
Visible-Light Activation of Persulfate or H2O2 by Fe2O3/TiO2 Immobilized on Glass Support for Photocatalytic Removal of Amoxicillin: Mechanism, Transformation Products, and Toxicity Assessment
 , ,
, ,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation and Imobilization of Fe2O3/TiO2 Nanocomposites
2.3. Characterization of Fe2O3/TiO2 Nanocomposites
2.4. Photoelectrochemical (PEC) Measurements
2.5. Photocatalytic Activity Evaluation
2.6. Analytical Methods
2.7. Calculations
3. Results and Discussion
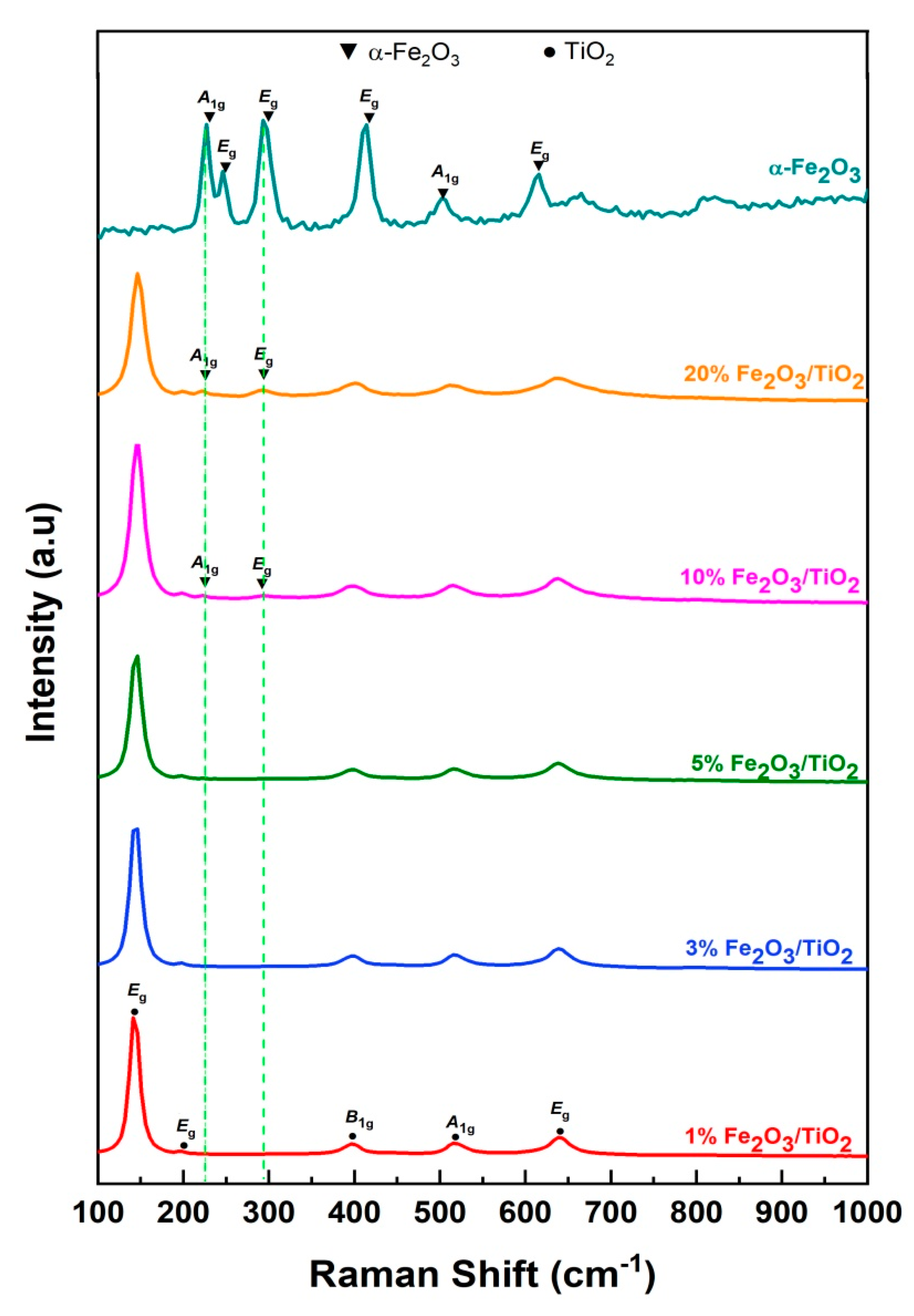
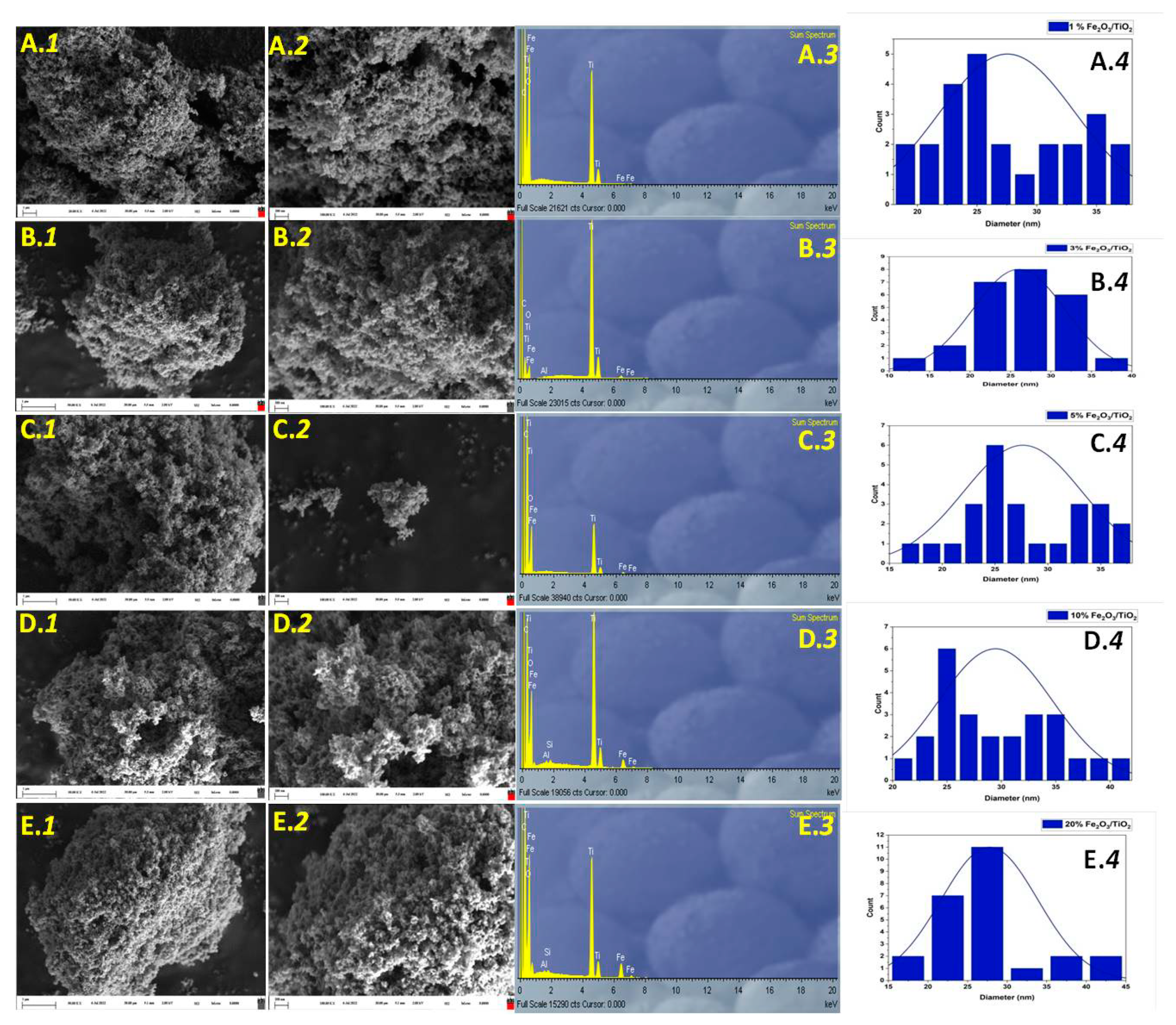
3.1. Material Characterization
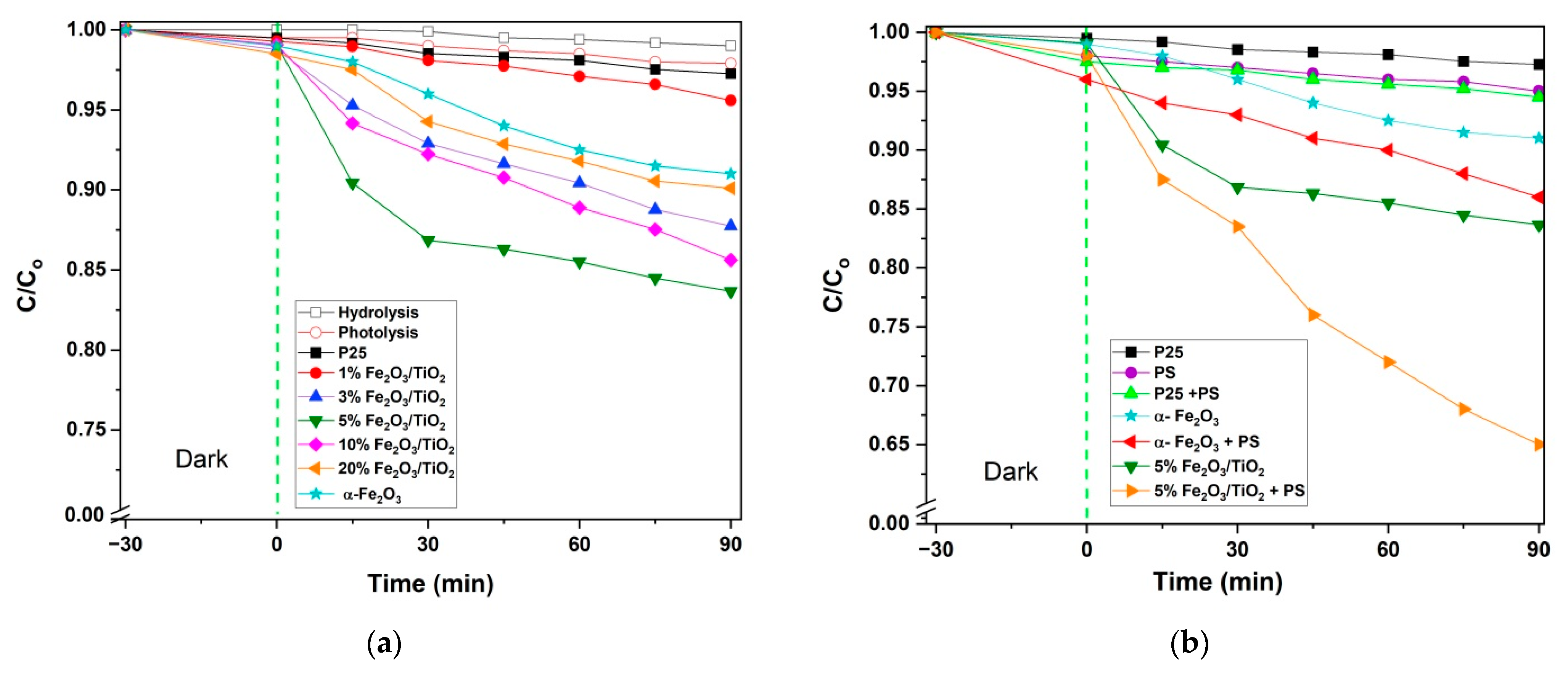
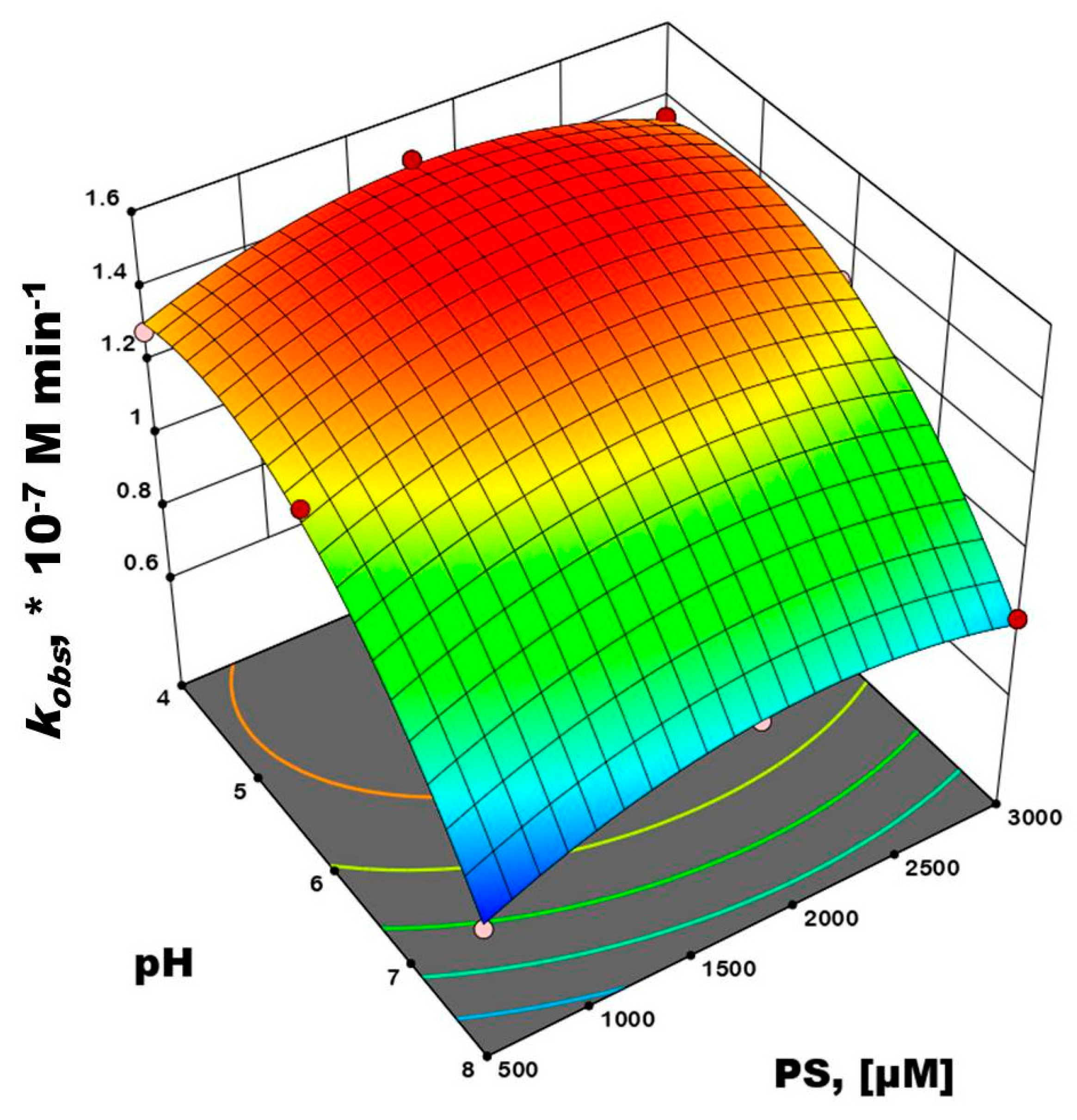
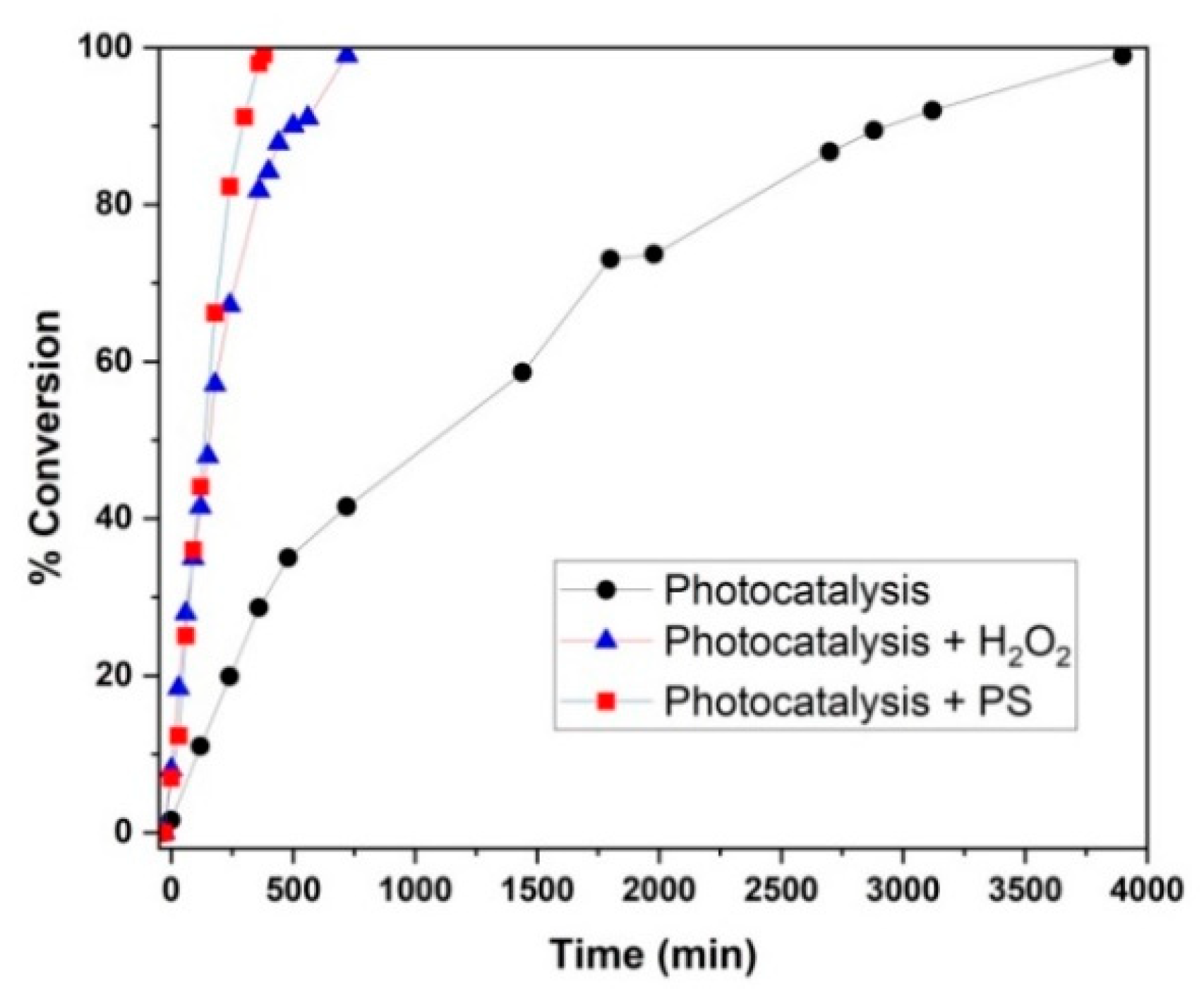
3.2. Photocatalytic Activity Tests
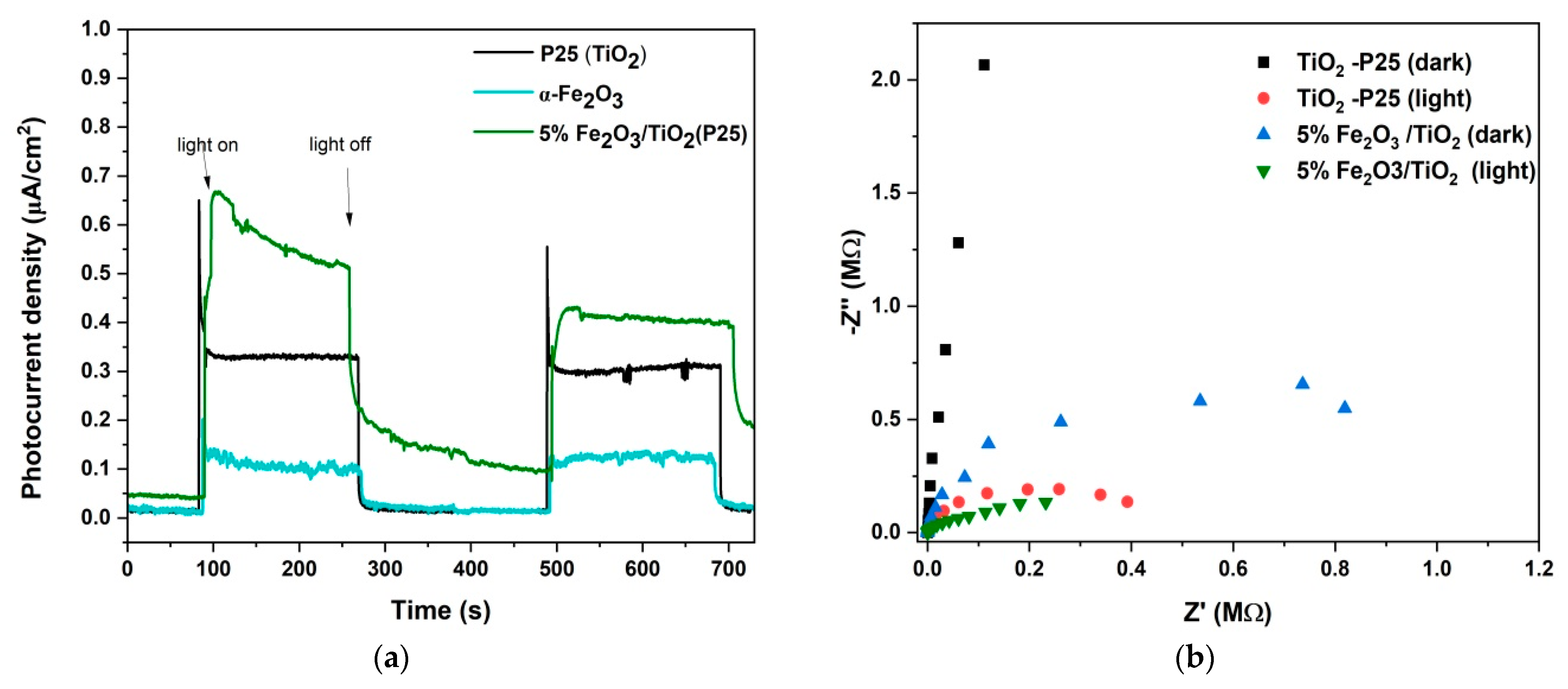
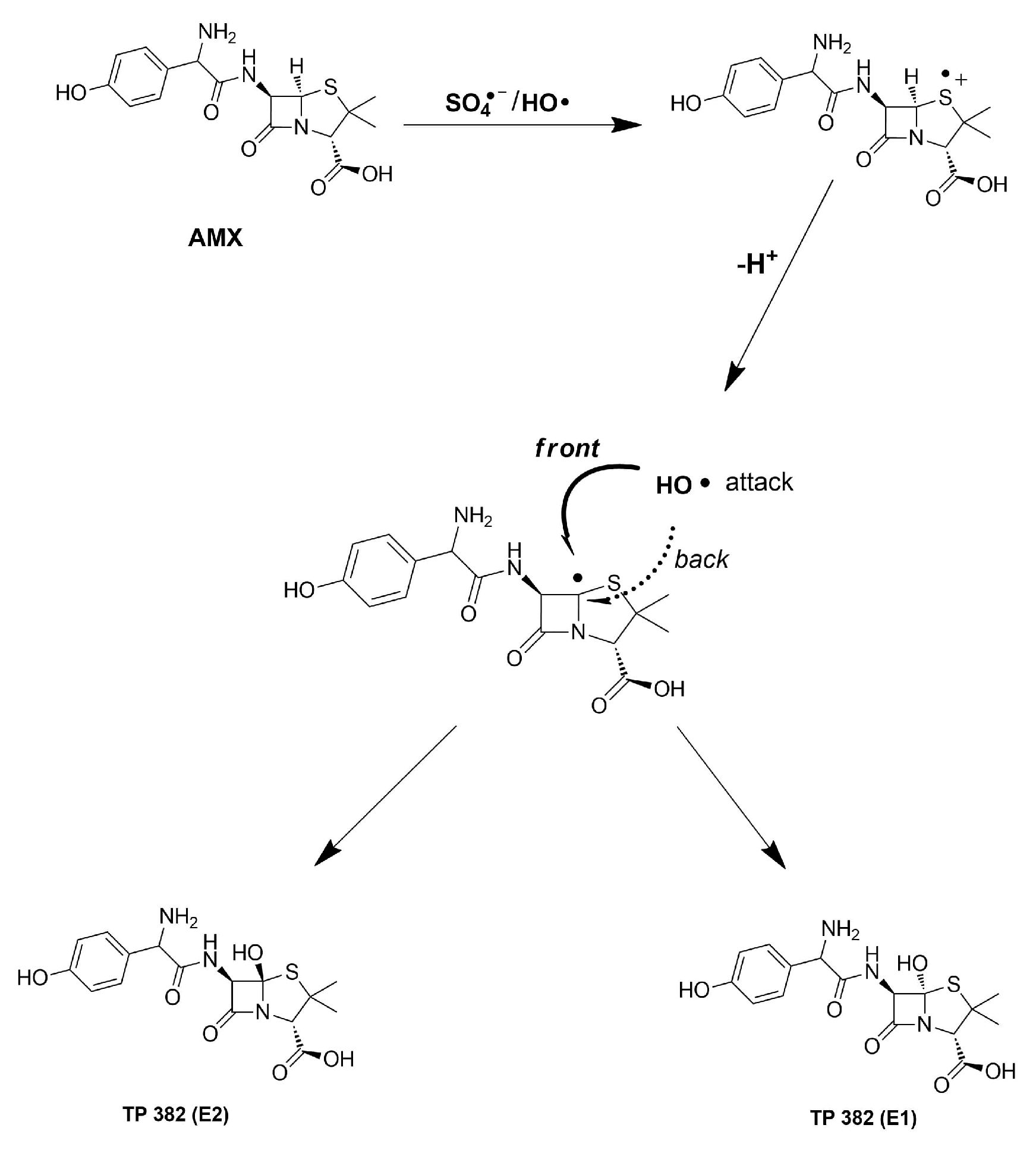
3.3. Mechanism
3.4. AMX Transformation Byproducts and Toxicity Evaluation
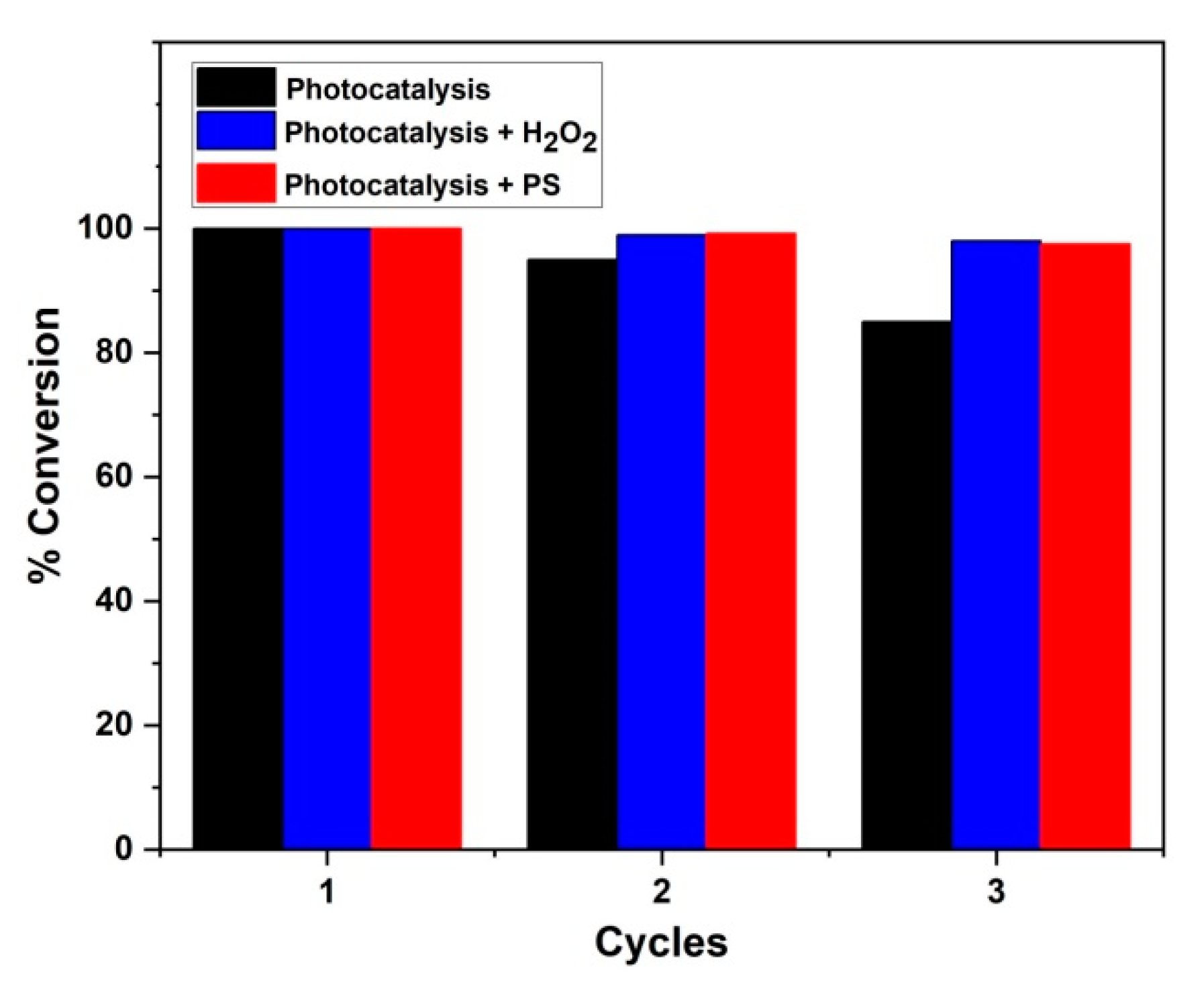
3.5. Stability Test
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Chong, M.N.; Jin, B.; Chow, C.W.K.; Saint, C. Recent developments in photocatalytic water treatment technology: A review. Water Res. 2010, 44, 2997–3027. [Google Scholar] [CrossRef] [PubMed]
- Pichat, P. Photocatalysis and Water Purification: From Fundamentals to Recent Applications; Wiley: Berlin, Germany, 2013. [Google Scholar]
- Schneider, J.; Matsuoka, M.; Takeuchi, M.; Zhang, J.; Horiuchi, Y.; Anpo, M.; Bahnemann, D.W. Understanding TiO2 photocatalysis: Mechanisms and materials. Chem. Rev. 2014, 114, 9919–9986. [Google Scholar] [CrossRef] [PubMed]
- Pelaez, M.; Nolan, N.T.; Pillai, S.C.; Seery, M.K.; Falaras, P.; Kontos, A.G.; Dunlop, P.S.M.; Hamilton, J.W.J.; Byrne, J.A.; O’Shea, K.; et al. A review on the visible light active titanium dioxide photocatalysts for environmental applications. Appl. Catal. B Environ. 2012, 125, 331–349. [Google Scholar] [CrossRef] [Green Version]
- Perović, K.; dela Rosa, F.M.; Kovačić, M.; Kušić, H.; Štangar, U.L.; Fresno, F.; Dionysiou, D.D.; Bozic, A.L. Recent achievements in development of TiO2-based composite photocatalytic materials for solar driven water purification and water splitting. Materials 2020, 13, 1338. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Chu, D. Energy band engineering of metal oxide for enhanced visible light absorption. In Multifunctional Photocatalytic Materials for Energy; Elsevier Ltd.: Amsterdam, The Netherlands, 2018; pp. 49–78. [Google Scholar]
- Lanzl, C.A.; Baltrusaitis, J.; Cwiertny, D.M. Dissolution of hematite nanoparticle aggregates: Influence of primary particle size, dissolution mechanism, and solution pH. Langmuir 2012, 28, 15797–15808. [Google Scholar] [CrossRef]
- Khasawneh, O.; Palaniandy, P. Removal of organic pollutants from water by Fe2O3/TiO2 based photocatalytic degradation: A review. Environ. Technol. Innov. 2021, 21, 101230. [Google Scholar] [CrossRef]
- dela Rosa, F.M.; Papac, J.; Garcia-Ballesteros, S.; Kovačić, M.; Katančić, Z.; Kušić, H.; Božić, A.L. Solar Light Activation of Persulfate by TiO2/Fe2O3 Layered Composite Films for Degradation of Amoxicillin: Degradation Mechanism, Matrix Effects, and Toxicity Assessments. Adv. Sustain. Syst. 2021, 5, 2100119. [Google Scholar] [CrossRef]
- García-Muñoz, P.; Zussblatt, N.P.; Pliego, G.; Zazo, J.A.; Fresno, F.; Chmelka, B.F.; Casas, J.A. Evaluation of photoassisted treatments for norfloxacin removal in water using mesoporous Fe2O3-TiO2 materials. J. Environ. Manage. 2019, 238, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Battula, V.R.; Jaryal, A.; Kaisalam, K. Visible light-driven simultaneous H2 production by water splitting coupled with selective oxidation of HMF to DFF catalyzed by porous carbon nitride. J. Mater. Chem. A 2019, 7, 5643–5649. [Google Scholar] [CrossRef]
- He, Y.; Qian, J.; Wang, P.; Wu, J.; Lu, B.; Tang, S. Acceleration of levofloxacin degradation by combination of multiple free radicals via MoS2 anchored in manganese ferrite doped perovskite activated PMS under visible light. Chem. Eng. J. 2022, 431, 133933. [Google Scholar] [CrossRef]
- Sakar, M.; Prakash, R.M.; Do, T.-O. Insights into the TiO2 -Based Photocatalytic Systems and Their Mechanisms. Catalysts 2019, 9, 680. [Google Scholar] [CrossRef] [Green Version]
- Herman, J.R.; Krotkov, N.; Celarier, E.; Larko, D.; Labow, G. Distribution from of UV radiation at the Earth ’ s surface from TOMS-measure UV-backscattered radiances. J. Geophys. Res. 1999, 104, 12059–12076. [Google Scholar] [CrossRef]
- Li, G.; Nie, X.; Gao, Y.; An, T. Can environmental pharmaceuticals be photocatalytically degraded and completely mineralized in water using g-C3N4/TiO2 under visible light irradiation?-Implications of persistent toxic intermediates. Appl. Catal. B Environ. 2016, 180, 726–732. [Google Scholar] [CrossRef]
- Kete, M.; Pavlica, E.; Fresno, F.; Bratina, G.; Štangar, U.L. Highly active photocatalytic coatings prepared by a low-temperature method. Environ. Sci. Pollut. Res. 2014, 21, 11238–11249. [Google Scholar] [CrossRef] [PubMed]
- Kovacic, M.; Kopcic, N.; Kusic, H.; Bozic, A.L. Solar driven degradation of 17β-estradiol using composite photocatalytic materials and artificial irradiation source: Influence of process and water matrix parameters. J. Photochem. Photobiol. A Chem. 2018, 361, 48–61. [Google Scholar] [CrossRef]
- EC Commission Implementing Decision (EU) 2018/840 of 5 June 2018 establishing a watch list of substances for Union-wide monitoring in the field of water policy pursuant to Directive 2008/105/EC of the European Parliament and of the Council and repealing Comm. Off. J. Eur. Union 2018, L 141, 9–12.
- Loos, R.; Marinov, D.; Sanseverino, I.; Napierska, D.; Lettieri, T. Review of the 1st Watch List under the Water Framework Directive and Recommendations for the 2nd Watch List; Publications Office of the European Union: Luxembourg, 2018. [Google Scholar]
- Andrews, D.; Lipson, R.; Nann, T. Comprehensive Nanoscience and Nanotechnology; Elsevier Ltd.: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Elbakkay, M.H.; El Rouby, W.M.A.; Mariño-López, A.; Sousa-Castillo, A.; Salgueiriño, V.; El-Dek, S.I.; Farghali, A.A.; Correa-Duarte, M.A.; Millet, P. One-pot synthesis of TiO2/Sb2S3/RGO complex multicomponent heterostructures for highly enhanced photoelectrochemical water splitting. Int. J. Hydrogen Energy 2021, 46, 31216–31227. [Google Scholar] [CrossRef]
- Moniz, S.J.A.; Shevlin, S.A.; An, X.; Guo, Z.X.; Tang, J. Fe2O3-TiO2 nanocomposites for enhanced charge separation and photocatalytic activity. Chem.-A Eur. J. 2014, 20, 15571–15579. [Google Scholar] [CrossRef]
- Dai, X.; Lu, G.; Hu, Y.; Xie, X.; Wang, X.; Sun, J. Reversible redox behavior of Fe2O3/TiO2 composites in the gaseous photodegradation process. Ceram. Int. 2019, 45, 13187–13192. [Google Scholar] [CrossRef]
- Lassoued, A.; Dkhil, B.; Gadri, A.; Ammar, S. Control of the shape and size of iron oxide (α-Fe2O3) nanoparticles synthesized through the chemical precipitation method. Results Phys. 2017, 7, 3007–3015. [Google Scholar] [CrossRef]
- Hua, J.; Gengsheng, J. Hydrothermal synthesis and characterization of monodisperse α-Fe2O3 nanoparticles. Mater. Lett. 2009, 63, 2725–2727. [Google Scholar] [CrossRef]
- Peña-flores, J.I.; Palomec-garfias, A.F.; Márquez-beltrán, C.; Sánchez-mora, E. Fe effect on the optical properties of TiO2: Fe2O3 nanostructured composites supported on SiO2 microsphere assemblies. Nanoscale Res. Lett. 2014, 9, 499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohsaka, T. Temperature Dependence of the Raman Spectrum in Anatase TiO2. J. Phys. Soc. Japan 1980, 48, 1661–1668. [Google Scholar] [CrossRef]
- Mansour, H.; Omri, K.; Bargougui, R.; Ammar, S. Novel α-Fe2O3/TiO2 nanocomposites with enhanced photocatalytic activity. Appl. Phys. A 2020, 126, 151. [Google Scholar] [CrossRef]
- De Faria, D.L.A.; Lopes, F.N. Heated goethite and natural hematite: Can Raman spectroscopy be used to differentiate them? Vib. Spectrosc. 2007, 45, 117–121. [Google Scholar] [CrossRef]
- Jubb, A.M.; Allen, H.C. Vibrational Spectroscopic Characterization of Hematite, Maghemite, and Magnetite Thin Films Produced by Vapor Deposition. ACS Appl. Mater. Interfaces 2010, 2, 2804–2812. [Google Scholar] [CrossRef]
- Cho, Y.; Huh, Y. Preparation of Hyperbranched Structures of α -Fe2O3. Bull. Korean Chem. Soc. 2009, 30, 1413–1415. [Google Scholar]
- Huang, R.; Liang, R.; Fan, H.; Ying, S.; Wu, L.; Wang, X. Enhanced Photocatalytic Fuel Denitrification over TiO2/α-Fe2O3 Nanocomposites under Visible Light irradiation. Sci. Rep. 2017, 7, 7858. [Google Scholar] [CrossRef]
- Lamm, B.; Trześniewski, B.J.; Döscher, H.; Smith, W.A.; Stefik, M. Emerging Postsynthetic Improvements of BiVO4 Photoanodes for Solar Water Splitting. ACS Energy Lett. 2018, 3, 112–124. [Google Scholar] [CrossRef]
- Singh, A.P.; Tossi, C.; Tittonen, I.; Hellman, A.; Wickman, B. Synergies of co-doping in ultra-thin hematite photoanodes for solar water oxidation: In and Ti as representative case. RSC Adv. 2020, 10, 33307–33316. [Google Scholar] [CrossRef]
- Akhavan, O. Thickness dependent activity of nanostructured TiO2 /α- Fe2O3 photocatalyst thin films. Appl. Surf. Sci. 2010, 257, 1724–1728. [Google Scholar] [CrossRef]
- Akhavan, O.; Azimirad, R.; Safa, S.; Larijani, M.M. Visible light photo-induced antibacterial activity of CNT-doped TiO2 thin films with various CNT contents. J. Mater. Chem. 2010, 20, 7386–7392. [Google Scholar] [CrossRef]
- Strens, R.G.J.; Wood, B.J. Diffuse reflectance spectra and optical properties of some iron and titanium oxides and oxyhydroxides. Mineral. Mag. 1979, 43, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Torrent, J.; Barron, V. Diffuse reflectance spectroscopy of iron oxides. Encycl. Surf. Colloid Sci. 2002, 1438–1446. [Google Scholar]
- Sayed, M.; Ren, B.; Ali, A.M.; Al-Anazi, A.; Nadagouda, M.N.; Ismail, A.A.; Dionysiou, D.D. Solar light induced photocatalytic activation of peroxymonosulfate by ultra-thin Ti3+ self-doped Fe2O3/TiO2 nanoflakes for the degradation of naphthalene. Appl. Catal. B Environ. 2022, 315, 121532. [Google Scholar] [CrossRef]
- Xie, W.; Li, R.; Xu, Q. Enhanced photocatalytic activity of Se-doped TiO2 under visible light irradiation. Sci. Rep. 2018, 8, 8752. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.Q.; Zi, T.Q.; Zhao, X.R.; Liu, C.; Ren, Q.; Fang, J.B.; Li, W.M.; Li, A.D. Enhanced visible light photocatalytic activity of Fe2O3 modified TiO2 prepared by atomic layer deposition. Sci. Rep. 2020, 10, 13437. [Google Scholar] [CrossRef]
- D’Amato, C.A.; Giovannetti, R.; Zannotti, M.; Rommozzi, E.; Ferraro, S.; Seghetti, C.; Minicucci, M.; Gunnella, R.; Di Cicco, A. Enhancement of visible-light photoactivity by polypropylene coated plasmonic Au/TiO2 for dye degradation in water solution. Appl. Surf. Sci. 2018, 441, 575–587. [Google Scholar] [CrossRef]
- Yuan, S.; Mu, J.; Mao, R.; Li, Y.; Zhang, Q.; Wang, H. All-Nanoparticle Self-assembly ZnO / TiO2 Heterojunction Thin Films with Remarkably Enhanced Photoelectrochemical Activity. ACS Appl. Mater. Interfaces 2014, 6, 5719–5725. [Google Scholar] [CrossRef]
- Mei, Q.; Zhang, F.; Wang, N.; Yang, Y.; Wu, R.; Wang, W. TiO2/Fe2O3 heterostructures with enhanced photocatalytic reduction of Cr(VI) under visible light irradiation. RSC Adv. 2019, 9, 22764–22771. [Google Scholar] [CrossRef] [Green Version]
- Zubair, M.; Svenum, I.; Rønning, M.; Yang, J. Core-Shell Nanostructures of Graphene-Wrapped CdS Nanoparticles and TiO2 (CdS @ G @ TiO2): The Role of Graphene in Enhanced Photocatalytic H2 Generation. Catalysts 2020, 10, 358. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Zong, X.; An, L.; Hua, S.; Miao, X.; Luan, S.; Wen, Y.; Tao, F.F.; Sun, Z. Consciously Constructing Heterojunction or Direct Z-Scheme Photocatalysts by Regulating Electron Flow Direction. ACS Catal. 2018, 8, 2209–2217. [Google Scholar] [CrossRef]
- Zhao, J.; Sun, Y.; Wu, F.; Shi, M.; Liu, X. Oxidative Degradation of Amoxicillin in Aqueous Solution by Thermally Activated Persulfate. J. Chem. 2019, 2019, 2505823. [Google Scholar] [CrossRef]
- Boncagni, N.T.; Otaegui, J.M.; Warner, E.; Curran, T.; Ren, J.; De Cortalezzi, M.M.F. Exchange of TiO2 nanoparticles between streams and streambeds. Environ. Sci. Technol. 2009, 43, 7699–7705. [Google Scholar] [CrossRef]
- Buscio, V.; Brosillon, S.; Mendret, J.; Crespi, M.; Gutiérrez-bouzán, C. Photocatalytic Membrane Reactor for the Removal of C.I. Disperse Red 73. Materials 2015, 8, 3633–3647. [Google Scholar] [CrossRef] [Green Version]
- Kritikos, D.E.; Xekoukoulotakis, N.P.; Psillakis, E.; Mantzavinos, D. Photocatalytic degradation of reactive black 5 in aqueous solutions: Effect of operating conditions and coupling with ultrasound irradiation. Water Res. 2007, 41, 2236–2246. [Google Scholar] [CrossRef]
- Shrimali, K.; Jin, J.; Hassas, B.V.; Wang, X.; Miller, J.D. The surface state of hematite and its wetting characteristics. J. Colloid Interface Sci. 2016, 477, 16–24. [Google Scholar] [CrossRef]
- Mendiola-Alvarez, S.Y.; Araña, J.; Doña Rodríguez, J.M.; Hernández-Ramírez, A.; Turnes Palomino, G.; Palomino Cabello, C.; Hinojosa-Reyes, L. Comparison of photocatalytic activity of α-Fe2O3-TiO2/P on the removal of pollutants on liquid and gaseous phase. J. Environ. Chem. Eng. 2021, 9, 104828. [Google Scholar] [CrossRef]
- Subramonian, W.; Wu, T.Y.; Chai, S.-P. Photocatalytic degradation of industrial pulp and paper mill effluent using synthesized magnetic Fe2O3-TiO2: Treatment efficiency and characterizations of reused photocatalyst. J. Environ. Manage. 2017, 187, 298–310. [Google Scholar] [CrossRef] [Green Version]
- Fang, G.; Wu, W.; Liu, C.; Dionysiou, D.D.; Deng, Y.; Zhou, D. Activation of persulfate with vanadium species for PCBs degradation: A mechanistic study. Appl. Catal. B Environ. 2017, 202, 1–11. [Google Scholar] [CrossRef]
- Matzek, L.W.; Carter, K.E. Sustained persulfate activation using solid iron: Kinetics and application to ciprofloxacin degradation. Chem. Eng. J. 2017, 307, 650–660. [Google Scholar] [CrossRef]
- Zhong, H.; Tian, Y.; Yang, Q.; Brusseau, M.L.; Yang, L.; Zeng, G. Degradation of landfill leachate compounds by persulfate for groundwater remediation. Chem. Eng. J. 2017, 307, 399–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furman, O.S.; Teel, A.M.Y.L.; Watts, R.J. Mechanism of Base Activation of Persulfate. Environ. Sci. Technol. 2010, 44, 6423–6428. [Google Scholar] [CrossRef] [PubMed]
- Saien, J.; Jafari, F. Chapter 1- Methods of Persulfate Activation for the Degradation of Pollutants: Fundamentals and Influencing Parameters. In Persulfate-Based Oxidation Processes in Environmental Remediation; The Royal Society of Chemistry: London, UK, 2022; pp. 1–59. [Google Scholar]
- Olmez-Hanci, T.; Arslan-Alaton, I. Comparison of sulfate and hydroxyl radical based advanced oxidation of phenol. Chem. Eng. J. 2013, 224, 10–16. [Google Scholar] [CrossRef]
- Kolthoff, I.M.; Miller, I.K. The Chemistry of Persulfate. I. The Kinetics and Mechanism of the Decomposition of the Persulfate Ion in Aqueous Medium. J. Am. Chem. Soc. 1951, 73, 3055–3059. [Google Scholar] [CrossRef]
- Kolthoff, I.M.; Miller, I.K. The Chemistry of Persulfate. II. The Reaction of Persulfate with Mercaptans Solubilized in Solutions of Saturated Fatty Acid Soaps. J. Am. Chem. Soc. 1951, 73, 5118–5122. [Google Scholar] [CrossRef]
- Gao, H.; Chen, J.; Zhang, Y.; Zhou, X. Sulfate radicals induced degradation of Triclosan in thermally activated persulfate system. Chem. Eng. J. 2016, 306, 522–530. [Google Scholar] [CrossRef]
- Kusic, H.; Peternel, I.; Ukic, S.; Koprivanac, N.; Bolanca, T.; Papic, S.; Bozic, A.L. Modeling of iron activated persulfate oxidation treating reactive azo dye in water matrix. Chem. Eng. J. 2011, 172, 109–121. [Google Scholar] [CrossRef]
- Xia, Y.; Yin, L. Core-shell structured α-Fe2O3@TiO2 nanocomposites with improved photocatalytic activity in the visible light region. Phys. Chem. Chem. Phys. 2013, 15, 18627–18634. [Google Scholar] [CrossRef]
- Liu, J.; Yang, S.; Wu, W.; Tian, Q.; Cui, S.; Dai, Z.; Ren, F.; Xiao, X.; Jiang, C. 3D Flowerlike α-Fe2O3@TiO2 Core–Shell Nanostructures: General Synthesis and Enhanced Photocatalytic Performance. ACS Sustain. Chem. Eng. 2015, 3, 2975–2984. [Google Scholar] [CrossRef]
- Monteagudo, J.M.; Durán, A.; Martín, I.S.; Vellón, B. Photocatalytic degradation of aniline by solar / TiO2 system in the presence of the electron acceptors Na2S2O8 and H2O2. Sep. Purif. Technol. 2020, 238, 116456. [Google Scholar] [CrossRef]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical Review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (⋅OH/⋅O− in Aqueous Solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886. [Google Scholar] [CrossRef] [Green Version]
- Rao, P.S.; Hayon, E. Redox potentials of free radicals. IV. Superoxide and hydroperoxy radicals O2− and HO2. J. Phys. Chem. 1975, 79, 397–402. [Google Scholar] [CrossRef]
- Matta, R.; Tlili, S.; Chiron, S.; Barbati, S. Removal of carbamazepine from urban wastewater by sulfate radical oxidation. Environ. Chem. Lett. 2011, 9, 347–353. [Google Scholar] [CrossRef]
- Anipsitakis, G.P.; Dionysiou, D.D. Radical generation by the interaction of transition metals with common oxidants. Environ. Sci. Technol. 2004, 38, 3705–3712. [Google Scholar] [CrossRef]
- Zhu, J.; Liu, S.; Ge, J.; Guo, X.; Wang, X.; Wu, H. Synthesis of Fe2O3-TiO2/fly-ash-cenosphere composite and its mechanism of photocatalytic oxidation under visible light. Res. Chem. Intermed. 2016, 42, 3637–3654. [Google Scholar] [CrossRef]
- Sabri, M.; Habibi-Yangjeh, A.; Chand, H.; Krishnan, V. Activation of persulfate by novel TiO2/FeOCl photocatalyst under visible light: Facile synthesis and high photocatalytic performance. Sep. Purif. Technol. 2020, 250, 117268. [Google Scholar] [CrossRef]
- Trovó, A.G.; Pupo Nogueira, R.F.; Agüera, A.; Fernandez-Alba, A.R.; Malato, S. Degradation of the antibiotic amoxicillin by photo-Fenton process-Chemical and toxicological assessment. Water Res. 2011, 45, 1394–1402. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xiao, Y.; Zhong, Y.; Lim, T.T. Comparison of amoxicillin photodegradation in the UV/H2O2 and UV/persulfate systems: Reaction kinetics, degradation pathways, and antibacterial activity. Chem. Eng. J. 2019, 372, 420–428. [Google Scholar] [CrossRef]
- Lamm, A.; Gozlan, I.; Rotstein, A.; Avisar, D. Detection of amoxicillin-diketopiperazine-2′, 5′ in wastewater samples. J. Environ. Sci. Heal. Part A 2009, 44, 1512–1517. [Google Scholar] [CrossRef]
- He, X.; Mezyk, S.P.; Michael, I.; Fatta-Kassinos, D.; Dionysiou, D.D. Degradation kinetics and mechanism of β-lactam antibiotics by the activation of H2O2 and Na2S2O8 under UV-254nm irradiation. J. Hazard. Mater. 2014, 279, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Liu, J.; Fang, W.; Xu, M.; Qin, Z.; Jiang, Z.; Shangguan, W. Photocatalytic hydrogen evolution with simultaneous antibiotic wastewater degradation via the visible-light-responsive bismuth spheres-g-C3N4 nanohybrid: Waste to energy insight. Chem. Eng. J. 2019, 358, 944–954. [Google Scholar] [CrossRef]
- Wei, Z.; Xu, M.; Liu, J.; Guo, W.; Jiang, Z.; Shangguan, W. Simultaneous visible-light-induced hydrogen production enhance- ment and antibiotic wastewater degradation using MoS2 @ ZnxCd1-xS: Solid-solution-assisted photocatalysis. Chinese J. Catal. 2020, 41, 103–113. [Google Scholar] [CrossRef]
- Ma, D.; Zhai, S.; Wang, Y.; Liu, A.; Chen, C. TiO2 Photocatalysis for Transfer Hydrogenation. Molecules 2019, 24, 330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomons, T.W.G. Organic Chemistry; Wiley: Berlin, Germany, 2011. [Google Scholar]
- Pérez-Parada, A.; Agüera, A.; del Mar Gómez-Ramos, M.; García-Reyes, J.F.; Heinzen, H.; Fernández-Alba, A.R. Behavior of amoxicillin in wastewater and river water: Identification of its main transformation products by liquid chromatography/electrospray quadrupole time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 2011, 25, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Arsand, J.B.; Hoff, R.B.; Jank, L.; Meirelles, L.N.; Silvia Díaz-Cruz, M.; Pizzolato, T.M.; Barceló, D. Transformation products of amoxicillin and ampicillin after photolysis in aqueous matrices: Identification and kinetics. Sci. Total Environ. 2018, 642, 954–967. [Google Scholar] [CrossRef]
- Gozlan, I.; Rotstein, A.; Avisar, D. Investigation of an amoxicillin oxidative degradation product formed under controlled environmental conditions. Environ. Chem. 2010, 7, 435–442. [Google Scholar] [CrossRef]
- Gozlan, I.; Rotstein, A.; Avisar, D. Amoxicillin-degradation products formed under controlled environmental conditions: Identification and determination in the aquatic environment. Chemosphere 2013, 91, 985–992. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Process Parameters | Model Variables/Coded Values | Level/Range | ||
|---|---|---|---|---|
| −1 | 0 | 1 | ||
| pH | X1 | 4 | 6 | 8 |
| [S2O82−] (µM) | X2 | 500 | 1750 | 3000 |
| Photocatalyst | TiO2 (P25) | 1% Fe2O3/TiO2 | 3% Fe2O3/TiO2 | 5% Fe2O3/TiO2 | 10% Fe2O3/TiO2 | 20% Fe2O3/TiO2 | α-Fe2O3 |
|---|---|---|---|---|---|---|---|
| Bandgap (eV) | 3.31 | 3.29 | 3.08 | 2.94 | 1.92 | 1.89 | 1.87 |
| Photocatalyst | Single-Point BET (m2/g) | Multipoint BET (m2/g) |
|---|---|---|
| TiO2–P25 | 46.6112 | 47.3512 ± 0.1206 |
| α-Fe2O3 | 25.2897 | 25.4548 ± 0.2408 |
| 1% Fe2O3/TiO2 | 49.7926 | 51.2769 ± 0.4552 |
| 3% Fe2O3/TiO2 | 39.3548 | 39.6042 ± 0.3677 |
| 5% Fe2O3/TiO2 | 40.4217 | 40.985 ± 0.2412 |
| 10% Fe2O3/TiO2 | 42.5486 | 43.4856 ± 0.1723 |
| 20% Fe2O3/TiO2 | 34.4068 | 34.7489 ± 0.3532 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
dela Rosa, F.M.; Popović, M.; Papac Zjačić, J.; Radić, G.; Kraljić Roković, M.; Kovačić, M.; Farré, M.J.; Genorio, B.; Lavrenčič Štangar, U.; Kušić, H.; et al. Visible-Light Activation of Persulfate or H2O2 by Fe2O3/TiO2 Immobilized on Glass Support for Photocatalytic Removal of Amoxicillin: Mechanism, Transformation Products, and Toxicity Assessment. Nanomaterials 2022, 12, 4328. https://doi.org/10.3390/nano12234328
dela Rosa FM, Popović M, Papac Zjačić J, Radić G, Kraljić Roković M, Kovačić M, Farré MJ, Genorio B, Lavrenčič Štangar U, Kušić H, et al. Visible-Light Activation of Persulfate or H2O2 by Fe2O3/TiO2 Immobilized on Glass Support for Photocatalytic Removal of Amoxicillin: Mechanism, Transformation Products, and Toxicity Assessment. Nanomaterials. 2022; 12(23):4328. https://doi.org/10.3390/nano12234328
Chicago/Turabian Styledela Rosa, Francis M., Marin Popović, Josipa Papac Zjačić, Gabrijela Radić, Marijana Kraljić Roković, Marin Kovačić, María José Farré, Boštjan Genorio, Urška Lavrenčič Štangar, Hrvoje Kušić, and et al. 2022. "Visible-Light Activation of Persulfate or H2O2 by Fe2O3/TiO2 Immobilized on Glass Support for Photocatalytic Removal of Amoxicillin: Mechanism, Transformation Products, and Toxicity Assessment" Nanomaterials 12, no. 23: 4328. https://doi.org/10.3390/nano12234328