
Exploiting Nanoscale Complexion in LATP Solid-State Electrolyte via Interfacial Mg2+ Doping


Abstract
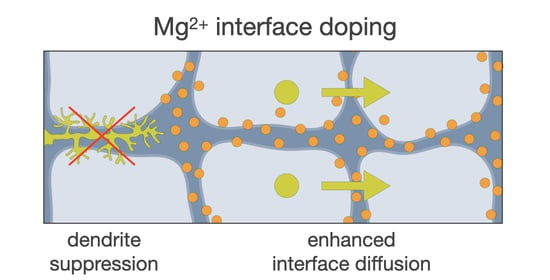
:
1. Introduction
2. Materials and Methods
2.1. Molecular Dynamics Simulations
2.2. Underlying Li1+xAlxTi2−x(PO4)3 Core-Shell Force Field
2.3. Force Field Extension by Mg2+
2.4. Monte-Carlo Swapping Protocol
2.5. Ion Dynamics Analysis
3. Results
3.1. Mg2+ as Interfacial Dopant Candidate
3.2. Atomistic Structures of Mg2+ Interfacially Doped LATP
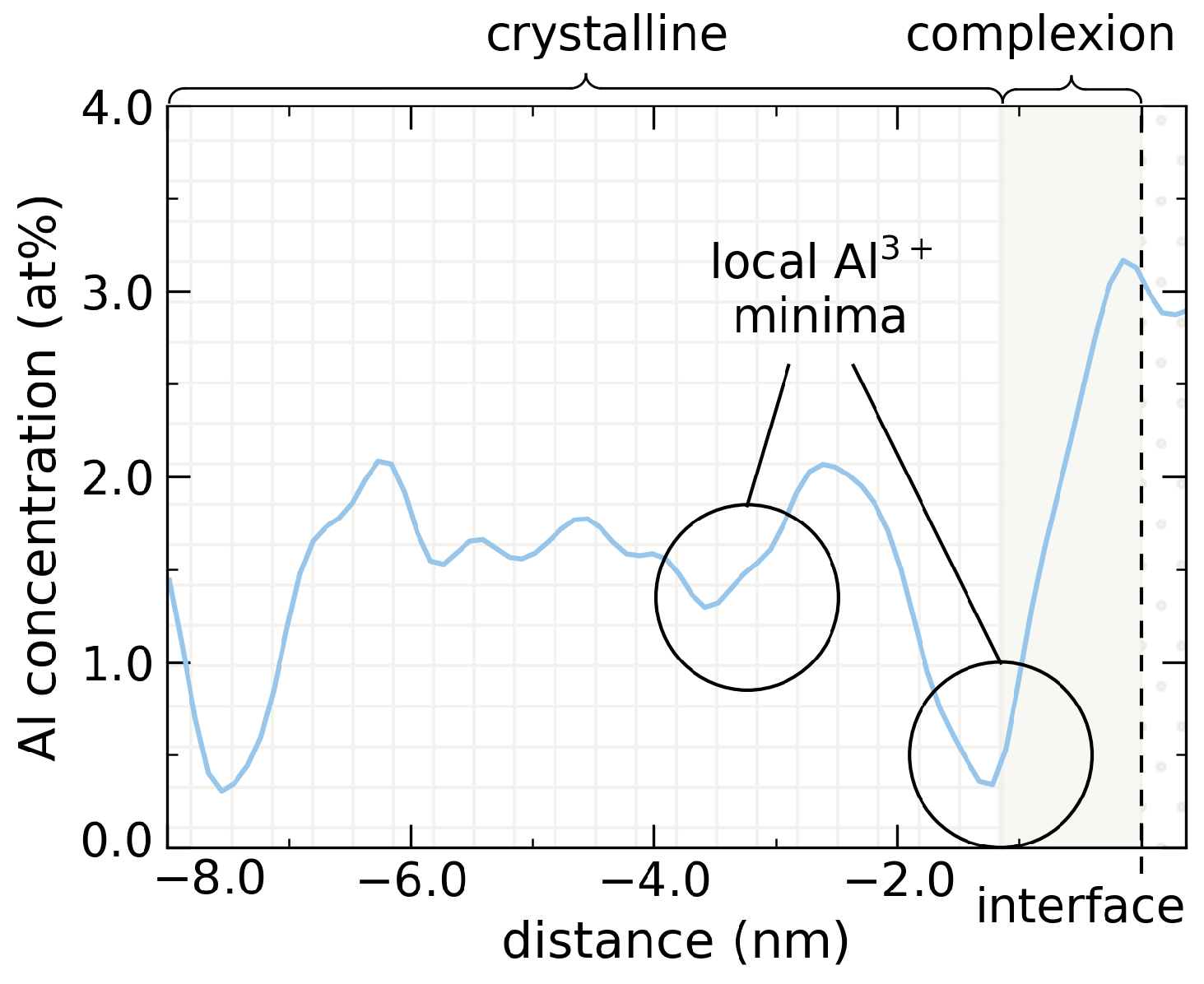
3.3. Dopant Bleeding into Grain Bulk Domain
3.4. Bleeding Implications on Crystalline Bulk
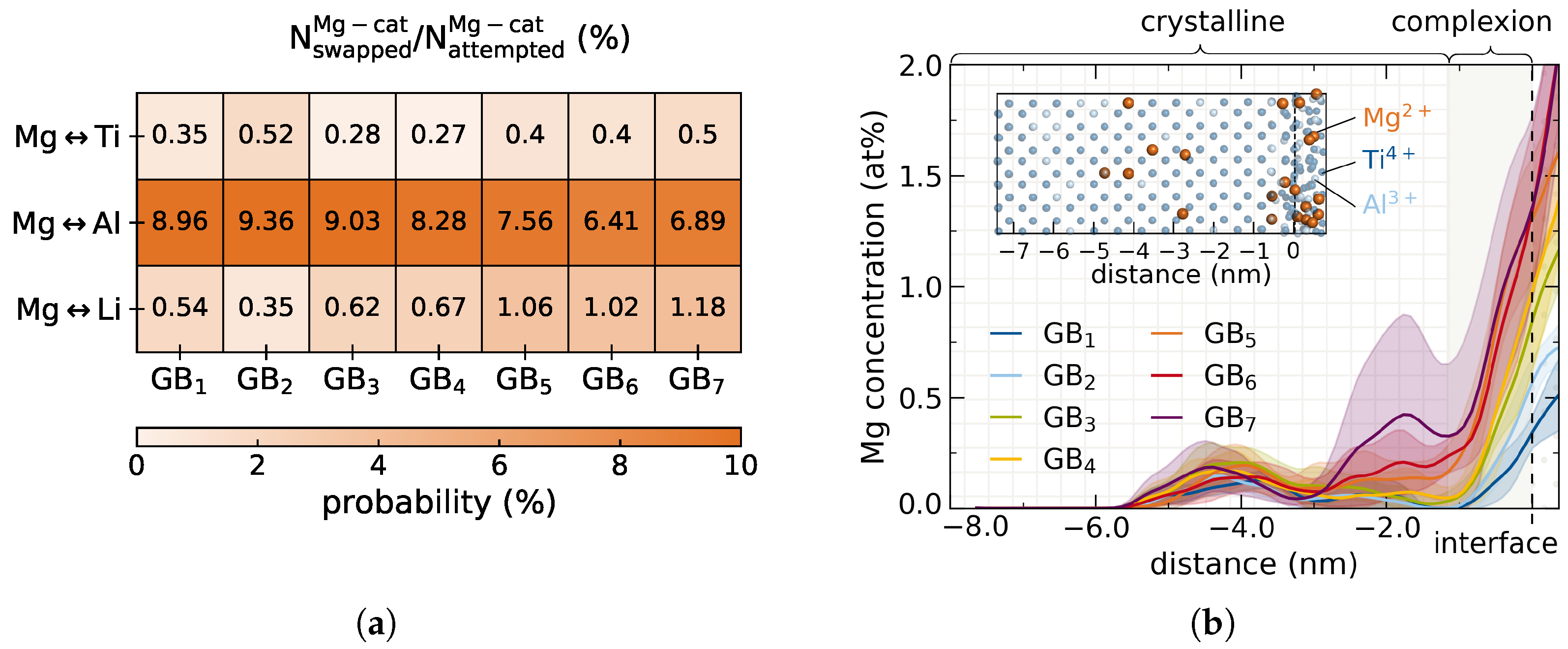
 Mg → Li), another one a former Al site (
Mg → Li), another one a former Al site (  Mg → Al) and the third a former Ti site (
Mg → Al) and the third a former Ti site (  Mg → Ti). Li positions of the full 2 ns trajectory are projected onto the same atomistic structure and the isosurface of the resulting Li density reveals the interconnected pathways.
Mg → Ti). Li positions of the full 2 ns trajectory are projected onto the same atomistic structure and the isosurface of the resulting Li density reveals the interconnected pathways. ). This is similar to the Ti (
). This is similar to the Ti (  ) and Al (
) and Al (  ) conductivity on the same lattice sites. For Mg doped into Li channels (
) conductivity on the same lattice sites. For Mg doped into Li channels (  ), the conductivity is 2–3 orders of magnitude lower as compared to Li in C. Even at elevated temperatures the Mg ions are virtually immobile and are thus not expected to penetrate further into the grain with electrolyte aging.
), the conductivity is 2–3 orders of magnitude lower as compared to Li in C. Even at elevated temperatures the Mg ions are virtually immobile and are thus not expected to penetrate further into the grain with electrolyte aging. ) in the doped crystal structures does not seem to be significantly affected. Bleeding of Mg does lead to a decrease in Li conductivity, more so for high doping concentrations C–C where Li channels are more heavily infiltrated. Yet, even the largest decrease is merely −0.51% of the observed reference. Therefore, due to the very little bleeding of the dopant, the crystalline bulk performance of LATP grains is not appreciably compromised, encouraging interfacial doping of LATP with the divalent Mg.
) in the doped crystal structures does not seem to be significantly affected. Bleeding of Mg does lead to a decrease in Li conductivity, more so for high doping concentrations C–C where Li channels are more heavily infiltrated. Yet, even the largest decrease is merely −0.51% of the observed reference. Therefore, due to the very little bleeding of the dopant, the crystalline bulk performance of LATP grains is not appreciably compromised, encouraging interfacial doping of LATP with the divalent Mg.3.5. Interphase Modifications via Aliovalent Doping
3.5.1. Dopant Impact on Structural Features
3.5.2. Non-Trivial Effects with Doping Concentration
3.6. Dopant Impact on Ion Dynamics
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| LIB | Lithium Ion Battery |
| ASSB | All Solid-State Battery |
| SSE | Solid-State Electrolyte |
| LATP | LiAlTi(PO) |
| TEM | Transmission Electron Microscopy |
| APT | Atom Probe Tomography |
| TM | Transition Metal |
| MD | Molecular Dynamics |
| DFT | Density Functional Theory |
| MC | Monte-Carlo |
| MSD | Mean Square Displacement |
| NASICON | NA Super Ionic CONductor |
| NN | Nearest Neighbor |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atom Types | |||||||||||
| Species | Mass (u) | Charge (e) | |||||||||
| Ti 1 | 47.867 | +2.196 | |||||||||
| Al | 26.982 | +1.647 | |||||||||
| Li 2 | 6.940 | +0.549 | |||||||||
| P | 30.974 | +2.745 | |||||||||
| O 1 | 15.698 | +0.500 | |||||||||
| O 1 | 0.301 | -1.598 | |||||||||
| Mg | 24.305 | +1.098 | |||||||||
| Buckingham Parameters | |||||||||||
| species | A (eV) | (Å) | C (eVÅ) | ||||||||
| Li-Li 2 | 38,533.955 | 0.100 | 0.000 | ||||||||
| Ti-Li 2 | 33,089.570 | 0.127 | 0.000 | ||||||||
| Ti-Ti 1 | 31,120.528 | 0.154 | 5.250 | ||||||||
| Li-O 2 | 15,465.549 | 0.167 | 0.000 | ||||||||
| O-O 1 | 11,782.885 | 0.234 | 30.220 | ||||||||
| Ti-O | 18,448.156 | 0.194 | 12.590 | ||||||||
| P-P | 53,210.800 | 0.284 | 0.000 | ||||||||
| P-O | 32,397.875 | 0.155 | 7.831 | ||||||||
| P-Li | 30,393.156 | 0.131 | 0.000 | ||||||||
| P-Ti | 10,469.346 | 0.139 | 136.835 | ||||||||
| Al-Al | 42,700.844 | 0.197 | 0.000 | ||||||||
| Al-Li | 32,315.936 | 0.127 | 0.000 | ||||||||
| Al-Ti | 10,489.082 | 0.131 | 6.862 | ||||||||
| Al-O | 17,491.787 | 0.179 | 7.920 | ||||||||
| Al-P | 10,580.062 | 0.137 | 114.906 | ||||||||
| Mg-Mg | 20,671.120 | 0.148 | 0.000 | ||||||||
| Mg-Li | 24,050.700 | 0.138 | 0.000 | ||||||||
| Mg-Ti | 12,947.270 | 0.117 | 6.200 | ||||||||
| Mg-O | 55,835.770 | 0.179 | 12.620 | ||||||||
| Mg-P | 56,162.410 | 0.130 | 120.000 | ||||||||
| Mg-Al | 21,485.920 | 0.230 | 0.580 | ||||||||
| Core-Shell Force Constants3 | |||||||||||
| species | k (eVÅ) | ||||||||||
| O-O | 88.6 | ||||||||||
References
- Ederer, B. BMW Group Strengthens Leadership Position in Battery Technology with Investment in Solid-State Innovator Solid Power. 2021. Available online: https://www.press.bmwgroup.com/global/article/detail/T0331495EN/bmw-group-strengthens-leadership-position-in-battery-technology-with-investment-in-solid-state-innovator-solid-power?language=en (accessed on 13 June 2022).
- Beltz, E.; Kulawik, J. Volkswagen Increases Stake in QuantumScape | Volkswagen Newsroom. 2020. Available online: https://www.volkswagen-newsroom.com/en/press-releases/volkswagen-increases-stake-in-quantumscape-6136 (accessed on 13 June 2022).
- Janek, J.; Zeier, W.G. A solid future for battery development. Nat. Energy 2016, 1, 16141. [Google Scholar] [CrossRef]
- Nanda, J.; Wang, C.; Liu, P. Frontiers of solid-state batteries. MRS Bull. 2018, 43, 740–745. [Google Scholar] [CrossRef]
- Bachman, J.C.; Muy, S.; Grimaud, A.; Chang, H.H.; Pour, N.; Lux, S.F.; Paschos, O.; Maglia, F.; Lupart, S.; Lamp, P.; et al. Inorganic Solid-State Electrolytes for Lithium Batteries: Mechanisms and Properties Governing Ion Conduction. Chem. Rev. 2016, 116, 140–162. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, J.; Lu, G.; Li, W.; Tao, Q.; Shi, C.; Jin, H.; Chen, G.; Wang, S. Fundamentals of Electrolytes for Solid-State Batteries: Challenges and Perspectives. Front. Mater. 2020, 7, 111. [Google Scholar] [CrossRef]
- Cao, D.; Sun, X.; Li, Q.; Natan, A.; Xiang, P.; Zhu, H. Lithium Dendrite in All-Solid-State Batteries: Growth Mechanisms, Suppression Strategies, and Characterizations. Matter 2020, 3, 57–94. [Google Scholar] [CrossRef]
- Byeon, Y.W.; Kim, H. Review on Interface and Interphase Issues in Sulfide Solid-State Electrolytes for All-Solid-State Li-Metal Batteries. Electrochem 2021, 2, 452–471. [Google Scholar] [CrossRef]
- Stegmaier, S.; Schierholz, R.; Povstugar, I.; Barthel, J.; Rittmeyer, S.P.; Yu, S.; Wengert, S.; Rostami, S.; Kungl, H.; Reuter, K.; et al. Nano-Scale Complexions Facilitate Li Dendrite-Free Operation in LATP Solid-State Electrolyte. Adv. Energy Mater. 2021, 11, 2100707. [Google Scholar] [CrossRef]
- Raj, V.; Venturi, V.; Kankanallu, V.R.; Kuiri, B.; Viswanathan, V.; Aetukuri, N.P.B. Direct correlation between void formation and lithium dendrite growth in solid-state electrolytes with interlayers. Nat. Mater. 2022. [Google Scholar] [CrossRef]
- Kim, K.J.; Balaish, M.; Wadaguchi, M.; Kong, L.; Rupp, J.L. Solid-State Li–Metal Batteries: Challenges and Horizons of Oxide and Sulfide Solid Electrolytes and Their Interfaces. Adv. Energy Mater. 2021, 11, 2002689. [Google Scholar] [CrossRef]
- Tang, M.; Carter, W.C.; Cannon, R.M. Diffuse interface model for structural transitions of grain boundaries. Phys. Rev. B 2006, 73, 024102. [Google Scholar] [CrossRef]
- Luo, J. Interfacial engineering of solid electrolytes. J. Materiomics 2015, 1, 22–32. [Google Scholar] [CrossRef]
- Luo, J. Let thermodynamics do the interfacial engineering of batteries and solid electrolytes. Energy Storage Mater. 2019, 21, 50–60. [Google Scholar] [CrossRef]
- Cantwell, P.R.; Frolov, T.; Rupert, T.J.; Krause, A.R.; Marvel, C.J.; Rohrer, G.S.; Rickman, J.M.; Harmer, M.P. Grain Boundary Complexion Transitions. Annu. Rev. Mater. Res. 2020, 50, 465–492. [Google Scholar] [CrossRef]
- Dillon, S.J.; Harmer, M.P. Demystifying the role of sintering additives with “complexion”. J. Eur. Ceram. Soc. 2008, 28, 1485–1493. [Google Scholar] [CrossRef]
- Kasap, S.; Capper, P. (Eds.) Springer Handbook of Electronic and Photonic Materials, 2nd ed.; Springer: Cham, Switzerland, 2017. [Google Scholar]
- Maier, J. Nanoionics: Ion transport and electrochemical storage in confined systems. Mater. Sustain. Energy 2010, 4, 160–170. [Google Scholar] [CrossRef]
- Maier, J. Pushing nanoionics to the limits: Charge carrier chemistry in extremely small systems. Chem. Mater. 2014, 26, 348–360. [Google Scholar] [CrossRef]
- Miao, X.; Wang, H.; Sun, R.; Wang, C.; Zhang, Z.; Li, Z.; Yin, L. Interface engineering of inorganic solid-state electrolytes for high-performance lithium metal batteries. Energy Environ. Sci. 2020, 13, 3780–3822. [Google Scholar] [CrossRef]
- Ding, Z.; Li, J.; Li, J.; An, C. Review—Interfaces: Key Issue to Be Solved for All Solid-State Lithium Battery Technologies. J. Electrochem. Soc. 2020, 167, 070541. [Google Scholar] [CrossRef]
- Du, M.; Liao, K.; Lu, Q.; Shao, Z. Recent advances in the interface engineering of solid-state Li-ion batteries with artificial buffer layers: Challenges, materials, construction, and characterization. Energy Environ. Sci. 2019, 12, 1780–1804. [Google Scholar] [CrossRef]
- Wang, S.; Wang, J.; Liu, J.; Song, H.; Liu, Y.; Wang, P.; He, P.; Xu, J.; Zhou, H. Ultra-fine surface solid-state electrolytes for long cycle life all-solid-state lithium-air batteries. J. Mater. Chem. A 2018, 6, 21248–21254. [Google Scholar] [CrossRef]
- Xu, B.; Li, W.; Duan, H.; Wang, H.; Guo, Y.; Li, H.; Liu, H. Li3PO4-added garnet-type Li6.5La3Zr1.5Ta0.5O12 for Li-dendrite suppression. J. Power Sources 2017, 354, 68–73. [Google Scholar] [CrossRef]
- Miara, L.J.; Richards, W.D.; Wang, Y.E.; Ceder, G. First-Principles Studies on Cation Dopants and Electrolyte|Cathode Interphases for Lithium Garnets. Chem. Mater. 2015, 27, 4040–4047. [Google Scholar] [CrossRef]
- Nikodimos, Y.; Abrha, L.H.; Weldeyohannes, H.H.; Shitaw, K.N.; Temesgen, N.T.; Olbasa, B.W.; Huang, C.J.; Jiang, S.K.; Wang, C.H.; Sheu, H.S.; et al. A new high-Li+-conductivity Mg-doped Li1.5Al0.5Ge1.5(PO4)3 solid electrolyte with enhanced electrochemical performance for solid-state lithium metal batteries. J. Mater. Chem. A 2020, 8, 26055–26065. [Google Scholar] [CrossRef]
- Richards, W.D.; Miara, L.J.; Wang, Y.; Kim, J.C.; Ceder, G. Interface Stability in Solid-State Batteries. Chem. Mater. 2016, 28, 266–273. [Google Scholar] [CrossRef]
- Han, X.; Gong, Y.; Fu, K.; He, X.; Hitz, G.T.; Dai, J.; Pearse, A.; Liu, B.; Wang, H.; Rubloff, G.; et al. Negating interfacial impedance in garnet-based solid-state Li metal batteries. Nat. Mater. 2017, 16, 572–579. [Google Scholar] [CrossRef]
- Camacho-Forero, L.E.; Balbuena, P.B. Elucidating Interfacial Phenomena between Solid-State Electrolytes and the Sulfur-Cathode of Lithium-Sulfur Batteries. Chem. Mater. 2020, 32, 360–373. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short–Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Hockney, R.W.; Eastwood, J.W. Computer Simulation Using Particles, 1st ed.; Taylor & Francis: New York, NY, USA, 1988. [Google Scholar]
- Shinoda, W.; Shiga, M.; Mikami, M. Rapid estimation of elastic constants by molecular dynamics simulation under constant stress. Phys. Rev. B 2004, 69, 134103. [Google Scholar] [CrossRef]
- Lewis, G.V.; Catlow, C.R. Potential models for ionic oxides. J. Phys. C Solid State Phys. 1985, 18, 1149–1161. [Google Scholar] [CrossRef]
- Kerisit, S.; Aaron Deskins, N.; Rosso, K.M.; Dupais, M. A shell model for atomistic simulation of charge transfer in titania. J. Phys. Chem. C 2008, 112, 7678–7688. [Google Scholar] [CrossRef]
- Tonda, A. Inspyred: Bio-inspired algorithms in Python. Genet. Program. Evolvable Mach. 2020, 21, 269–272. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Kristallogr. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Garrity, K.F.; Bennett, J.W.; Rabe, K.M.; Vanderbilt, D. Pseudopotentials for high-throughput DFT calculations. Comput. Mater. Sci. 2014, 81, 446–452. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Türk, H.; Schmidt, F.P.; Götsch, T.; Girgsdies, F.; Hammud, A.; Ivanov, D.; Vinke, I.C.; de Haart, L.G.; Eichel, R.A.; Reuter, K.; et al. Complexions at the Electrolyte/Electrode Interface in Solid Oxide Cells. Adv. Mater. Interfaces 2021, 8, 2100967. [Google Scholar] [CrossRef]
- Geyer, C.J.; Thompson, E.A. Annealing Markov chain monte carlo with applications to ancestral inference. J. Am. Stat. Assoc. 1995, 90, 909–920. [Google Scholar] [CrossRef]
- Earl, D.J.; Deem, M.W. Parallel tempering: Theory, applications, and new perspectives. Phys. Chem. Chem. Phys. 2005, 7, 3910–3916. [Google Scholar] [CrossRef]
- Mehrer, H. Diffusion in Solids, 1st ed.; Springer: Berlin, Germany, 2007. [Google Scholar] [CrossRef]
- He, X.; Zhu, Y.; Epstein, A.; Mo, Y. Statistical variances of diffusional properties from ab initio molecular dynamics simulations. npj Comput. Mater. 2018, 4, 18. [Google Scholar] [CrossRef]
- Cheung, E.A.; Nguyen, H.; Avdeev, M.; De Souza, N.R.; Meng, Y.S.; Sharma, N. Insights into the Fast Sodium Conductor NASICON and the Effects of Mg2+ Doping on Na+ Conductivity. Chem. Mater. 2021, 33, 8768–8774. [Google Scholar] [CrossRef]
- Choi, S.H.; Lee, S.J.; Yoo, D.J.; Park, J.H.; Park, J.H.; Ko, Y.N.; Park, J.; Sung, Y.E.; Chung, S.Y.; Kim, H.; et al. Marginal Magnesium Doping for High-Performance Lithium Metal Batteries. Adv. Energy Mater. 2019, 9, 1902278. [Google Scholar] [CrossRef]
- Gao, P.; Wu, H.; Zhang, X.; Jia, H.; Kim, J.M.; Engelhard, M.H.; Niu, C.; Xu, Z.; Zhang, J.G.; Xu, W. Optimization of Magnesium-Doped Lithium Metal Anode for High Performance Lithium Metal Batteries through Modeling and Experiment. Angew. Chem. Int. Ed. 2021, 60, 16506–16513. [Google Scholar] [CrossRef] [PubMed]
- Shannon, R.D. Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides. Acta Crystallogr. 1976, A32, 751–767. [Google Scholar] [CrossRef]
- Squires, A.; Dean, J.M.; Morgan, B.J. Aliovalent doping response and impact on ionic conductivity in the antiperovskite solid electrolyte Li3OCl. ChemRxiv 2021. [Google Scholar] [CrossRef]
- Aono, H.; Sugimoto, E.; Sadaoka, Y.; Imanaka, N.; Adachi, G.y. Ionic Conductivity of the Lithium Titanium Phosphate (Li1+XMXTi2−X(PO4)3, M=Al, Sc, Y and La) Systems. J. Electrochem. Soc. 1989, 136, 590–591. [Google Scholar] [CrossRef]
- Aono, H.; Sugimoto, E.; Sadaoka, Y.; Imanaka, N.; Adachi, G.y. Ionic Conductivity of Solid Electrolytes Based on Lithium Titanium Phosphate. J. Electrochem. Soc. 1990, 137, 1023–1027. [Google Scholar] [CrossRef]
- Redhammer, G.J.; Rettenwander, D.; Pristat, S.; Dashjav, E.; Kumar, C.M.; Topa, D.; Tietz, F. A single crystal X-ray and powder neutron diffraction study on NASICON-type Li1+xAlxTi2−x(PO4)3 (0≤x≤0.5) crystals: Implications on ionic conductivity. Solid State Sci. 2016, 60, 99–107. [Google Scholar] [CrossRef]
- Monchak, M.; Hupfer, T.; Senyshyn, A.; Boysen, H.; Chernyshov, D.; Hansen, T.; Schell, K.G.; Bucharsky, E.C.; Hoffmann, M.J.; Ehrenberg, H. Lithium Diffusion Pathway in Li1.3Al0.3Ti1.7(PO4)3 (LATP) Superionic Conductor. Inorg. Chem. 2016, 55, 2941–2945. [Google Scholar] [CrossRef]
- Scheurer, C. Atomistic Grain-Boundary Model of the Solid-State-Electrolyte LATP. Max Planck Digit. Libr. Edmond—Open Res. Data Repos. Max Planck Soc. 2022. Available online: https://edmond.mpdl.mpg.de/dataset.xhtml?persistentId=doi:10.17617/3.KJWDRB (accessed on 10 August 2022).
- Kick, M.; Grosu, C.; Schuderer, M.; Scheurer, C.; Oberhofer, H. Mobile Small Polarons Qualitatively Explain Conductivity in Lithium Titanium Oxide Battery Electrodes. J. Phys. Chem. Lett. 2020, 11, 2535–2540. [Google Scholar] [CrossRef]
- Yu, J.; Sushko, M.L.; Kerisit, S.; Rosso, K.M.; Liu, J. Kinetic Monte Carlo study of ambipolar lithium ion and electron-polaron diffusion into nanostructured TiO2. J. Phys. Chem. Lett. 2012, 3, 2076–2081. [Google Scholar] [CrossRef]
- Mertens, A.; Yu, S.; Schön, N.; Gunduz, D.C.; Tempel, H.; Schierholz, R.; Hausen, F.; Kungl, H.; Granwehr, J.; Eichel, R.A. Superionic bulk conductivity in Li1.3Al0.3Ti1.7(PO4)3 solid electrolyte. Solid State Ionics 2017, 309, 180–186. [Google Scholar] [CrossRef]
- Stegmaier, S.; Voss, J.; Reuter, K.; Luntz, A.C. Li+ Defects in a Solid-State Li Ion Battery: Theoretical Insights with a Li3OCl Electrolyte. Chem. Mater. 2017, 29, 4330–4340. [Google Scholar] [CrossRef]
- Grillon, N.; Bouyssou, É.; Jacques, S.; Gautier, G. Cycle life and statistical predictive reliability model for all-solid-state thin film microbatteries. Microelectron. Reliab. 2019, 93, 102–108. [Google Scholar] [CrossRef]
- Fang, X.; He, Y.; Fan, X.; Zhang, D.; Hu, H. Modeling and simulation in capacity degradation and control of all-solid-state lithium battery based on time-aging polymer electrolyte. Polymers 2021, 13, 1206. [Google Scholar] [CrossRef]
- Guo, J.; Li, Y.; Pedersen, K.; Stroe, D.i. Lithium-Ion Battery Operation, Degradation, and Aging Mechanism in Electric Vehicles: An Overview. Energies 2021, 14, 5220. [Google Scholar] [CrossRef]
- Waetzig, K.; Rost, A.; Heubner, C.; Coeler, M.; Nikolowski, K.; Wolter, M.; Schilm, J. Synthesis and sintering of Li1.3Al0.3Ti1.7(PO4)3 (LATP) electrolyte for ceramics with improved Li+ conductivity. J. Alloys Compd. 2020, 818, 153237. [Google Scholar] [CrossRef]
- Castillo, A.; Yagoubi, S.; Rapaud, O.; Pradeilles, N.; Charpentier, T.; Foy, E.; Khodja, H. Mg2+ Doping Effect on Ionic Conductivity of Li1.15Zr1.85Y0.15(PO4)3 Nasicon-Type Solid Electrolyte for All-Solid-State Lithium Ions Batteries. 1st Int. Symp. Solid State Batter. Available online: https://hal.archives-ouvertes.fr/cea-02340786 (accessed on 13 June 2022).
- Li, J.; Liu, C.; Miao, C.; Kou, Z.; Xiao, W. Enhanced ionic conductivity and electrochemical stability of Indium doping Li1.3Al0.3Ti1.7(PO4)3 solid electrolytes for all-solid-state lithium-ion batteries. Ionics 2022, 28, 63–72. [Google Scholar] [CrossRef]
- Epp, V.; Ma, Q.; Hammer, E.M.; Tietz, F.; Wilkening, M. Very fast bulk Li ion diffusivity in crystalline Li1.5Al0.5Ti1.5(PO4)3 as seen using NMR relaxometry. Phys. Chem. Chem. Phys. 2015, 17, 32115–32121. [Google Scholar] [CrossRef]
- Francisco, B.E.; Stoldt, C.R.; M’Peko, J.C. Lithium-ion trapping from local structural distortions in sodium super ionic conductor (NASICON) electrolytes. Chem. Mater. 2014, 26, 4741–4749. [Google Scholar] [CrossRef]
- Lang, B.; Ziebarth, B.; Elsässer, C. Lithium Ion Conduction in LiTi2(PO4)3 and Related Compounds Based on the NASICON Structure: A First-Principles Study. Chem. Mater. 2015, 27, 5040–5048. [Google Scholar] [CrossRef]
- Krauskopf, T.; Richter, F.H.; Zeier, W.G.; Janek, J. Physicochemical Concepts of the Lithium Metal Anode in Solid-State Batteries. Chem. Rev. 2020, 120, 7745–7794. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Westover, A.S.; Yue, J.; Fan, X.; Wang, F.; Chi, M.; Leonard, D.N.; Dudney, N.J.; Wang, H.; Wang, C. High electronic conductivity as the origin of lithium dendrite formation within solid electrolytes. Nat. Energy 2019, 4, 187–196. [Google Scholar] [CrossRef]
- Lv, Q.; Jiang, Y.; Wang, B.; Chen, Y.; Jin, F.; Wu, B.; Ren, H.; Zhang, N.; Xu, R.; Li, Y.; et al. Suppressing lithium dendrites within inorganic solid-state electrolytes. Cell Rep. Phys. Sci. 2022, 3, 100706. [Google Scholar] [CrossRef]
- Yu, S.; Mertens, A.; Tempel, H.; Schierholz, R.; Kungl, H.; Eichel, R.A. Monolithic All-Phosphate Solid-State Lithium-Ion Battery with Improved Interfacial Compatibility. ACS Appl. Mater. Interfaces 2018, 10, 22264–22277. [Google Scholar] [CrossRef]
- Yu, S.; Mertens, A.; Gao, X.; Gunduz, D.C.; Schierholz, R.; Benning, S.; Hausen, F.; Mertens, J.; Kungl, H.; Tempel, H.; et al. Influence of microstructure and AlPO4 secondary-phase on the ionic conductivity of Li1.3Al0.3Ti1.7(PO4)3 solid-state electrolyte. Funct. Mater. Lett. 2016, 9, 1650066. [Google Scholar] [CrossRef]
- Gunduz, D.C.; Schierholz, R.; Yu, S.; Tempel, H.; Kungl, H.; Eichel, R.A. Combined quantitative microscopy on the microstructure and phase evolution in Li1.3Al0.3Ti1.7(PO4)3 ceramics. J. Adv. Ceram. 2020, 9, 149–161. [Google Scholar] [CrossRef]
- Welsch, A.M.; Behrens, H.; Murawski, D.; Horn, I. Lithium Mobility in Borate and Phosphate Glass Networks. Z. Phys. Chem. 2017, 231, 1303–1321. [Google Scholar] [CrossRef]
- Wang, D.; Yang, J.; Liu, J.; Li, X.; Li, R.; Cai, M.; Sham, T.K.; Sun, X. Atomic layer deposited coatings to significantly stabilize anodes for Li ion batteries: Effects of coating thickness and the size of anode particles. J. Mater. Chem. A 2014, 2, 2306–2312. [Google Scholar] [CrossRef]
- Li, X.; Liu, J.; Banis, M.N.; Lushington, A.; Li, R.; Cai, M.; Sun, X. Atomic layer deposition of solid-state electrolyte coated cathode materials with superior high-voltage cycling behavior for lithium ion battery application. Energy Environ. Sci. 2014, 7, 768–778. [Google Scholar] [CrossRef]
- Han, L.; Hsieh, C.T.; Chandra Mallick, B.; Li, J.; Ashraf Gandomi, Y. Recent progress and future prospects of atomic layer deposition to prepare/modify solid-state electrolytes and interfaces between electrodes for next-generation lithium batteries. Nanoscale Adv. 2021, 3, 2728–2740. [Google Scholar] [CrossRef]
- Jiang, S.P. A review of wet impregnation—An alternative method for the fabrication of high performance and nano-structured electrodes of solid oxide fuel cells. Mater. Sci. Eng. A 2006, 418, 199–210. [Google Scholar] [CrossRef]
- Mehrabadi, B.A.; Eskandari, S.; Khan, U.; White, R.D.; Regalbuto, J.R. A Review of Preparation Methods for Supported Metal Catalysts. Adv. Catal. 2017, 61, 1–35. [Google Scholar] [CrossRef]
- Zhu, C.; Byrd, R.H.; Lu, P.; Nocedal, J. Algorithm 778: L-BFGS-B: Fortran Subroutines for Large-Scale Bound-Constrained Optimization. ACM Trans. Math. Softw. 1997, 23, 550–560. [Google Scholar] [CrossRef]
- Jain, A.; Ong, S.P.; Hautier, G.; Chen, W.; Richards, W.D.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G.; et al. Commentary: The materials project: A materials genome approach to accelerating materials innovation. APL Mater. 2013, 1, 011002. [Google Scholar] [CrossRef]
- Allen, F.H. The Development, Status and Scientific Impact of Crystallographic Databases. Acta Crystallogr. 1998, A54, 758–771. [Google Scholar] [CrossRef]
- Zhang, M.; Huang, Z.; Cheng, J.; Yamamoto, O.; Imanishi, N.; Chi, B.; Pu, J.; Li, J. Solid state lithium ionic conducting thin film Li1.4Al0.4Ge1.6(PO4)3 prepared by tape casting. J. Alloys Compd. 2014, 590, 147–152. [Google Scholar] [CrossRef]
- Vijayakumar, M.; Kerisit, S.; Rosso, K.M.; Burton, S.D.; Sears, J.A.; Yang, Z.; Graff, G.L.; Liu, J.; Hu, J. Lithium diffusion in Li4Ti5O12 at high temperatures. J. Power Sources 2011, 196, 2211–2220. [Google Scholar] [CrossRef]
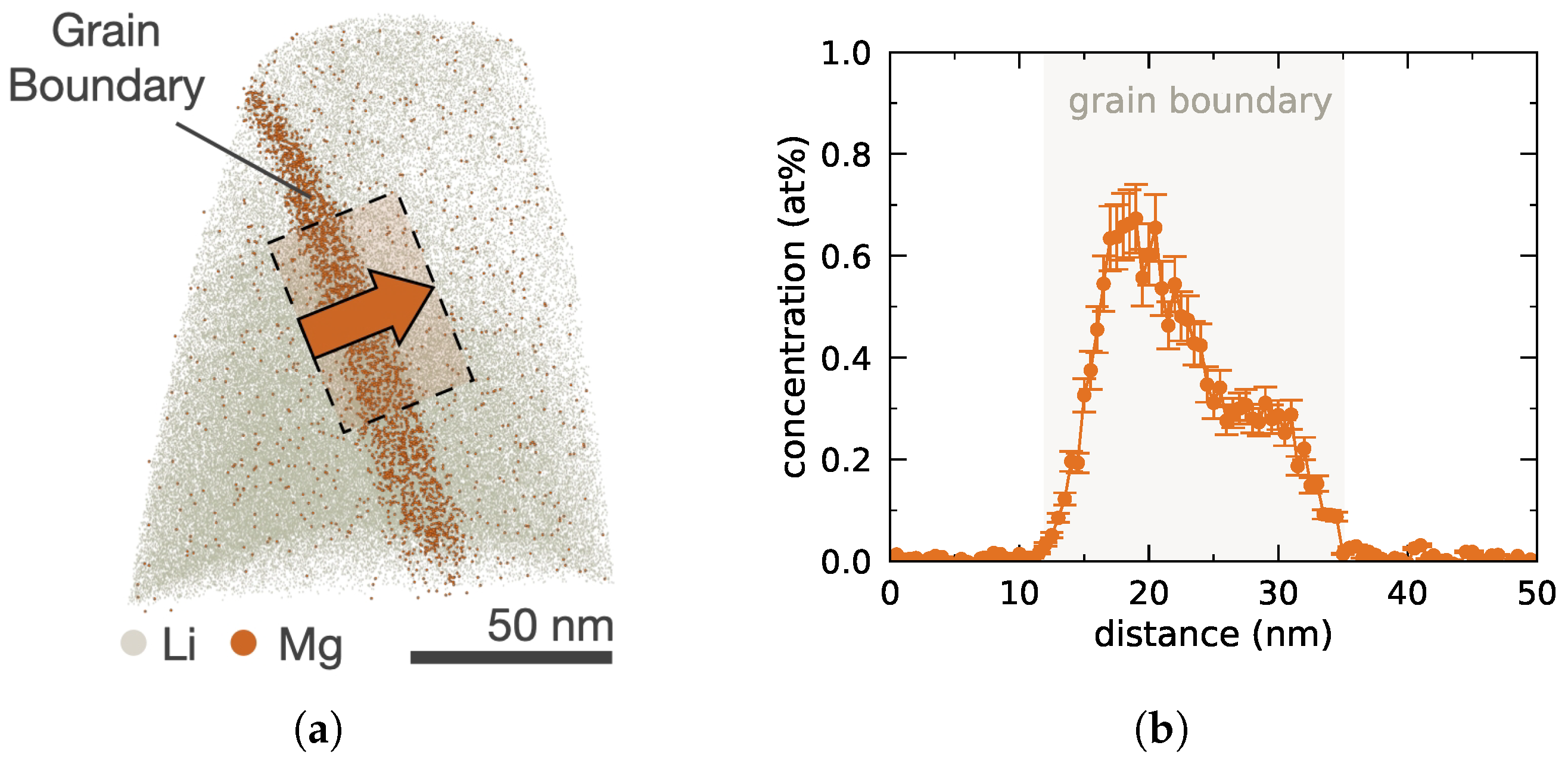
 visualizing Mg accumulation in the grain boundary of the APT sample and Li atom map
visualizing Mg accumulation in the grain boundary of the APT sample and Li atom map  as reference. (b) Elemental Mg profile across the grain boundary, where the overlayed rectangle
as reference. (b) Elemental Mg profile across the grain boundary, where the overlayed rectangle  in (a) indicates the selected subvolume used for averaging, with the arrow indicating the direction used for positive distances. Adapted with permission from Ref. [9] under CC BY 4.0.
visualizing Mg accumulation in the grain boundary of the APT sample and Li atom map as reference. (b) Elemental Mg profile across the grain boundary, where the overlayed rectangle in (a) indicates the selected subvolume used for averaging, with the arrow indicating the direction used for positive distances. Adapted with permission from Ref. [9] under CC BY 4.0.
in (a) indicates the selected subvolume used for averaging, with the arrow indicating the direction used for positive distances. Adapted with permission from Ref. [9] under CC BY 4.0.
visualizing Mg accumulation in the grain boundary of the APT sample and Li atom map as reference. (b) Elemental Mg profile across the grain boundary, where the overlayed rectangle in (a) indicates the selected subvolume used for averaging, with the arrow indicating the direction used for positive distances. Adapted with permission from Ref. [9] under CC BY 4.0.
 , Al
, Al  , Ti
, Ti  , O
, O  , and P
, and P  . The atomistic grain boundary reference structure is adapted with permission from Ref. [9] under CC BY 4.0.
, Al , Ti , O , and P . The atomistic grain boundary reference structure is adapted with permission from Ref. [9] under CC BY 4.0.
. The atomistic grain boundary reference structure is adapted with permission from Ref. [9] under CC BY 4.0.
, Al , Ti , O , and P . The atomistic grain boundary reference structure is adapted with permission from Ref. [9] under CC BY 4.0.
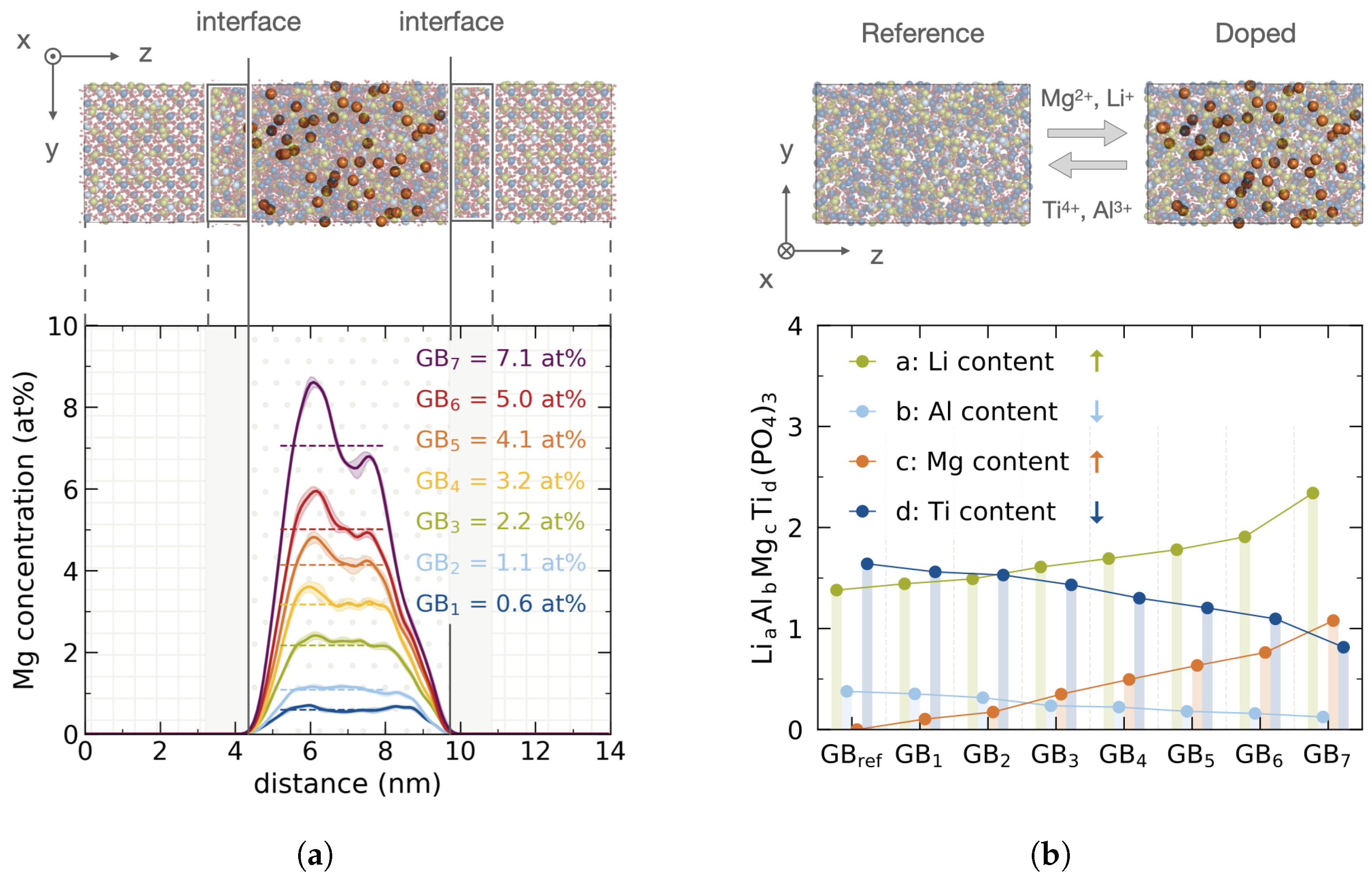
 , Mg
, Mg  , Al
, Al  , Ti
, Ti  , O
, O  , and P
, and P  ; (for better visualization Mg is depicted larger and other ions are transparent). Bottom: Corresponding Mg profiles across the grain boundary for different doping concentrations GB–GB. (b) Top: Amorphous bulk reference structure and exemplary doped structure. Elemental colors, radii and opacity are adopted from (a). Bottom: Cationic composition normalized to phosphate content with LiAlMgTi(PO).
, Mg , Al , Ti , O , and P ; (for better visualization Mg is depicted larger and other ions are transparent). Bottom: Corresponding Mg profiles across the grain boundary for different doping concentrations GB–GB. (b) Top: Amorphous bulk reference structure and exemplary doped structure. Elemental colors, radii and opacity are adopted from (a). Bottom: Cationic composition normalized to phosphate content with LiAlMgTi(PO).
; (for better visualization Mg is depicted larger and other ions are transparent). Bottom: Corresponding Mg profiles across the grain boundary for different doping concentrations GB–GB. (b) Top: Amorphous bulk reference structure and exemplary doped structure. Elemental colors, radii and opacity are adopted from (a). Bottom: Cationic composition normalized to phosphate content with LiAlMgTi(PO).
, Mg , Al , Ti , O , and P ; (for better visualization Mg is depicted larger and other ions are transparent). Bottom: Corresponding Mg profiles across the grain boundary for different doping concentrations GB–GB. (b) Top: Amorphous bulk reference structure and exemplary doped structure. Elemental colors, radii and opacity are adopted from (a). Bottom: Cationic composition normalized to phosphate content with LiAlMgTi(PO).


 , Mg Ti
, Mg Ti  , and Mg Li
, and Mg Li  . On the abscissa, the relative volume change ()/ in percent, for the correspondingly doped crystal structures C–C is shown.
, Mg Ti , and Mg Li . On the abscissa, the relative volume change ()/ in percent, for the correspondingly doped crystal structures C–C is shown.
. On the abscissa, the relative volume change ()/ in percent, for the correspondingly doped crystal structures C–C is shown.
, Mg Ti , and Mg Li . On the abscissa, the relative volume change ()/ in percent, for the correspondingly doped crystal structures C–C is shown. , Al , and Ti site. Charge carrier 3D pathways are illustrated from collapsing Li ions of a 2 ns MD trajectory at 700 K into the same structure
, Al , and Ti site. Charge carrier 3D pathways are illustrated from collapsing Li ions of a 2 ns MD trajectory at 700 K into the same structure  . Right: Li density difference of doped system and undoped reference structure showing Li avoiding the channel with incorporated Mg at a number density of
. Right: Li density difference of doped system and undoped reference structure showing Li avoiding the channel with incorporated Mg at a number density of  = −0.4 10Å and Li being trapped around Mg on Ti/Al sites with a number density of
= −0.4 10Å and Li being trapped around Mg on Ti/Al sites with a number density of  = +0.4 10Å.
, Al , and Ti site. Charge carrier 3D pathways are illustrated from collapsing Li ions of a 2 ns MD trajectory at 700 K into the same structure . Right: Li density difference of doped system and undoped reference structure showing Li avoiding the channel with incorporated Mg at a number density of = −0.4 10Å and Li being trapped around Mg on Ti/Al sites with a number density of = +0.4 10Å.
= +0.4 10Å.
, Al , and Ti site. Charge carrier 3D pathways are illustrated from collapsing Li ions of a 2 ns MD trajectory at 700 K into the same structure . Right: Li density difference of doped system and undoped reference structure showing Li avoiding the channel with incorporated Mg at a number density of = −0.4 10Å and Li being trapped around Mg on Ti/Al sites with a number density of = +0.4 10Å.

 , Al
, Al  , Ti
, Ti  , O
, O  , and P
, and P  . (b) Ternary plot showing relative composition for A, A–A, and a completely Ti depleted T according to .
, Al , Ti , O , and P . (b) Ternary plot showing relative composition for A, A–A, and a completely Ti depleted T according to .
. (b) Ternary plot showing relative composition for A, A–A, and a completely Ti depleted T according to .
, Al , Ti , O , and P . (b) Ternary plot showing relative composition for A, A–A, and a completely Ti depleted T according to .
 and O
and O  . (b) as a function of doping concentration A–A and respective Ti atom density averaged over six lowest energy walkers defined as .
and O . (b) as a function of doping concentration A–A and respective Ti atom density averaged over six lowest energy walkers defined as .
. (b) as a function of doping concentration A–A and respective Ti atom density averaged over six lowest energy walkers defined as .
and O . (b) as a function of doping concentration A–A and respective Ti atom density averaged over six lowest energy walkers defined as .


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stegmaier, S.; Reuter, K.; Scheurer, C. Exploiting Nanoscale Complexion in LATP Solid-State Electrolyte via Interfacial Mg2+ Doping. Nanomaterials 2022, 12, 2912. https://doi.org/10.3390/nano12172912
Stegmaier S, Reuter K, Scheurer C. Exploiting Nanoscale Complexion in LATP Solid-State Electrolyte via Interfacial Mg2+ Doping. Nanomaterials. 2022; 12(17):2912. https://doi.org/10.3390/nano12172912
Chicago/Turabian StyleStegmaier, Sina, Karsten Reuter, and Christoph Scheurer. 2022. "Exploiting Nanoscale Complexion in LATP Solid-State Electrolyte via Interfacial Mg2+ Doping" Nanomaterials 12, no. 17: 2912. https://doi.org/10.3390/nano12172912