
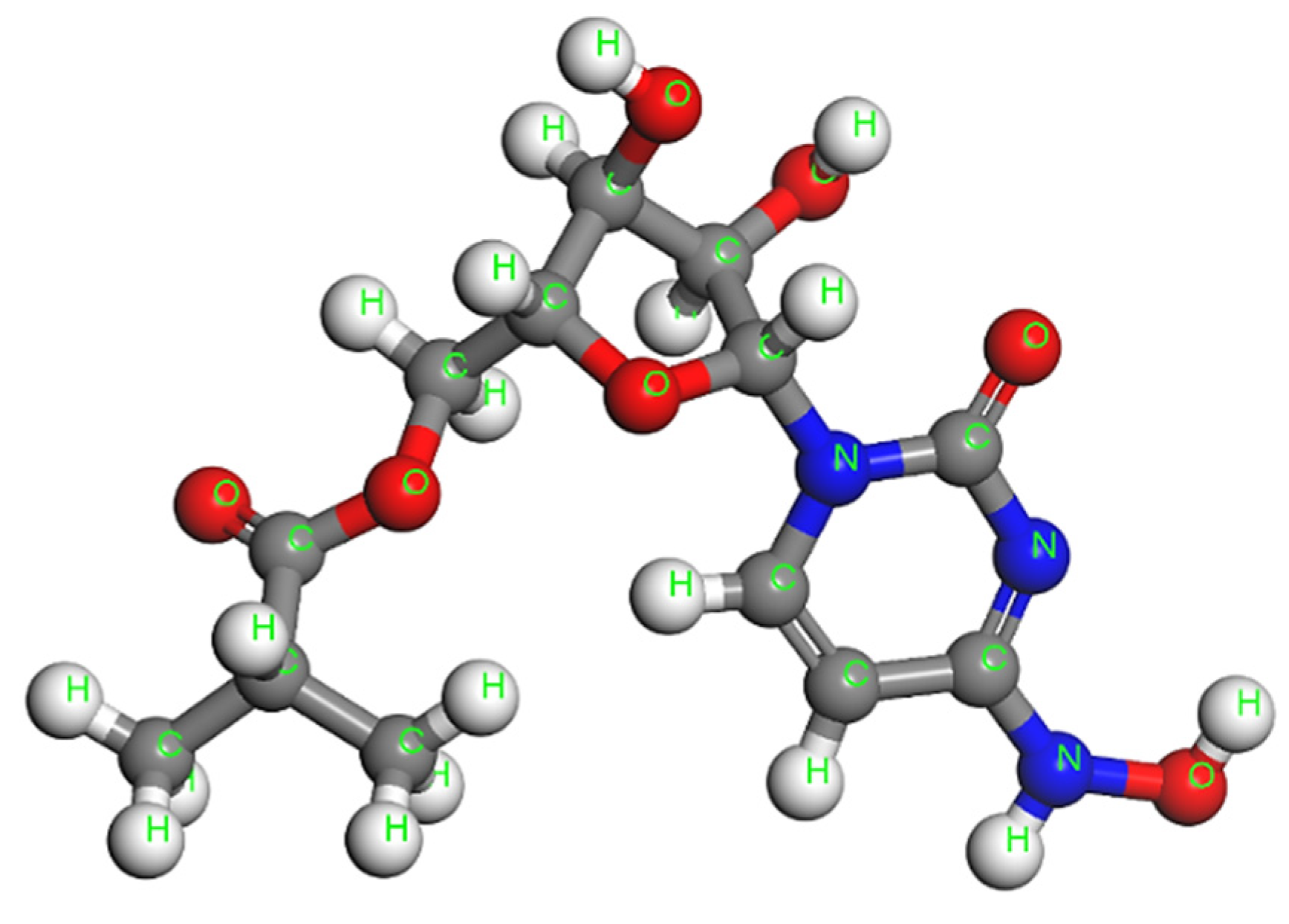
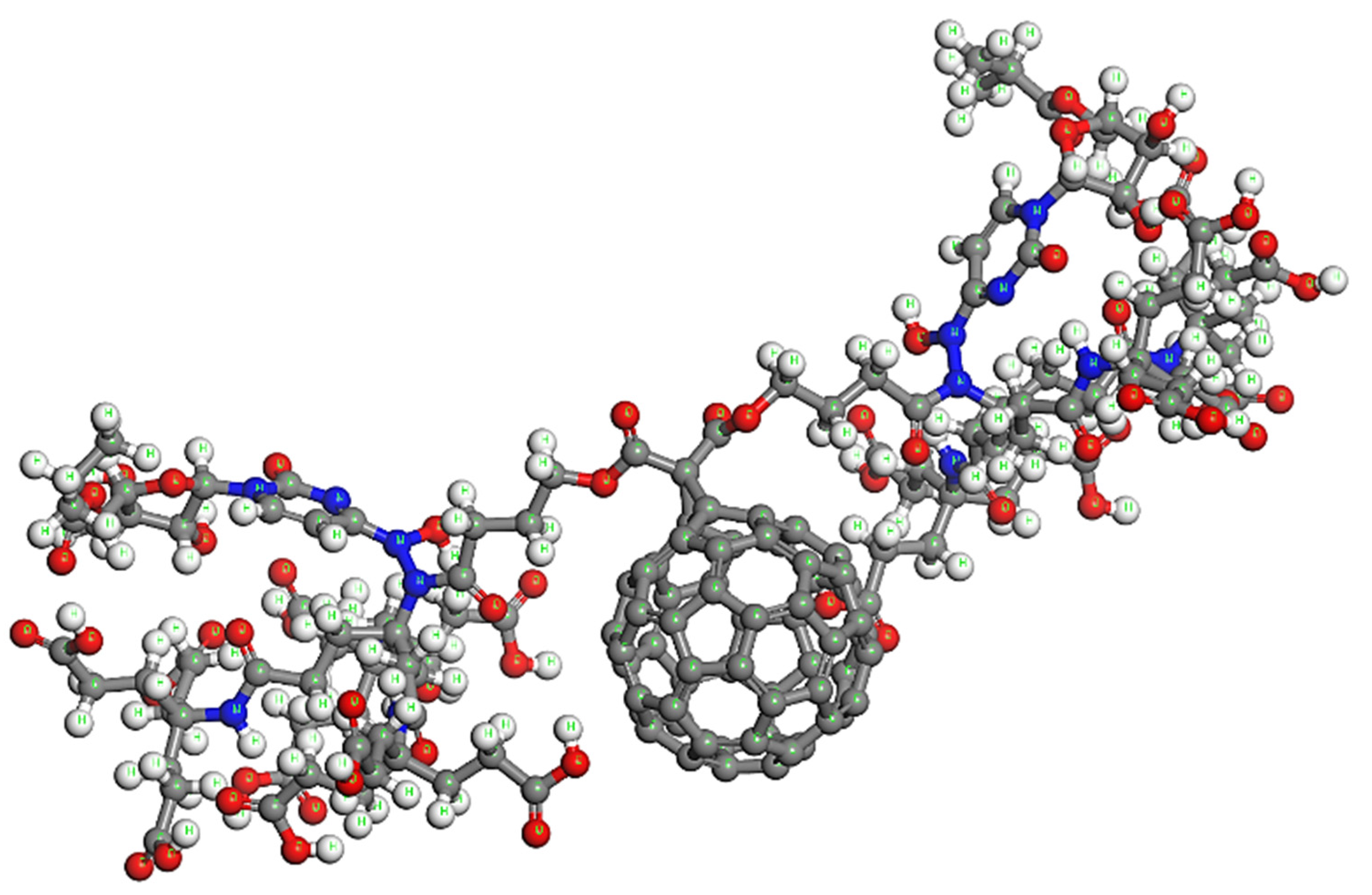
Fullerene Derivatives for Drug Delivery against COVID-19: A Molecular Dynamics Investigation of Dendro[60]fullerene as Nanocarrier of Molnupiravir

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
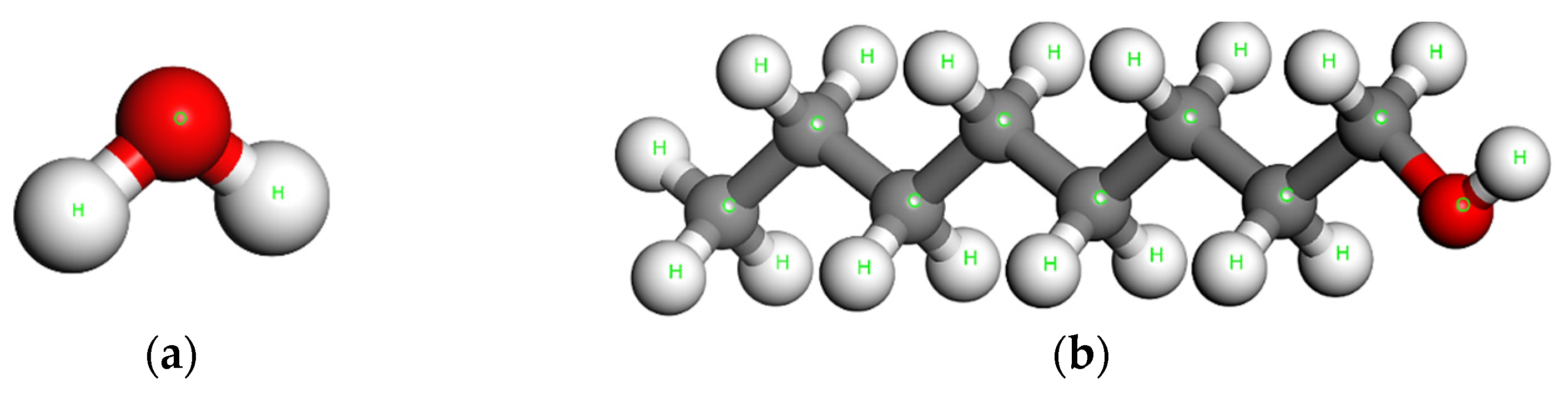
2. Solubility and Partition Coefficient
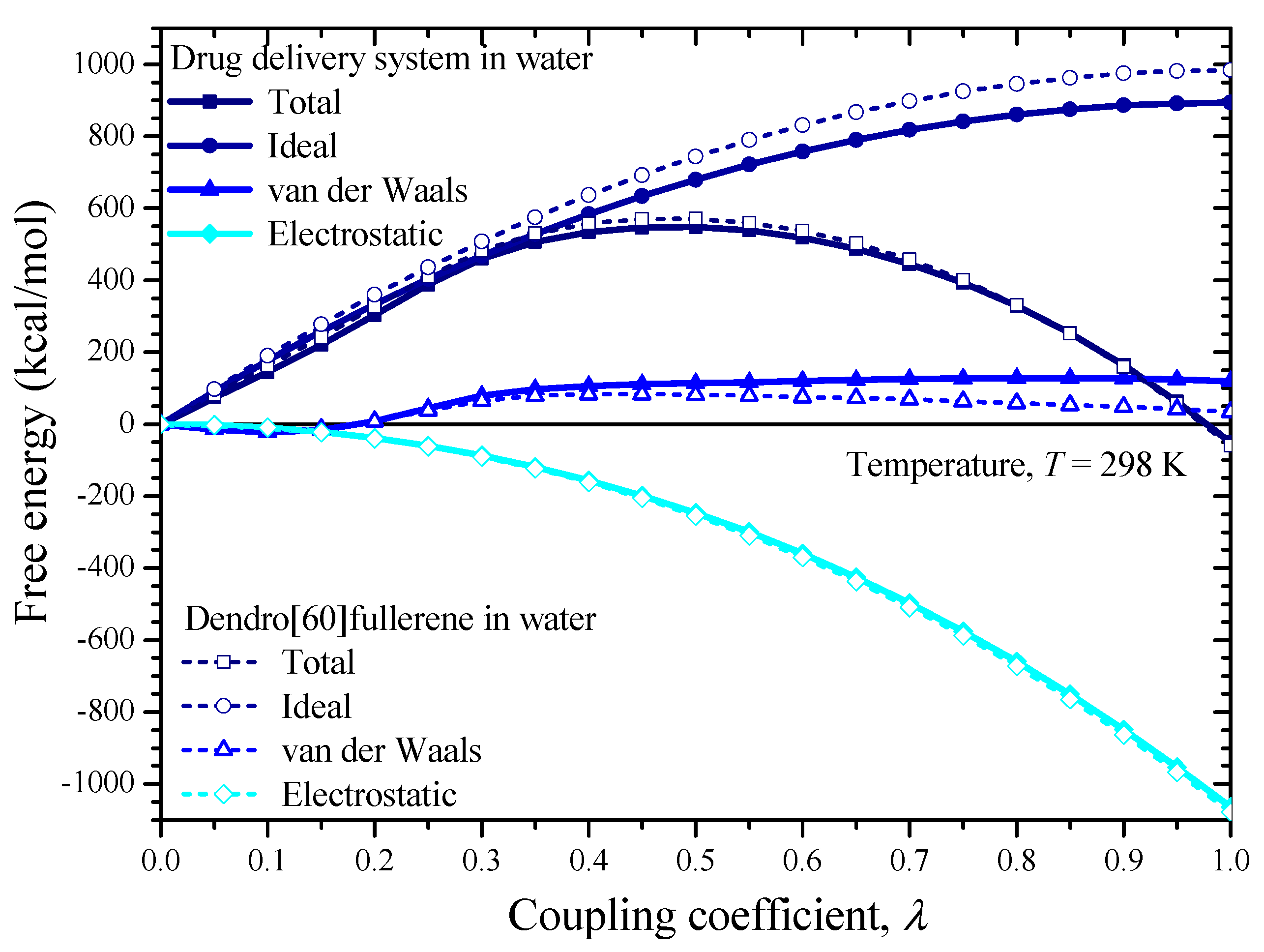
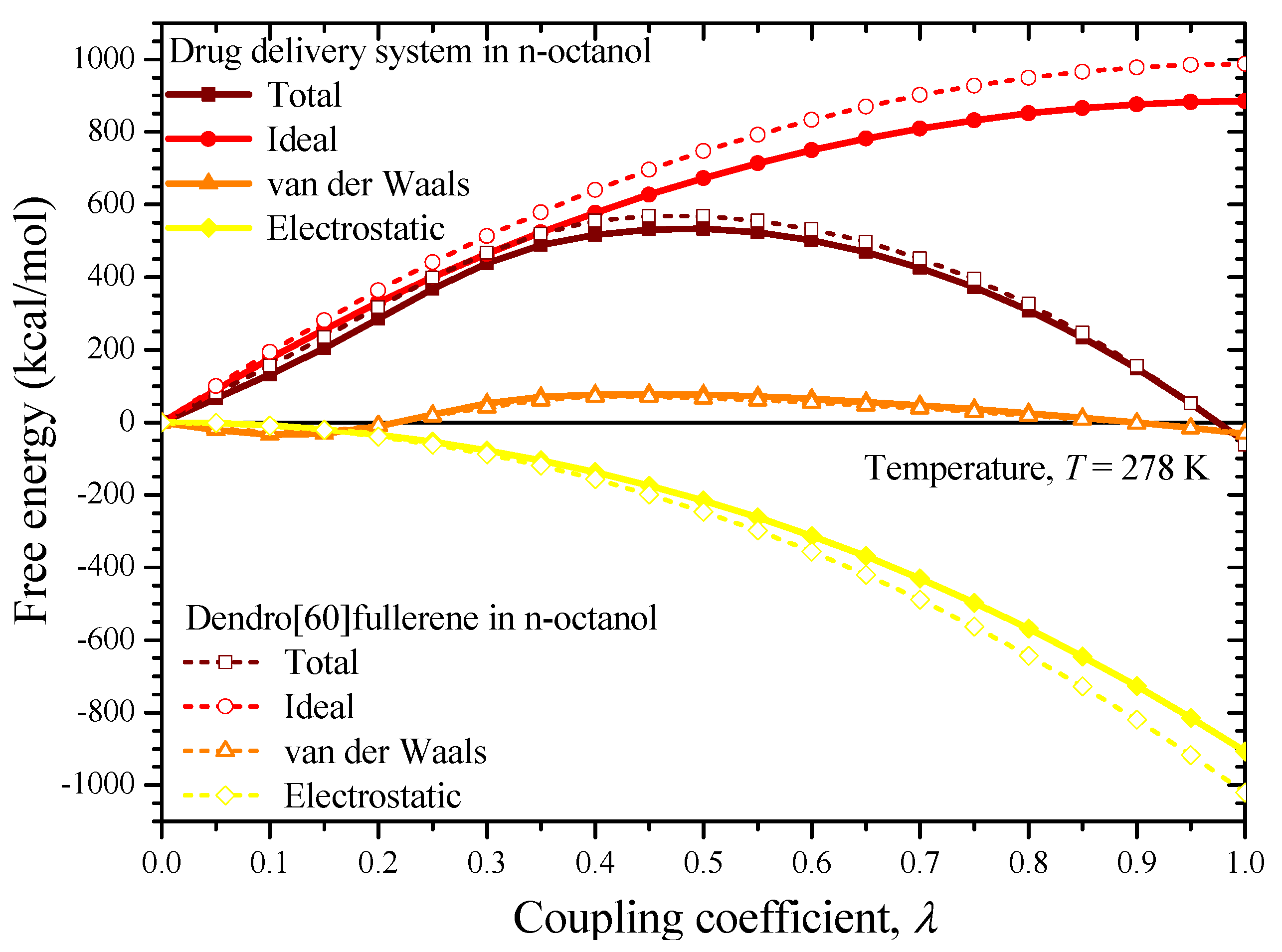
3. Solubility and Solvation Free Energy
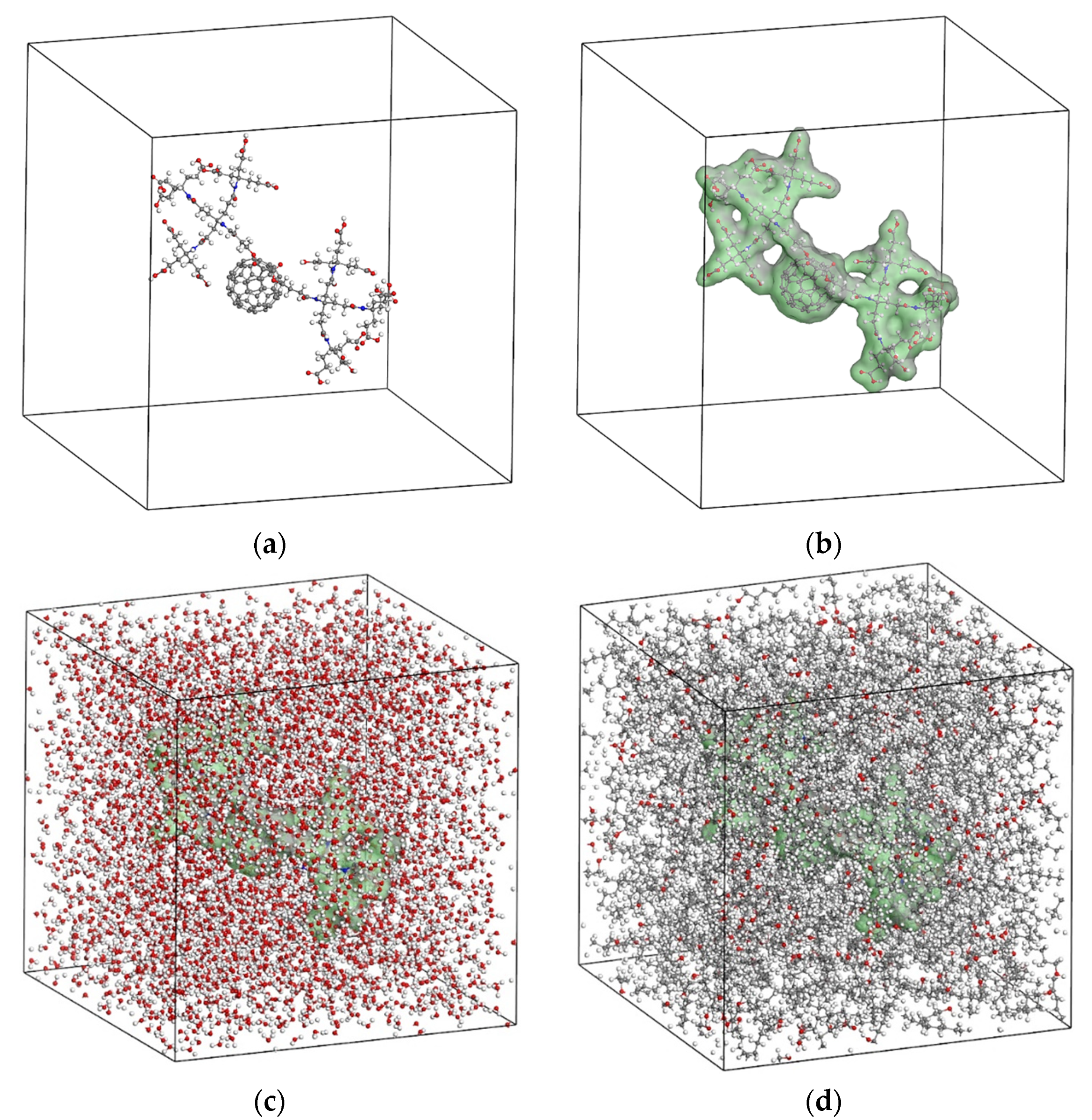
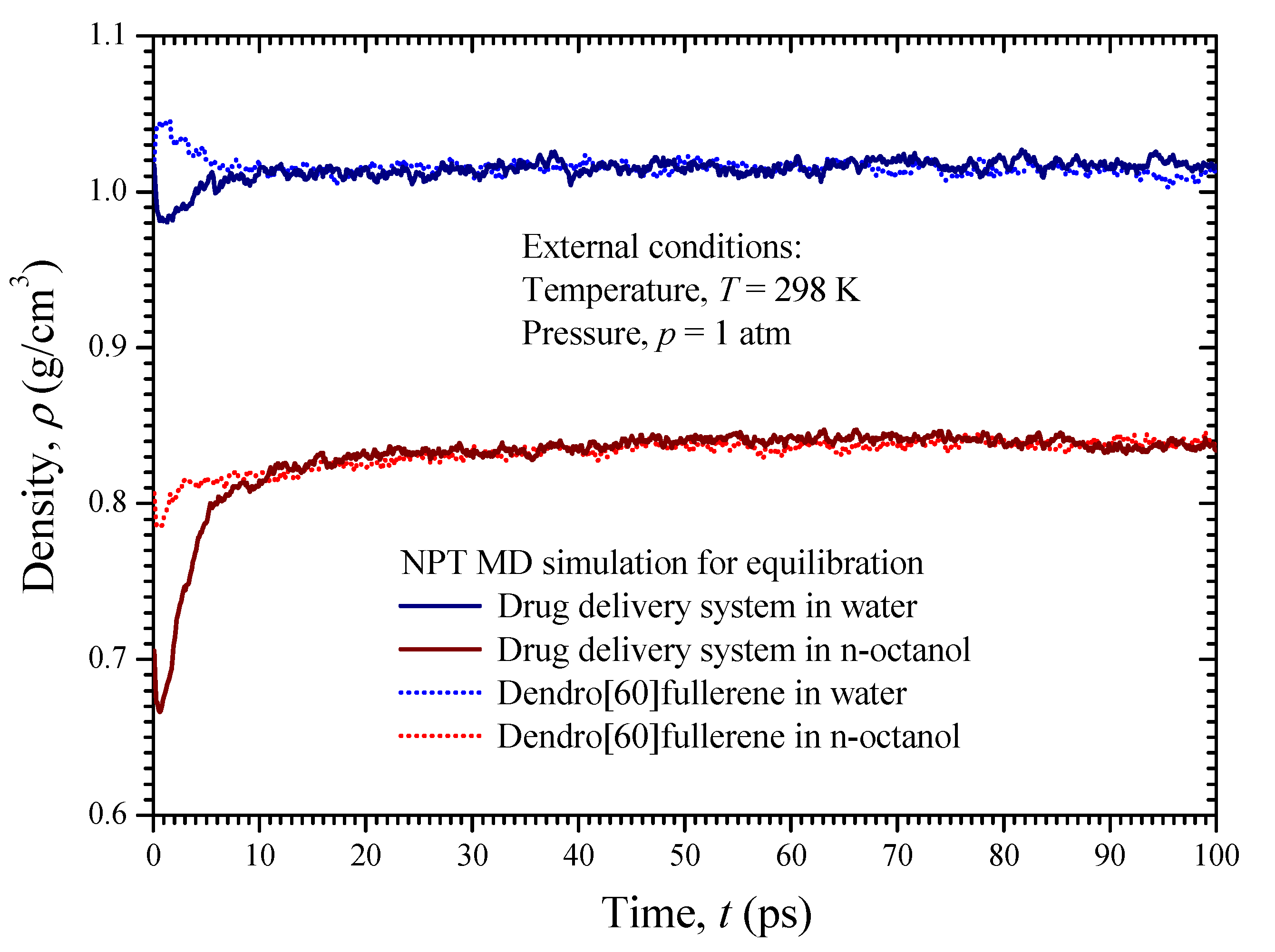
4. Simulation Details
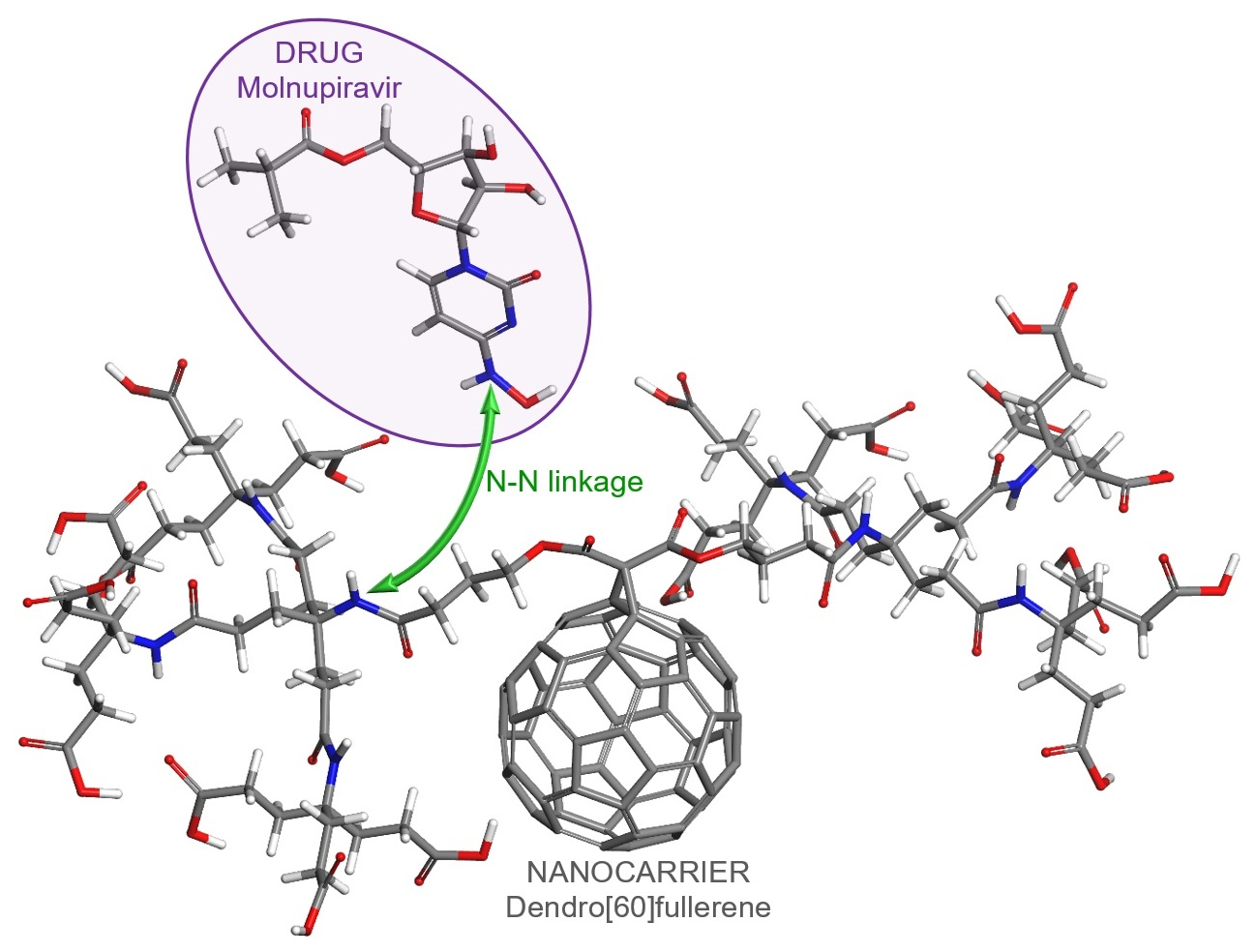
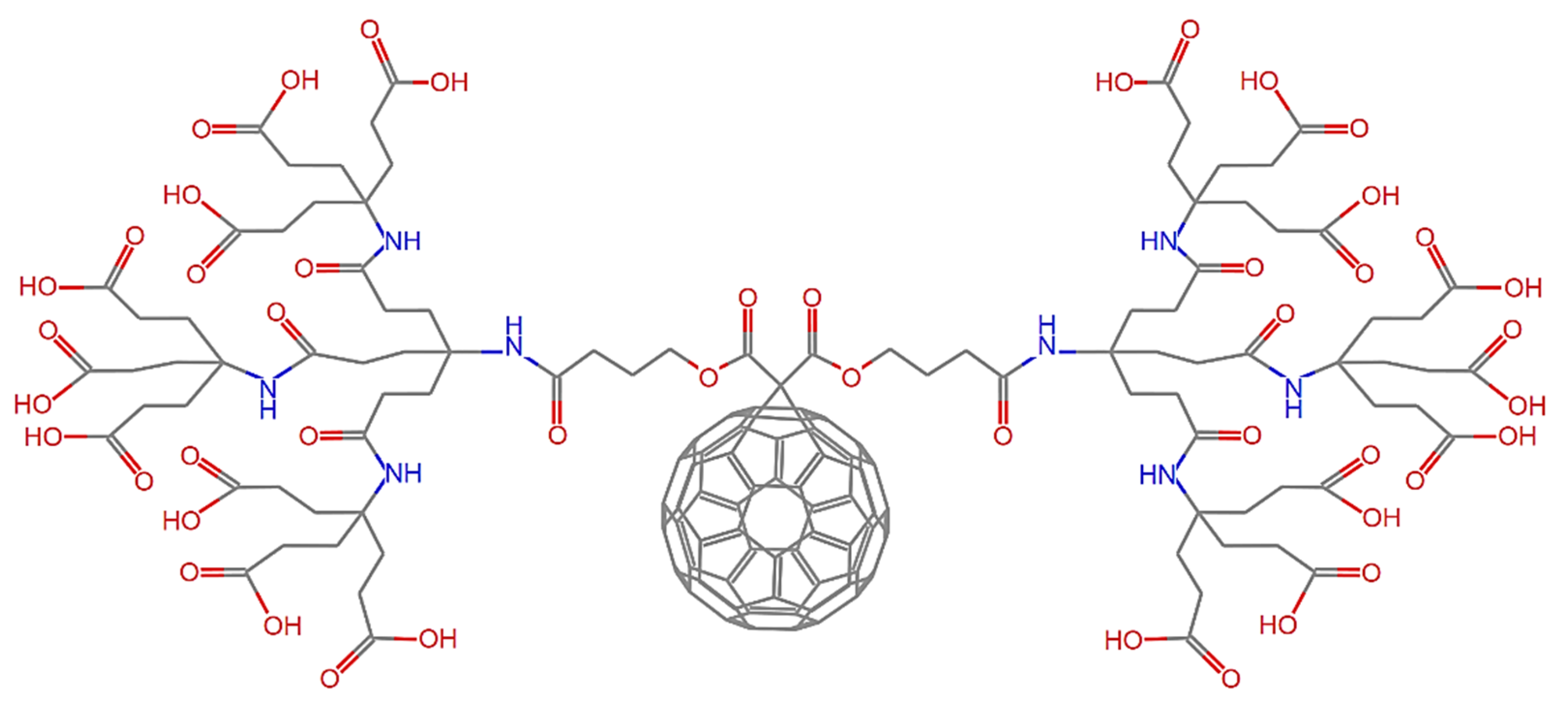
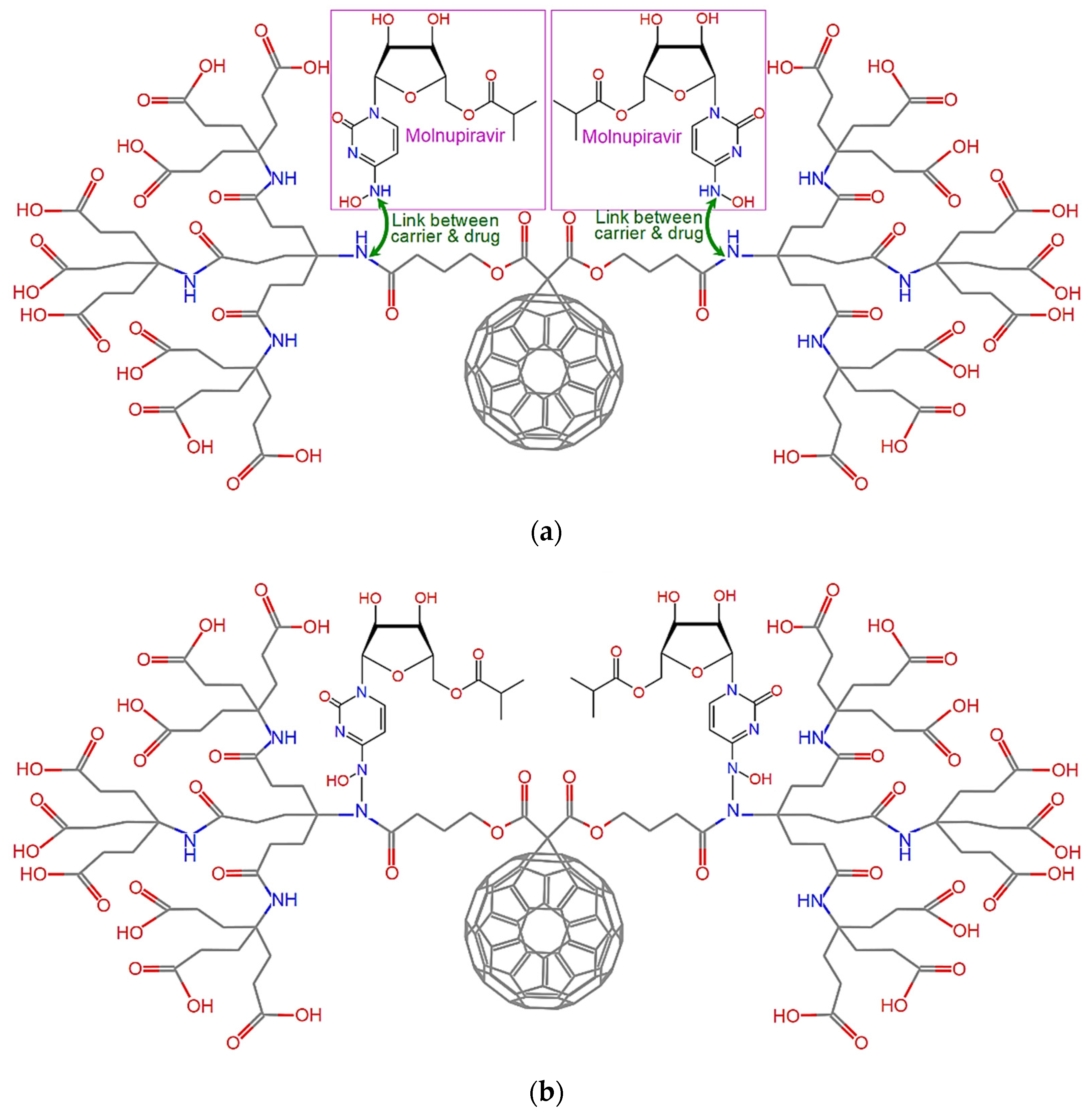
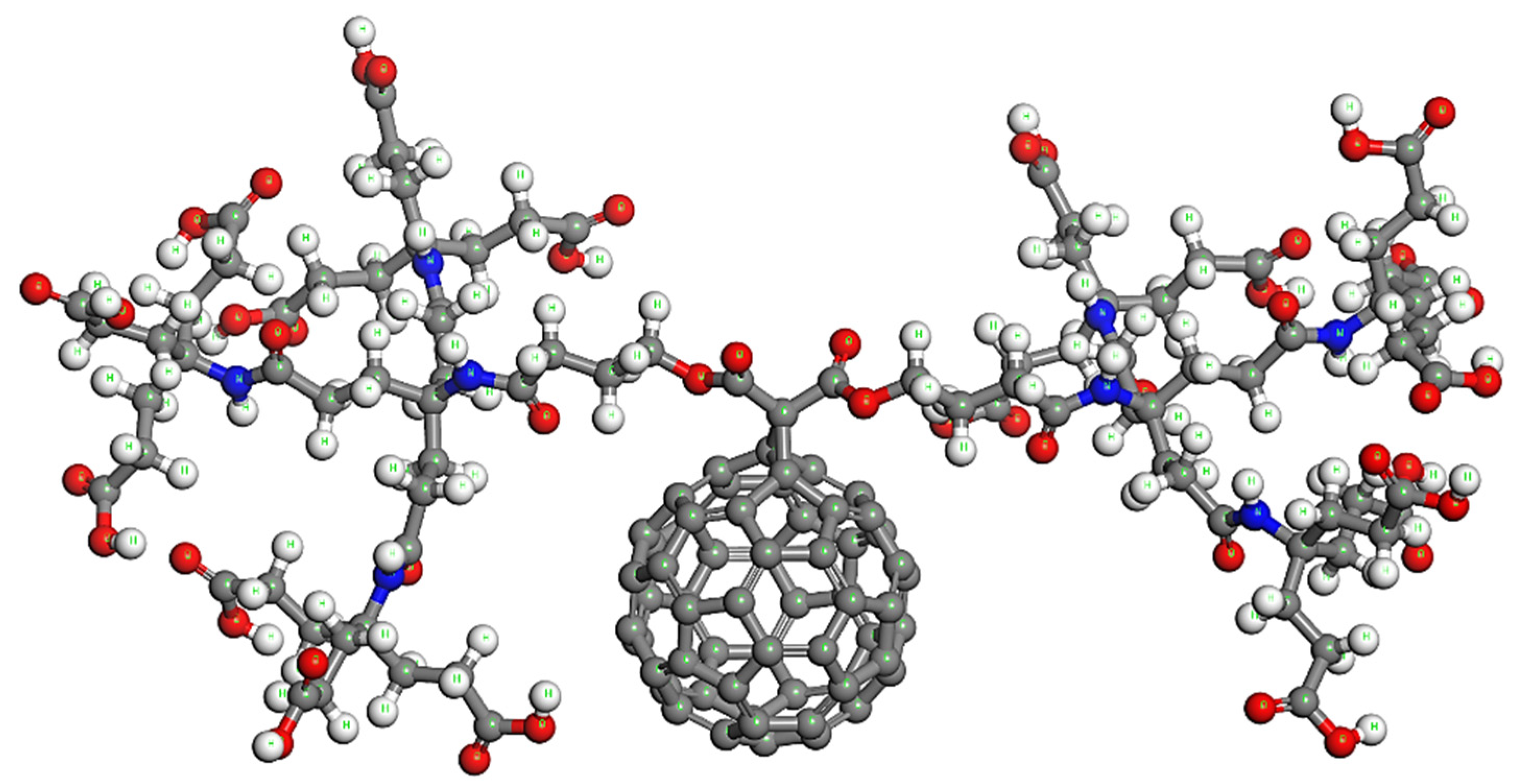
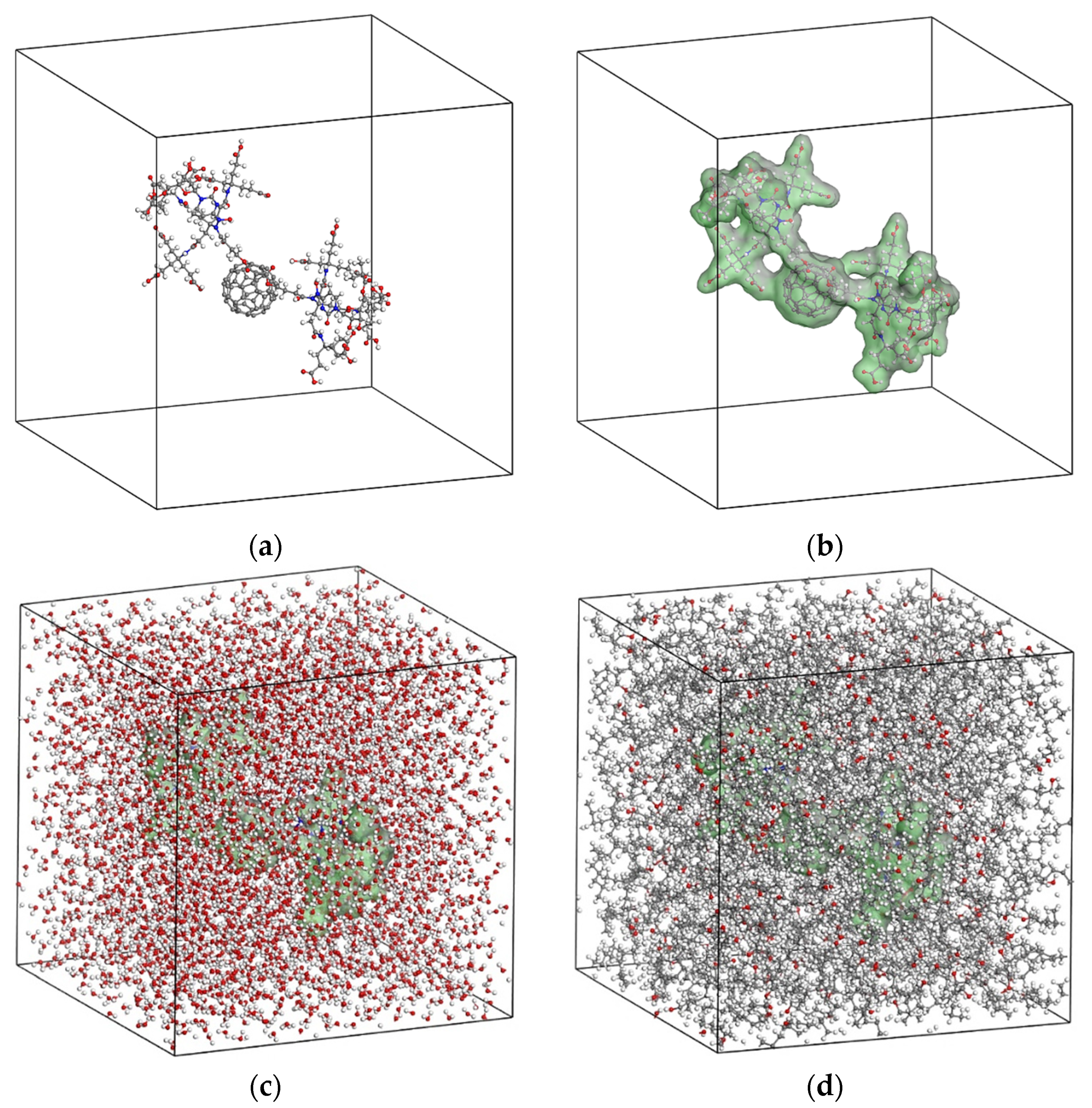
5. Proposed Drug Delivery System
6. Results and Discussion
7. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
References
- Lurie, N.; Saville, M.; Hatchett, R.; Halton, J. Developing COVID-19 vaccines at pandemic speed. N. Engl. J. Med. 2020, 382, 1969–1973. [Google Scholar] [CrossRef]
- Ucciferri, C.; Vecchiet, J.; Falasca, K. Role of monoclonal antibody drugs in the treatment of COVID-19. World J. Clin. Cases 2020, 8, 4280–4285. [Google Scholar] [CrossRef]
- Mei, M.; Tan, X. Current strategies of antiviral drug discovery for COVID-19. Front. Mol. Biosci. 2021, 8, 671263. [Google Scholar] [CrossRef]
- Weiss, C.; Carriere, M.; Fusco, L.; Capua, I.; Regla-Nava, J.A.; Pasquali, M.; Scott, J.A.; Vitale, F.; Unal, M.A.; Mattevi, C.; et al. Toward nanotechnology-enabled approaches against the COVID-19 pandemic. ACS Nano 2020, 14, 6383–6406. [Google Scholar] [CrossRef]
- Pourkarim, F.; Pourtaghi-Anvarian, S.; Rezaee, H. Molnupiravir: A new candidate for COVID-19 treatment. Pharmacol. Res. Perspect. 2022, 10, e00909. [Google Scholar] [CrossRef]
- Kabinger, F.; Stiller, C.; Schmitzová, J.; Dienemann, C.; Kokic, G.; Hillen, H.S.; Höbartner, C.; Cramer, P. Mechanism of molnupiravir-induced SARS-CoV-2 mutagenesis. Nat. Struct. Mol. Biol. 2021, 28, 740–746. [Google Scholar] [CrossRef]
- Sharov, A.V.; Burkhanova, T.M.; Tok, T.T.; Babashkina, M.G.; Safin, D.A. Computational analysis of molnupiravir. Int. J. Mol. Sci. 2022, 23, 1508. [Google Scholar] [CrossRef]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef]
- Polidoro, R.B.; Hagan, R.S.; de Santis Santiago, R.; Schmidt, N.W. Overview: Systemic inflammatory response derived from lung injury caused by SARS-CoV-2 infection explains severe outcomes in COVID-19. Front. Immunol. 2020, 11, 1626. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, L. Lung tissue distribution of drugs as a key factor for COVID-19 treatment. Br. J. Pharmacol. 2020, 177, 4995–4996. [Google Scholar] [CrossRef]
- Ziegler, C.G.K.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M.; et al. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell 2020, 181, 1016–1035.e19. [Google Scholar] [CrossRef]
- Webster, D.M.; Sundaram, P.; Byrne, M.E. Injectable nanomaterials for drug delivery: Carriers, targeting moieties, and therapeutics. Eur. J. Pharm. Biopharm. 2013, 84, 1–20. [Google Scholar] [CrossRef]
- Abd Elkodous, M.; Olojede, S.O.; Morsi, M.; El-Sayyad, G.S. Nanomaterial-based drug delivery systems as promising carriers for patients with COVID-19. RSC Adv. 2021, 11, 26463–26480. [Google Scholar] [CrossRef]
- Witika, B.A.; Makoni, P.A.; Mweetwa, L.L.; Ntemi, P.V.; Chikukwa, M.T.R.; Matafwali, S.K.; Mwila, C.; Mudenda, S.; Katandula, J.; Walker, R.B. Nano-biomimetic drug delivery vehicles: Potential approaches for COVID-19 treatment. Molecules 2020, 25, 5952. [Google Scholar] [CrossRef]
- Thakur, V.; Ratho, R.K.; Panda, J.J. Respiratory delivery of favipiravir-tocilizumab combination through mucoadhesive protein-lipidic nanovesicles: Prospective therapeutics against COVID-19. VirusDisease 2021, 32, 31–136. [Google Scholar] [CrossRef]
- Castro, E.; Garcia, A.H.; Zavala, G.; Echegoyen, L. Fullerenes in biology and medicine. J. Mater. Chem. B 2017, 5, 6523–6535. [Google Scholar] [CrossRef]
- Serda, M.; Gawecki, R.; Dulski, M.; Sajewicz, M.; Talik, E.; Szubka, M.; Zubko, M.; Malarz, K.; Mrozek-Wilczkiewicz, A.; Musiol, R. Synthesis and applications of [60]fullerene nanoconjugate with 5-aminolevulinic acid and its glycoconjugate as drug delivery vehicles. RSC Adv. 2022, 12, 6377–6388. [Google Scholar] [CrossRef]
- Zhao, L.; Li, H.; Tan, L. A novel fullerene-based drug delivery system delivering doxorubicin for potential lung cancer therapy. J. Nanosci. Nanotechnol. 2017, 17, 5147–5154. [Google Scholar] [CrossRef]
- Tan, L.; Wu, T.; Tang, Z.-W.; Xiao, J.-Y.; Zhuo, R.-X.; Shi, B.; Liu, C.-J. Water-soluble photoluminescent fullerene capped mesoporous silica for pH-responsive drug delivery and bioimaging. Nanotechnology 2016, 27, 315104. [Google Scholar] [CrossRef]
- Shi, J.; Liu, Y.; Wang, L.; Gao, J.; Zhang, J.; Yu, X.; Ma, R.; Liu, R.; Zhang, Z. A tumoral acidic pH-responsive drug delivery system based on a novel photosensitizer (fullerene) for in vitro and in vivo chemo-photodynamic therapy. Acta Biomater. 2014, 10, 1280–1291. [Google Scholar] [CrossRef]
- Shi, J.; Yu, X.; Wang, L.; Liu, Y.; Gao, J.; Zhang, J.; Ma, R.; Liu, R.; Zhang, Z. PEGylated fullerene/iron oxide nanocomposites for photodynamic therapy, targeted drug delivery and MR imaging. Biomaterials 2013, 34, 9666–9677. [Google Scholar] [CrossRef]
- Shi, J.; Zhang, H.; Wang, L.; Li, L.; Wang, H.; Wang, Z.; Li, Z.; Chen, C.; Hou, L.; Zhang, C.; et al. PEI-derivatized fullerene drug delivery using folate as a homing device targeting to tumor. Biomaterials 2013, 34, 251–261. [Google Scholar] [CrossRef]
- Lucafò, M.; Pacor, S.; Fabbro, C.; Da Ros, T.; Zorzet, S.; Prato, M.; Sava, G. Study of a potential drug delivery system based on carbon nanoparticles: Effects of fullerene derivatives in MCF7 mammary carcinoma cells. J. Nanopart. Res. 2012, 14, 830. [Google Scholar] [CrossRef]
- Ganji, M.D.; Yazdani, H.; Mirnejad, A. B36N36 fullerene-like nanocages: A novel material for drug delivery. Phys. E Low-Dimens. Syst. Nanostruct. 2010, 42, 2184–2189. [Google Scholar] [CrossRef]
- Hazrati, M.K.; Javanshir, Z.; Bagheri, Z. B24N24 fullerene as a carrier for 5-fluorouracil anti-cancer drug delivery: DFT studies. J. Mol. Graph. Model. 2017, 77, 17–24. [Google Scholar] [CrossRef]
- Mahani, N.M.; Sabermahani, F.; Shamsolmaali, A. C24 fullerene as drug delivery for anticancer activity of pyridine derivatives: A density functional theory approach. Pak. J. Pharm. Sci. 2019, 32, 2741–2744. [Google Scholar]
- Xu, H.; Tu, X.; Fan, G.; Wang, Q.; Wang, X.; Chu, X. Adsorption properties study of boron nitride fullerene for the application as smart drug delivery agent of anti-cancer drug hydroxyurea by density functional theory. J. Mol. Liq. 2020, 318, 114315. [Google Scholar] [CrossRef]
- Alipour, E.; Alimohammady, F.; Yumashev, A.; Maseleno, A. Fullerene C60 containing porphyrin-like metal center as drug delivery system for ibuprofen drug. J. Mol. Model. 2020, 26, 7. [Google Scholar] [CrossRef]
- Li, W.; Zhao, T. Hydroxyurea anticancer drug adsorption on the pristine and doped C70 fullerene as potential carriers for drug delivery. J. Mol. Liq. 2021, 340, 117226. [Google Scholar] [CrossRef]
- Tavakoli, S.; Ahmadi, S.A.; Ghazanfari, D.; Sheikhhosseini, E. Theoretical investigation of functionalized fullerene nano carrier drug delivery of fluoxetine. J. Indian Chem. Soc. 2022, 99, 100561. [Google Scholar] [CrossRef]
- Vuong, B.X.; Hajali, N.; Asadi, A.; Baqer, A.A.; Hachim, S.K.; Canli, G. Drug delivery assessment of an iron-doped fullerene cage towards thiotepa anticancer drug. Inorg. Chem. Commun. 2022, 141, 109558. [Google Scholar] [CrossRef]
- Azamat, J.; Heravi, M.R.P.; Habibzadeh, S.; Ebadi, A.G.; Shoaei, S.M.; Vessally, E. Hetero Diels–alder cycloadduct of anti-tumor (E)-3-X-indoline-2-thiones with C20 fullerene as drug delivery in solution vs. gas phase: A DFT survey. Inorg. Chem. Commun. 2022, 139, 109353. [Google Scholar] [CrossRef]
- Yousefi, M.; Rad, M.S.; Shakibazadeh, R.; Ghodrati, L.; Kachoie, M.A. Simulating a heteroatomic CBN fullerene-like nanocage towards the drug delivery of fluorouracil. Mol. Simul. 2022. [Google Scholar] [CrossRef]
- Shahabi, M.; Raissi, H. A new insight into the transfer and delivery of anti-SARS-CoV-2 drug Carmofur with the assistance of graphene oxide quantum dot as a highly efficient nanovector toward COVID-19 by molecular dynamics simulation. RSC Adv. 2022, 12, 14167–14174. [Google Scholar] [CrossRef]
- Bibi, S.; Urrehman, S.; Khalid, L.; Yaseen, M.; Khan, A.Q.; Jia, R. Metal doped fullerene complexes as promising drug delivery materials against COVID-19. Chem. Pap. 2021, 75, 6487–6497. [Google Scholar] [CrossRef]
- Yao, C.; Xiang, F.; Xu, Z. Metal oxide nanocage as drug delivery systems for Favipiravir, as an effective drug for the treatment of COVID-19: A computational study. J. Mol. Model. 2022, 28, 64. [Google Scholar] [CrossRef]
- Parlak, C.; Alver, Ö.; Bağlayan, Ö. Quantum mechanical simulation of Molnupiravir drug interaction with Si-doped C60 fullerene. Comput. Theor. Chem. 2021, 1202, 113336. [Google Scholar] [CrossRef]
- Brettreich, M.; Hirsch, A. A highly water-soluble dendro[60]fullerene. Tetrahedron Lett. 1998, 39, 2731–2734. [Google Scholar] [CrossRef]
- Pourbasheer, E.; Aalizadeh, R.; Ardabili, J.S.; Ganjali, M.R. QSPR study on solubility of some fullerenes derivatives using the genetic algorithms—Multiple linear regression. J. Mol. Liq. 2015, 204, 162–169. [Google Scholar] [CrossRef]
- Marcus, Y.; Kamlet, M.J.; Taft, R.W. Linear solvation energy relationships. Standard molar Gibbs free energies and enthalpies of transfer of ions from water into nonaqueous solvents. J. Phys. Chem. 1988, 92, 3613–3622. [Google Scholar] [CrossRef]
- Suárez, M.; Makowski, K.; Lemos, R.; Almagro, L.; Rodríguez, H.; Herranz, M.Á.; Molero, D.; Ortiz, O.; Maroto, E.; Albericio, F.; et al. An androsterone-H2@C60 hybrid: Synthesis, properties and molecular docking simulations with SARS-Cov-2. ChemPlusChem 2021, 86, 970–971. [Google Scholar] [CrossRef]
- Ghaemi, F.; Amiri, A.; Bajuri, M.Y.; Yuhana, N.Y.; Ferrara, M. Role of different types of nanomaterials against diagnosis, prevention and therapy of COVID-19. Sustain. Cities Soc. 2021, 72, 103046. [Google Scholar] [CrossRef]
- Hurmach, V.V.; Platonov, M.O.; Prylutska, S.V.; Scharff, P.; Prylutskyy, Y.I.; Ritter, U. C60 fullerene against SARS-CoV-2 coronavirus: An in silico insight. Sci. Rep. 2021, 11, 17748. [Google Scholar] [CrossRef]
- Marforio, T.D.; Mattioli, E.J.; Zerbetto, F.; Calvaresi, M. Fullerenes against COVID-19: Repurposing C60 and C70 to clog the active site of SARS-CoV-2 protease. Molecules 2022, 27, 1916. [Google Scholar] [CrossRef]
- Serrano-Aroca, Á.; Takayama, K.; Tuñón-Molina, A.; Seyran, M.; Hassan, S.S.; Pal Choudhury, P.; Uversky, V.N.; Lundstrom, K.; Adadi, P.; Palù, G.; et al. Carbon-based nanomaterials: Promising antiviral agents to combat COVID-19 in the microbial-resistant era. ACS Nano 2021, 15, 8069–8086. [Google Scholar] [CrossRef]
- van Gunsteren, W.F.; Berendsen, H.J.C. Thermodynamic cycle integration by computer simulation as a tool for obtaining free energy differences in molecular chemistry. J. Comput. Aided Mol. Des. 1987, 1, 171–176. [Google Scholar] [CrossRef]
- Salo-Ahen, O.M.H. Molecular dynamics simulations in drug discovery and pharmaceutical development. Processes 2021, 9, 71. [Google Scholar] [CrossRef]
- Sun, H.; Jin, Z.; Yang, C.; Akkermans, R.L.C.; Robertson, S.H.; Spenley, N.A.; Miller, S.; Todd, S.M. COMPASS II: Extended coverage for polymer and drug-like molecule databases. J. Mol. Model. 2016, 22, 47. [Google Scholar] [CrossRef]
- Accelrys Software Inc. Materials Studio; Accelrys Software Inc.: San Diego, CA, USA, 2009. [Google Scholar]
- Marx, D.; Hutter, J. Ab Initio Molecular Dynamics: Basic Theory and Advanced Methods, 1st ed.; Cambridge University Press: New York, NY, USA, 2009. [Google Scholar]
- Chen, Z.; Mao, R.; Liu, Y. Fullerenes for cancer diagnosis and therapy: Preparation, biological and clinical perspectives. Curr. Drug Metab. 2012, 13, 1035–1045. [Google Scholar] [CrossRef]
- Peng, L.; Liu, S.; Feng, A.; Yuan, J. Polymeric nanocarriers based on cyclodextrins for drug delivery: Host-guest interaction as stimuli responsive linker. Mol. Pharm. 2017, 14, 2475–2486. [Google Scholar] [CrossRef]
- McMurry, J.E.; Gay, R.C.; Robinson, J.K. Chemistry, 7th ed.; Pearson: Edimbourg, UK, 2016. [Google Scholar]
- Chen, L.; Deng, Z.; Zhao, C. Nitrogen-nitrogen bond formation reactions involved in natural product biosynthesis. ACS Chem. Biol. 2021, 16, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Jafvert, C.T.; Kulkarni, P.P. Buckminsterfullerene’s (C60) octanol-water partition coefficient (Kow) and aqueous solubility. Environ. Sci. Technol. 2008, 42, 5945–5950. [Google Scholar] [CrossRef] [PubMed]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giannopoulos, G.I. Fullerene Derivatives for Drug Delivery against COVID-19: A Molecular Dynamics Investigation of Dendro[60]fullerene as Nanocarrier of Molnupiravir. Nanomaterials 2022, 12, 2711. https://doi.org/10.3390/nano12152711
Giannopoulos GI. Fullerene Derivatives for Drug Delivery against COVID-19: A Molecular Dynamics Investigation of Dendro[60]fullerene as Nanocarrier of Molnupiravir. Nanomaterials. 2022; 12(15):2711. https://doi.org/10.3390/nano12152711
Chicago/Turabian StyleGiannopoulos, Georgios I. 2022. "Fullerene Derivatives for Drug Delivery against COVID-19: A Molecular Dynamics Investigation of Dendro[60]fullerene as Nanocarrier of Molnupiravir" Nanomaterials 12, no. 15: 2711. https://doi.org/10.3390/nano12152711