renal transplant patients after multiple oral dose administration. J. Clin. Pharmacol. 1997, 37, 405–415. [Google Scholar] [CrossRef]
- Brattstrom, C.; Tyden, G.; Sawe, J.; Herlenius, G.; Claesson, K.; Groth, C.G. A randomized, double-blind, placebo-controlled study to determine safety, tolerance, and preliminary pharmacokinetics of ascending single doses of orally administered sirolimus (rapamycin) in stable renal transplant recipients. Transplant. Proc. 1996, 28, 985–986. [Google Scholar]
- MacDonald, A.; Scarola, J.; Burke, J.T.; Zimmerman, J.J. Clinical pharmacokinetics and therapeutic drug monitoring of sirolimus. Clin. Ther. 2000, 22 (Suppl. B), B101–B121. [Google Scholar] [CrossRef]
- Trepanier, D.J.; Gallant, H.; Legatt, D.F.; Yatscoff, R.W. Rapamycin: Distribution, pharmacokinetics and therapeutic range investigations: An update. Clin. Biochem. 1998, 31, 345–351. [Google Scholar] [CrossRef]
- Schreiber, K.H.; Arriola Apelo, S.I.; Yu, D.; Brinkman, J.A.; Velarde, M.C.; Syed, F.A.; Liao, C.Y.; Baar, E.L.; Carbajal, K.A.; Sherman, D.S.; et al. A novel rapamycin analog is highly selective for mTORC1 in vivo. Nat. Commun. 2019, 10, 3194. [Google Scholar] [CrossRef]
- Arriola Apelo, S.I.; Neuman, J.C.; Baar, E.L.; Syed, F.A.; Cummings, N.E.; Brar, H.K.; Pumper, C.P.; Kimple, M.E.; Lamming, D.W. Alternative rapamycin treatment regimens mitigate the impact of rapamycin on glucose homeostasis and the immune system. Aging Cell 2016, 15, 28–38. [Google Scholar] [CrossRef]
- Bottiger, Y.; Sawe, J.; Brattstrom, C.; Tollemar, J.; Burke, J.T.; Hass, G.; Zimmerman, J.J. Pharmacokinetic interaction between single oral doses of diltiazem and sirolimus in healthy volunteers. Clin. Pharmacol. Ther. 2001, 69, 32–40. [Google Scholar] [CrossRef]
- Hu, Y.; Sun, B.; Zhao, B.; Mei, D.; Gu, Q.; Tian, Z. Cisplatin-induced cardiotoxicity with midrange ejection fraction: A case report and review of the literature. Medicine 2018, 97, e13807. [Google Scholar] [CrossRef]
- Raja, W.; Mir, M.H.; Dar, I.; Banday, M.A.; Ahmad, I. Cisplatin induced paroxysmal supraventricular tachycardia. Indian J. Med. Paediatr. Oncol. 2013, 34, 330–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, A.C.; McMahon, K.; Hall, M.; Neves, K.B.; Rios, F.J.; Montezano, A.C.; Welsh, P.; Waterston, A.; White, J.; Mark, P.B.; et al. Comprehensive Characterization of the Vascular Effects of Cisplatin-Based Chemotherapy in Patients with Testicular Cancer. JACC Cardio Oncol. 2020, 2, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Vaughn, D.J.; Palmer, S.C.; Carver, J.R.; Jacobs, L.A.; Mohler, E.R. Cardiovascular risk in long-term survivors of testicular cancer. Cancer 2008, 112, 1949–1953. [Google Scholar] [CrossRef]
- Bjerring, A.W.; Fossa, S.D.; Haugnes, H.S.; Nome, R.; Stokke, T.M.; Haugaa, K.H.; Kiserud, C.E.; Edvardsen, T.; Sarvari, S.I. The cardiac impact of cisplatin-based chemotherapy in survivors of testicular cancer: A 30-year follow-up. Eur. Heart J. Cardiovasc. Imaging 2021, 22, 443–450. [Google Scholar] [CrossRef] [PubMed]
- McMullen, J.R.; Sherwood, M.C.; Tarnavski, O.; Zhang, L.; Dorfman, A.L.; Shioi, T.; Izumo, S. Inhibition of mTOR signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation 2004, 109, 3050–3055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, G.; Chen, W.; Yan, M.; Liu, J.; Luo, H.; Wang, C.; Yang, P. Rapamycin regulates the balance between cardiomyocyte apoptosis and autophagy in chronic heart failure by inhibiting mTOR signaling. Int. J. Mol. Med. 2020, 45, 195–209. [Google Scholar] [CrossRef] [Green Version]
- Quarles, E.; Basisty, N.; Chiao, Y.A.; Merrihew, G.; Gu, H.; Sweetwyne, M.T.; Fredrickson, J.; Nguyen, N.H.; Razumova, M.; Kooiker, K.; et al. Rapamycin persistently improves cardiac function in aged, male and female mice, even following cessation of treatment. Aging Cell 2020, 19, e13086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, J.; Hu, W.; Song, Z.P.; Chen, Y.G.; Zhang, D.D.; Wang, C.Q. Rapamycin Inhibits Cardiac Hypertrophy by Promoting Autophagy via the MEK/ERK/Beclin-1 Pathway. Front. Physiol. 2016, 7, 104. [Google Scholar] [CrossRef] [PubMed]
- Vallabhapurapu, S.; Powolny-Budnicka, I.; Riemann, M.; Schmid, R.M.; Paxian, S.; Pfeffer, K.; Korner, H.; Weih, F. Rel/NF-kappaB family member RelA regulates NK1.1− to NK1.1+ transition as well as IL-15-induced expansion of NKT cells. Eur. J. Immunol. 2008, 38, 3508–3519. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, E.; Xie, X.; Wang, J.; Zheng, H.; Ju, Y.; Chen, L.; Li, C.; Zhou, X.; Li, Z.; et al. Induction of Foxp3 and activation of Tregs by HSP gp96 for treatment of autoimmune diseases. Iscience 2021, 24, 1034–1045. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H. FOXP3 and its role in the immune system. Adv. Exp. Med. Biol. 2009, 665, 17–29. [Google Scholar] [CrossRef]
- Smale, S.T. Selective transcription in response to an inflammatory stimulus. Cell 2010, 140, 833–844. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test Name | Free Rapamycin | Rapamycin NP |
|---|---|---|
| WBC (×103/μL) | 2.12 ± 0.38 | 2.12 ± 0.38 |
| RBC (×106/μL) | 7.76 ± 0.10 | 7.79 ± 0.27 |
| Hemoglobin (g/dL) | 11.16 ± 0.22 | 11.14 ± 0.27 |
| Hematocrit (%) | 38.60 ± 1.04 | 37.94 ± 1.49 |
| MCV (fL) | 49.70 ± 0.73 | 48.68 ± 0.69 |
| MCH (pg) | 14.38 ± 0.30 | 14.32 ± 0.24 |
| MCHC (g/dL) | 28.96 ± 0.82 | 29.44 ± 0.83 |
| Platelets (×103/μL) | 703.00 ± 57.56 | 794.00 ± 30.84 |
| Blood Urea Nitrogen (mg/dL) | 22.00 ± 9.84 | 16.80 ± 0.49 |
| Creatinine (mg/dL) | 0.48 ± 0.21 | 0.43 ± 0.02 |
| Total Protein (g/dL) | 7.20 ± 3.22 | 7.16 ± 0.15 |
| AST (U/L) | 71.6 ± 32.02 | 72.00 ± 13.22 |
| ALT (U/L) | 60.40 ± 27.01 | 70.80 ± 6.53 |
| Sodium (Na+) (mmol/L) | 140.25 ± 1.03 | 138.60 ± 0.24 |
| Potassium (K+) (mmol/L) | 4.35 ± 0.29 | 4.24 ± 0.11 |
| Chloride (Cl−) (mmol/L) | 107.00 ± 0.71 | 106.40 ± 0.40 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Q.; Doherty, J.; Akk, A.; Springer, L.E.; Fan, P.; Spasojevic, I.; Halade, G.V.; Yang, H.; Pham, C.T.N.; Wickline, S.A.; et al. Safety Profile of Rapamycin Perfluorocarbon Nanoparticles for Preventing Cisplatin-Induced Kidney Injury. Nanomaterials 2022, 12, 336. https://doi.org/10.3390/nano12030336
Zhou Q, Doherty J, Akk A, Springer LE, Fan P, Spasojevic I, Halade GV, Yang H, Pham CTN, Wickline SA, et al. Safety Profile of Rapamycin Perfluorocarbon Nanoparticles for Preventing Cisplatin-Induced Kidney Injury. Nanomaterials. 2022; 12(3):336. https://doi.org/10.3390/nano12030336
Chicago/Turabian StyleZhou, Qingyu, Justin Doherty, Antonina Akk, Luke E. Springer, Ping Fan, Ivan Spasojevic, Ganesh V. Halade, Huanghe Yang, Christine T. N. Pham, Samuel A. Wickline, and et al. 2022. "Safety Profile of Rapamycin Perfluorocarbon Nanoparticles for Preventing Cisplatin-Induced Kidney Injury" Nanomaterials 12, no. 3: 336. https://doi.org/10.3390/nano12030336