
A Novel Nanocomposite of Zn(II)-Protoporphyrin-Chitosan-Multi Walled Carbon Nanotubes and the Application to Caffeic Acid Sensing

Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Apparatus and Measurements
2.3. Preparation of CS-Functionalized MWCNTs
2.4. Synthesis of the PPIX–CS–MWCNTs
2.5. Synthesis of Me–PPIX–CS–MWCNTs
2.6. Preparation of Me–PPIX–CS–MWCNTs/GCE
3. Results and Discussion
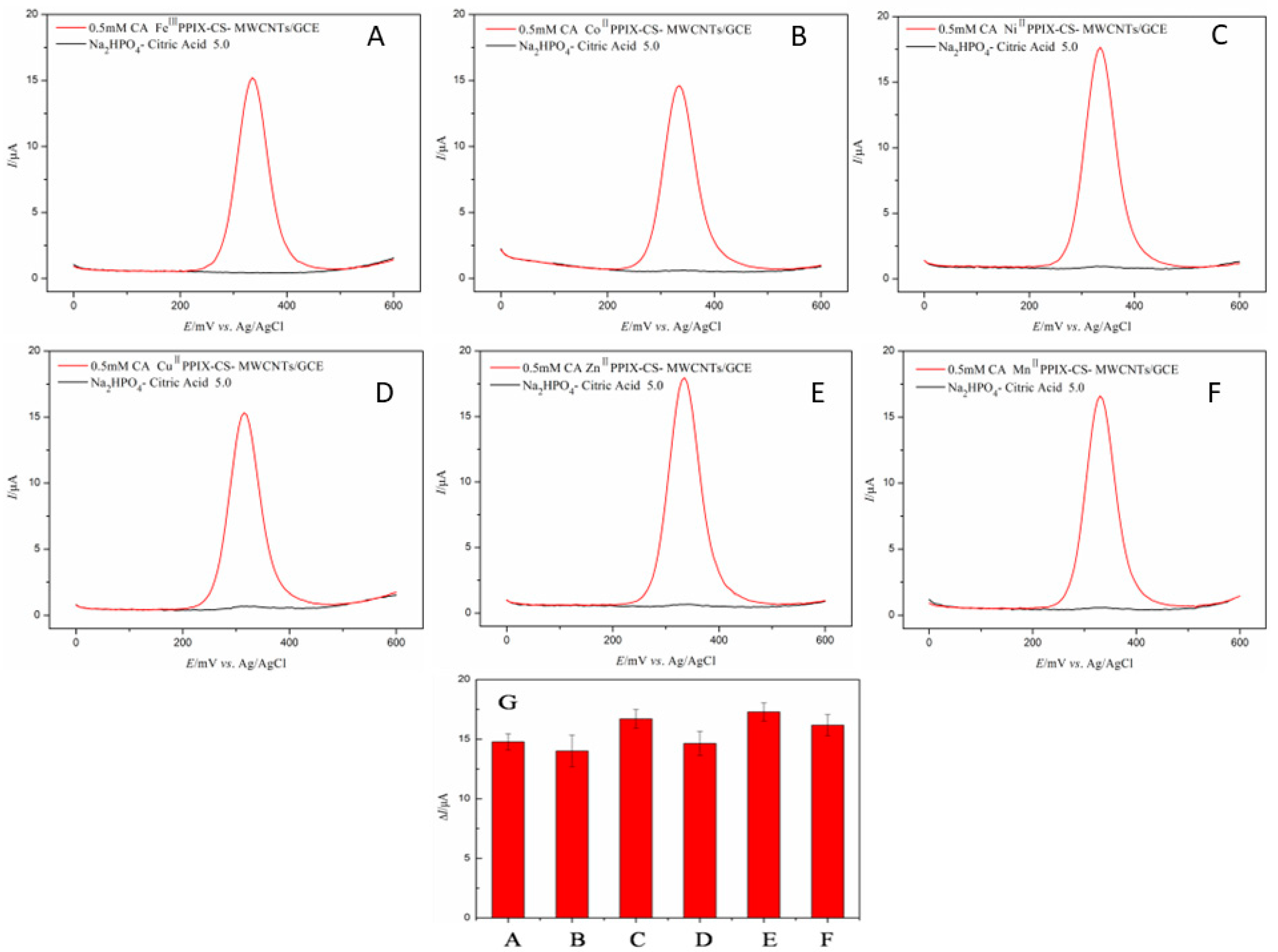
3.1. Screening of Transition Metal Ions of Me–PPIX–CS–MWCNTs
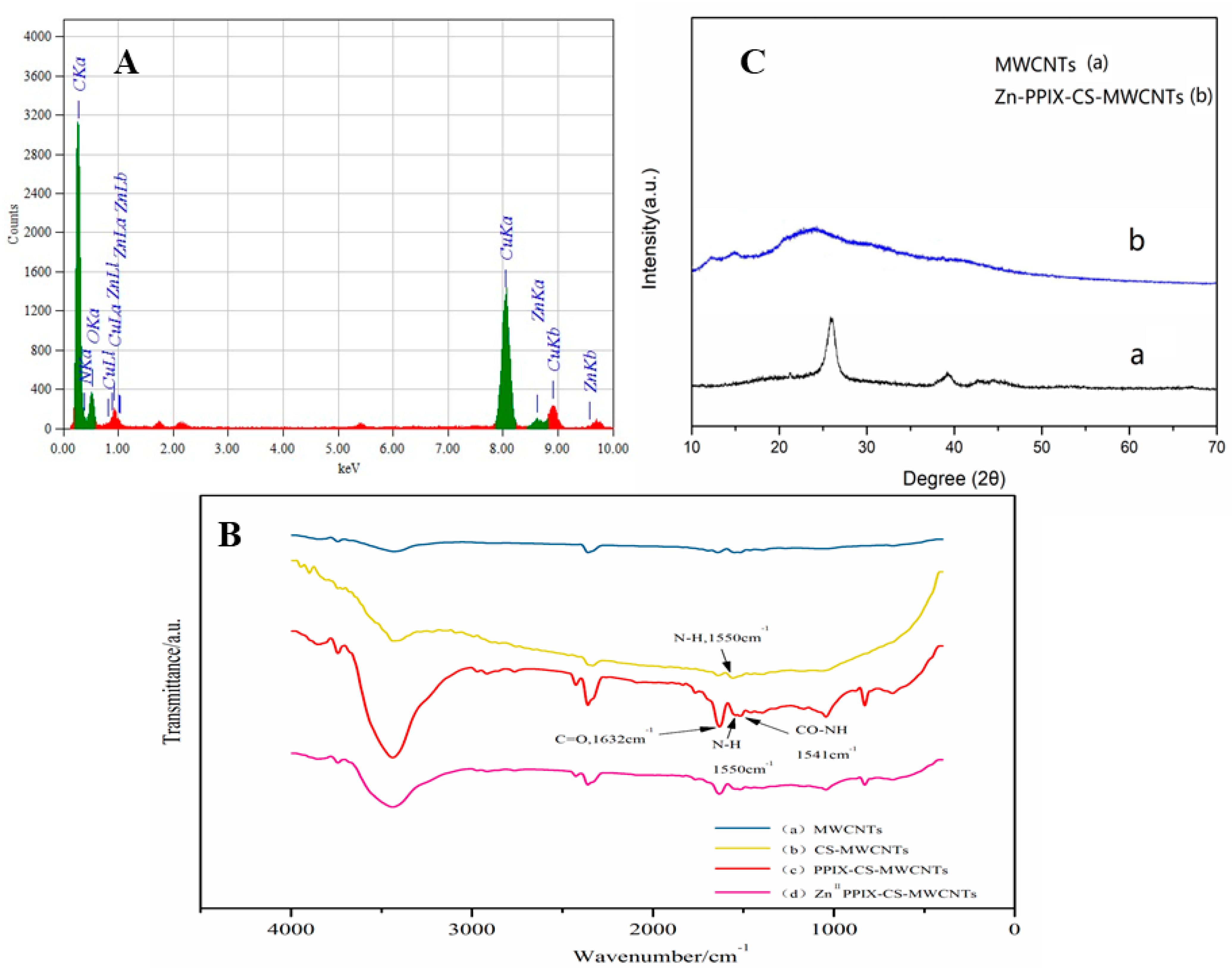
3.2. Characterization of Zn–PPIX–CS–MWCNTs
3.3. Electrochemical Performance and Electrocatalytic Activity of Zn–PPIX–CS–MWCNTs
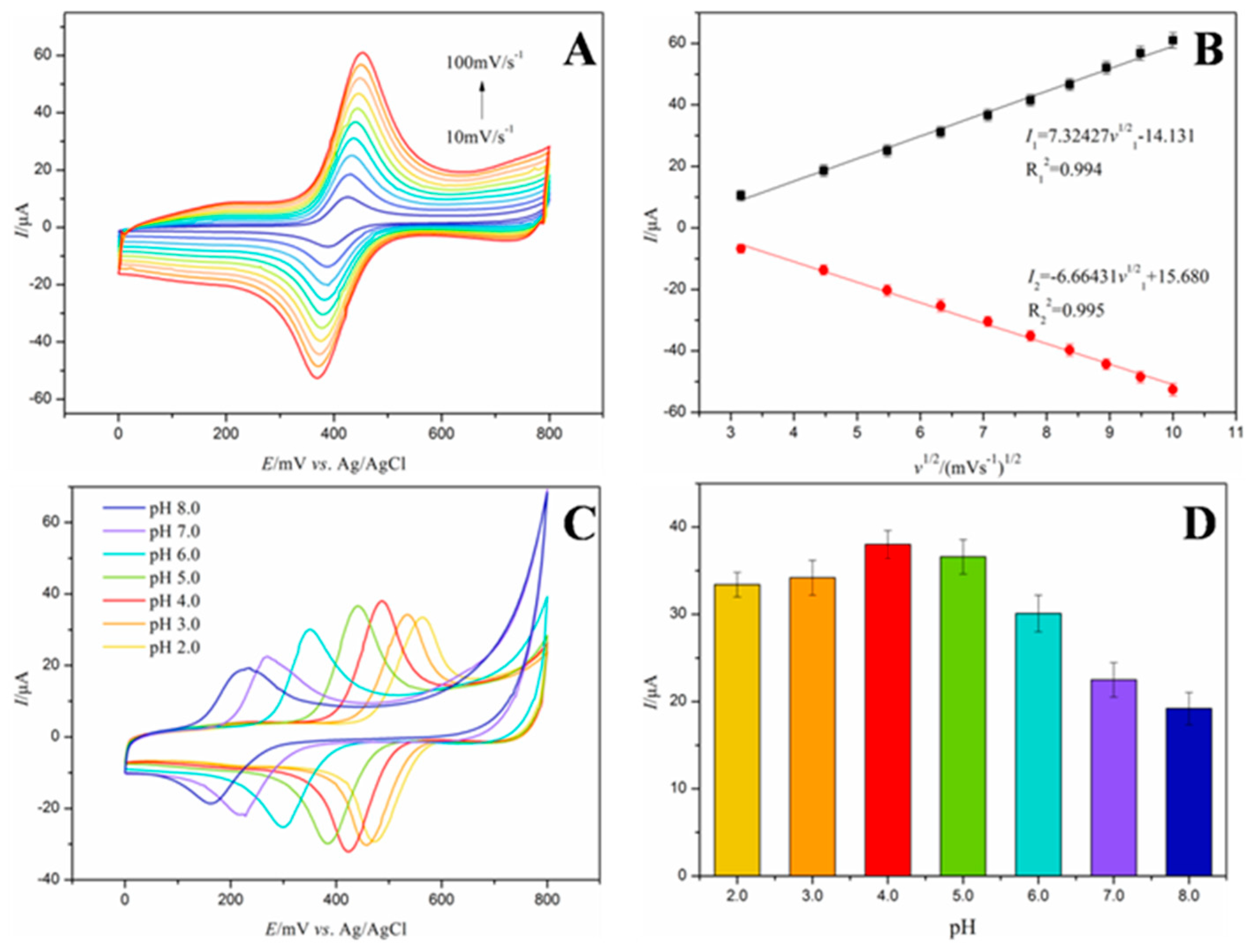
3.4. Scanning-Rate Optimization and pH-Gradient Optimization of the Zn–PPIX–CS–MWCNTs/GCE Electrode
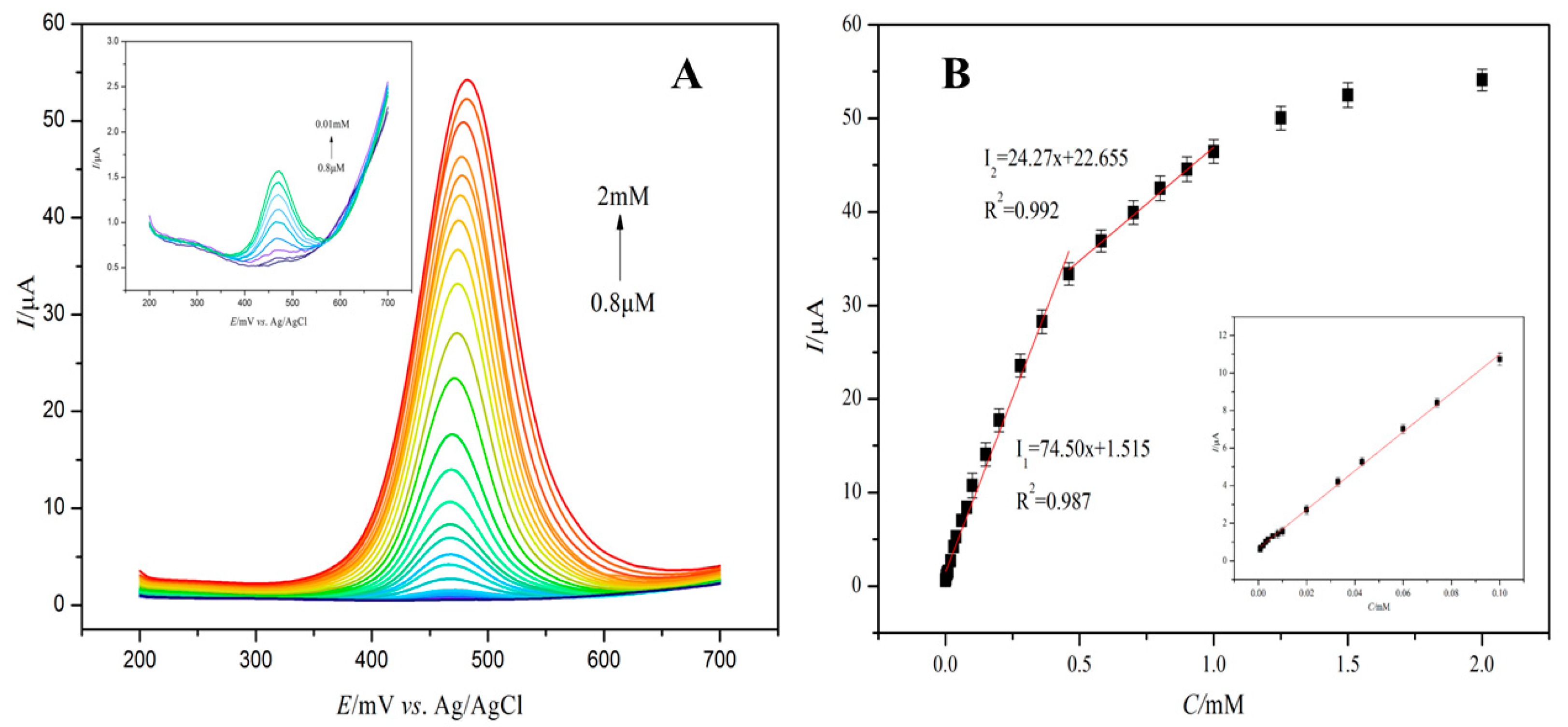
3.5. Electrochemical Detection of Caffeic Acid by Zn–PPIX–CS–MWCNT-Modified GC Electrode
3.6. Study on Repeatability, Stability, Selectivity, and Applicability of Caffeic-Acid Sensors
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Leite, F.R.F.; Santos, W.D.J.R.; Kubota, L.T. Selective determination of caffeic acid in wines with electrochemical sensor based on molecularly imprinted siloxanes. Sens. Actuators B Chem. 2014, 193, 238–246. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, L.; Bei, J.; Zhao, Q.; Li, X.; He, J.; Yao, Y. An enhanced photo-electrochemical sensor constructed from pillar [5]arene functionalized Au NPs for ultrasensitive detection of caffeic acid. Talanta 2022, 243, 123322. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Qiu, S.; Wang, L.; Zhang, N.; Shi, Y.; Zhou, H.; Liu, X.; Shao, L.; Liu, X.; Chen, J.; et al. Reproductive and developmental toxicity study of caffeic acid in mice. Food Chem. Toxicol. 2019, 123, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.C.; Ou, T.T.; Wu, C.H.; Wang, C.J. Prevention of diet-induced hyperlipidemia and obesity by caffeic acid in C57BL/6 mice through regulation of hepatic lipogenesis gene expression. J. Agric. Food Chem. 2013, 61, 11082–11088. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.S.; Park, S.H.; Kim, M.S.; Han, S.Y.; Kim, H.S.; Kang, Y.H. Caffeic acid disturbs monocyte adhesion onto cultured endothelial cells stimulated by adipokine resistin. J. Agric. Food Chem. 2012, 60, 2730–2739. [Google Scholar] [CrossRef]
- Wan, Y.; Wang, F.; Zou, B.; Shen, Y.; Li, Y.; Zhang, A.; Fu, G. Molecular mechanism underlying the ability of caffeic acid to decrease uric acid levels in hyperuricemia rats. J. Funct. Foods 2019, 57, 150–156. [Google Scholar] [CrossRef]
- Santos, N.A.; Damasceno, S.S.; de Araújo, P.H.M.; Marques, V.C.; Rosenhaim, R.; Fernandes, V.J.; Queiroz, N.; Santos, I.M.; Maia, A.S.; Souza, A.G. Caffeic Acid: An Efficient Antioxidant for Soybean Biodiesel Contaminated with Metals. Energy Fuels 2011, 25, 4190–4194. [Google Scholar] [CrossRef]
- Xiang, X.; Shi, J.; Huang, F.; Zheng, M.; Deng, Q. Quantum dots-based label-free fluorescence sensor for sensitive and non-enzymatic detection of caffeic acid. Talanta 2015, 141, 182–187. [Google Scholar] [CrossRef]
- Gültekin, A.; Karanfil, G.; Kuş, M.; Sönmezoğlu, S.; Say, R. Preparation of MIP-based QCM nanosensor for detection of caffeic acid. Talanta 2014, 119, 533–537. [Google Scholar] [CrossRef]
- Tang, C.; Sojinu, O.S. Simultaneous determination of caffeic acid phenethyl ester and its metabolite caffeic acid in dog plasma using liquid chromatography tandem mass spectrometry. Talanta 2012, 94, 232–239. [Google Scholar] [CrossRef]
- Ye, L.H.; Du, L.J.; Cao, J. Fatty acids-based microemulsion liquid chromatographic determination of multiple caffeoylquinic acid isomers and caffeic acid in honeysuckle sample. J. Pharm. Biomed. Anal. 2019, 171, 22–29. [Google Scholar] [CrossRef]
- Filik, H.; Cetintas, G.; Avan, A.A.; Aydar, S.; Koc, S.N.; Boz, I. Square-wave stripping voltammetric determination of caffeic acid on electrochemically reduced graphene oxide-Nafion composite film. Talanta 2013, 116, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, C.; Curulli, A.; Pasquali, M.; Zane, D. Determination of caffeic acid in wine using PEDOT film modified electrode. Food Chem. 2014, 156, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Bharath, G.; Alhseinat, E.; Madhu, R.; Mugo, S.M.; Alwasel, S.; Harrath, A.H. Facile synthesis of Au@α-Fe2O3@RGO ternary nanocomposites for enhanced electrochemical sensing of caffeic acid toward biomedical applications. J. Alloy. Compd. 2018, 750, 819–827. [Google Scholar] [CrossRef]
- Satyanarayana, M.; Goud, K.Y.; Reddy, K.K.; Kumar, V.S.; Gobi, K.V. Silver nanoparticles impregnated chitosan layered carbon nanotube as sensor interface for electrochemical detection of clopidogrel in-vitro. Mater. Sci. Eng. C 2019, 101, 103–110. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, C.; Chen, Y.; Wang, Z.; Lan, L.; Wang, Y.; Han, B.; Pan, M.; Jiao, J.; Chen, Q. A simple immunosensor for alpha-fetoprotein determination based on gold nanoparticles-dextran-reduced graphene oxide. J. Electroanal. Chem. 2019, 833, 126–132. [Google Scholar] [CrossRef]
- Chahardoli, A.; Qalekhani, F.; Shokoohinia, Y.; Fattahi, A. Caffeic acid based titanium dioxide nanoparticles: Blood compatibility, anti-inflammatory, and cytotoxicity. J. Mol. Liq. 2022, 361, 119674. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, Z.; Liu, H.; Guo, J.; Long, S. Hybrid molecules based on caffeic acid as potential therapeutics: A focused review. European. J. Med. Chem. 2022, 243, 114745. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Feng, S.; Khan, M.A.; Lv, J.; Chen, S.-M.; Hao, Q.; Lei, W. Highly sensitivity electrochemical sensor based on ErGO/MWCNTs nanohybrid for 2,4-dinitroanisole electroanalysis. Microchem. J. 2019, 151, 104226. [Google Scholar] [CrossRef]
- Kozonoe, C.E.; de Paiva FloroBonfim, R.; Brito Alves, R.M.; Schmal, M. The Fe-Co-Cu supported on MWCNT as catalyst for the tri-reforming of methane—Investigating the structure changes of the catalysts. Fuel 2019, 256, 115917. [Google Scholar] [CrossRef]
- Song, Y.; Zhao, M.; Wang, X.; Qu, H.; Liu, Y.; Chen, S. Simultaneous electrochemical determination of catechol and hydroquinone in seawater using Co3O4/MWCNTs/GCE. Mater. Chem. Phys. 2019, 234, 217–223. [Google Scholar] [CrossRef]
- Al-Husseini, A.; Al-sammarraie, A.; Rashid Saleh, W. Specific NH3 Gas Sensor Worked at Room Temperature Based on MWCNTs-OH Network. Nano Hybrids Compos. 2018, 23, 8–16. [Google Scholar] [CrossRef]
- Faraji, M.; Abedini, A. Fabrication of electrochemically interconnected MoO3/GO/MWCNTs/graphite sheets for high performance all-solid-state symmetric supercapacitor. Int. J. Hydrogen Energy 2019, 44, 2741–2751. [Google Scholar] [CrossRef]
- Gholivand, M.B.; Shamsipur, M.; Ehzari, H. Cetirizine dihydrochloride sensor based on nano composite chitosan, MWCNTs and ionic liquid. Microchem. J. 2019, 146, 692–700. [Google Scholar] [CrossRef]
- Trani, A.; Petrucci, R.; Marrosu, G.; Zane, D.; Curulli, A. Selective electrochemical determination of caffeine at a gold-chitosan nanocomposite sensor: May little change on nanocomposites synthesis affect selectivity? J. Electroanal. Chem. 2017, 788, 99–106. [Google Scholar] [CrossRef]
- Yang, X.; Tu, Y.; Li, L.; Shang, S.; Tao, X.M. Well-dispersed chitosan/graphene oxide nanocomposites. ACS Appl. Mater. Interfaces 2010, 2, 1707–1713. [Google Scholar] [CrossRef]
- Abdolrahimi, M.; Seifi, M.; Ramezanzadeh, M.H. Study the effect of acetic acid on structural, optical and mechanical properties of PVA/chitosan/MWCNT films. Chin. J. Phys. 2018, 56, 221–230. [Google Scholar] [CrossRef]
- Sun, L.; Yuan, Z.; Gong, W.; Xu, Z.; Lu, J.; Zou, Q.; Wu, Y.; Su, G.; Wang, H. Removal of trace Cr(VI) from water using chitosan-iron nanowires in porous anodic alumina. Sci. China Chem. 2015, 59, 383–386. [Google Scholar] [CrossRef]
- Liu, Y.; Yan, S.; Li, M.; Wang, K.; Zeng, D.; Yang, H. A new polyethyleneinmine-gold nanoparticles-protoporphyrin cobalt (II) nanocomposite for electrochemical hydrogen peroxide biosensing. Colloids Surf. A Physicochem. Eng. Asp. 2019, 572, 345–351. [Google Scholar] [CrossRef]
- Castro, K.A.D.F.; Rodrigues, J.M.M.; Mendes, R.F.; Neves, M.d.G.P.M.S.; Simões, M.M.Q.; Cavaleiro, J.A.S.; Almeida Paz, F.A.; Tomé, J.P.C.; Nakagaki, S. New copper porphyrins as functional models of catechol oxidase. J. Catal. 2016, 344, 303–312. [Google Scholar] [CrossRef]
- Zhang, M.; Smith, A.; Gorski, W. Carbon Nanotube—Chitosan System for Electrochemical Sensing Based on Dehydrogenase Enzymes. Anal. Chem. 2004, 76, 5045–5050. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, M.; Patil, M.; Epur, R.; Yun, Y.; Shanov, V.; Schulz, M.; Heineman, W.R.; Datta, M.K.; Kumta, P.N. Gold-coated carbon nanotube electrode arrays: Immunosensors for impedimetric detection of bone biomarkers. Biosens. Bioelectron. 2016, 77, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Lu, H. Mesoporous structured MIPs@CDs fluorescence sensor for highly sensitive detection of TNT. Biosens. Bioelectron. 2016, 85, 950–956. [Google Scholar] [CrossRef] [PubMed]
- Di Carlo, G.; Curulli, A.; Toro, R.G.; Bianchini, C.; De Caro, T.; Padeletti, G.; Zane, D.; Ingo, G.M. Green synthesis of gold-chitosan nanocomposites for caffeic acid sensing. Langmuir 2012, 28, 5471–5479. [Google Scholar] [CrossRef]
- Diaconu, M.; Litescu, S.C.; Radu, G.L. Laccase–MWCNT–chitosan biosensor—A new tool for total polyphenolic content evaluation from in vitro cultivated plants. Sens. Actuators B Chem. 2010, 145, 800–806. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrode Materials | Linear Range (μM) | Detection Limit (μM) | Reference |
|---|---|---|---|
| CDs@MIPs | 0.5–200 | 0.11 | [33] |
| Au@a-Fe2O3@RGO | 19–1869 | 0.098 | [14] |
| Nafion/ER-GO | 0.1–10 | 0.091 | [12] |
| AuNP−CHIT | 0.05–2000 | 0.025 | [34] |
| laccase-MWCNT-chitosan | 0.735–500 | 0.275 | [35] |
| Zn-PPIX-CS-MWCNTs | 0.8–1600 | 0.022 | This work |
| Sample | CA Added (μM) | HPLC Found (µM) | CA Found (µM) | Recovery (%) |
|---|---|---|---|---|
| 1 | 0 | 3.15 | 3.17 | 100.6 |
| 2 | 10 | 13.15 | 13.30 | 101.0 |
| 3 | 20 | 23.10 | 22.80 | 98.7 |
| 4 | 30 | 34.80 | 34.10 | 98.0 |
| 5 | 40 | 42.50 | 42.90 | 100.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, B.; Wen, X.; Wang, J.; Sun, Y. A Novel Nanocomposite of Zn(II)-Protoporphyrin-Chitosan-Multi Walled Carbon Nanotubes and the Application to Caffeic Acid Sensing. Nanomaterials 2022, 12, 3412. https://doi.org/10.3390/nano12193412
Han B, Wen X, Wang J, Sun Y. A Novel Nanocomposite of Zn(II)-Protoporphyrin-Chitosan-Multi Walled Carbon Nanotubes and the Application to Caffeic Acid Sensing. Nanomaterials. 2022; 12(19):3412. https://doi.org/10.3390/nano12193412
Chicago/Turabian StyleHan, Bingkai, Xin Wen, Jinneng Wang, and Yingrui Sun. 2022. "A Novel Nanocomposite of Zn(II)-Protoporphyrin-Chitosan-Multi Walled Carbon Nanotubes and the Application to Caffeic Acid Sensing" Nanomaterials 12, no. 19: 3412. https://doi.org/10.3390/nano12193412