
Ag Atom Anchored on Defective Hexagonal Boron Nitride Nanosheets As Single Atom Adsorbents for Enhanced Adsorptive Desulfurization via S-Ag Bonds

 ,
,
Abstract
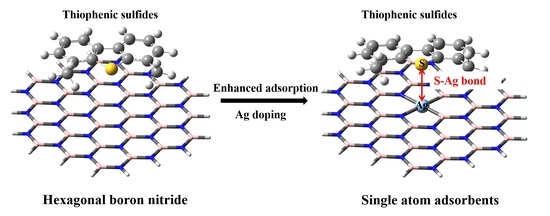
:
1. Introduction
2. Computational Details
2.1. Theory
2.2. Model
3. Results and Discussion
3.1. Monoatomic Ag-Doped h-BN
3.2. Adsorption Complexes of Ag-Doped h-BN Nanosheets and Aromatics or Thiophenic Compounds
3.2.1. Structures
3.2.2. Energetics
3.3. Analysis of the Nature of Thiophenic Compounds Adsorption
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, Y.X.; Shen, J.X.; Peng, S.S.; Zhang, J.K.; Wu, J.; Liu, X.Q.; Sun, L.B. Enhancing oxidation resistance of Cu(I) by tailoring microenvironment in zeolites for efficient adsorptive desulfurization. Nat. Commun. 2020, 11, 3206. [Google Scholar] [CrossRef] [PubMed]
- Saleh, T.A. Characterization, determination and elimination technologies for sulfur from petroleum: Toward cleaner fuel and a safe environment. Trends Environ. Anal. Chem. 2020, 25, e00080. [Google Scholar] [CrossRef]
- Zhang, M.; Zhu, W.; Li, H.; Xun, S.; Li, M.; Li, Y.; Wei, Y.; Li, H. Fabrication and characterization of tungsten-containing mesoporous silica for heterogeneous oxidative desulfurization. Chin. J. Catal. 2016, 37, 971–978. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, J.; Li, H.; Wei, Y.; Fu, Y.; Liao, W.; Zhu, L.; Chen, G.; Zhu, W.; Li, H. Tuning the electrophilicity of vanadium-substituted polyoxometalate based ionic liquids for high-efficiency aerobic oxidative desulfurization. Appl. Catal. B 2020, 271, 118936. [Google Scholar] [CrossRef]
- Jiang, W.; Zhu, K.; Li, H.; Zhu, L.; Hua, M.; Xiao, J.; Wang, C.; Yang, Z.; Chen, G.; Zhu, W.; et al. Synergistic effect of dual Brønsted acidic deep eutectic solvents for oxidative desulfurization of diesel fuel. Chem. Eng. J. 2020, 394, 124831. [Google Scholar] [CrossRef]
- Li, H.; Zhu, W.; Zhu, S.; Xia, J.; Chang, Y.; Jiang, W.; Zhang, M.; Zhou, Y.; Li, H. The selectivity for sulfur removal from oils: An insight from conceptual density functional theory. AlChE J. 2016, 62, 2087–2100. [Google Scholar] [CrossRef]
- Triantafyllidis, K.S.; Deliyanni, E.A. Desulfurization of diesel fuels: Adsorption of 4,6-DMDBT on different origin and surface chemistry nanoporous activated carbons. Chem. Eng. J. 2014, 236, 406–414. [Google Scholar] [CrossRef]
- Saha, B.; Vedachalam, S.; Dalai, A.K. Review on recent advances in adsorptive desulfurization. Fuel Process. Technol. 2021, 214, 106685. [Google Scholar] [CrossRef]
- Saleh, T.A. Simultaneous adsorptive desulfurization of diesel fuel over bimetallic nanoparticles loaded on activated carbon. J. Cleaner Prod. 2018, 172, 2123–2132. [Google Scholar] [CrossRef]
- Saleh, T.A.; Sulaiman, K.O.; Al-Hammadi, S.A.; Dafalla, H.; Danmaliki, G.I. Adsorptive desulfurization of thiophene, benzothiophene and dibenzothiophene over activated carbon manganese oxide nanocomposite: With column system evaluation. J. Cleaner Prod. 2017, 154, 401–412. [Google Scholar] [CrossRef]
- Wu, P.; Lu, L.; He, J.; Chen, L.; Chao, Y.; He, M.; Zhu, F.; Chu, X.; Li, H.; Zhu, W. Hexagonal boron nitride: A metal-free catalyst for deep oxidative desulfurization of fuel oils. Green Energy Environ. 2020, 5, 166–172. [Google Scholar] [CrossRef]
- Lu, L.J.; He, J.; Wu, P.W.; Wu, Y.C.; Chao, Y.H.; Li, H.P.; Tao, D.J.; Fan, L.; Li, H.M.; Zhu, W.S. Taming electronic properties of boron nitride nanosheets as metal-free catalysts for aerobic oxidative desulfurization of fuels. Green Chem. 2018, 20, 4453–4460. [Google Scholar] [CrossRef]
- Mu, L.P.; Luo, J.; Wang, C.; Liu, J.X.; Zou, Y.R.; Li, X.W.; Huang, Y.; Wu, P.W.; Ji, H.Y.; Zhu, W.S. BN/ZIF-8 derived carbon hybrid materials for adsorptive desulfurization: Insights into adsorptive property and reaction kinetics. Fuel 2021, 288, 119685. [Google Scholar] [CrossRef]
- Lv, N.; Sun, L.; Chen, L.; Li, Y.; Zhang, J.; Wu, P.; Li, H.; Zhu, W.; Li, H. The mechanism of thiophene oxidation on metal-free two-dimensional hexagonal boron nitride. Phys. Chem. Chem. Phys. 2019, 21, 21867–21874. [Google Scholar] [CrossRef]
- Zhu, W.S.; Dai, B.L.; Wu, P.W.; Chao, Y.H.; Xiong, J.; Xun, S.H.; Li, H.P.; Li, H.M. Graphene-Analogue Hexagonal BN Supported with Tungsten-based Ionic Liquid for Oxidative Desulfurization of Fuels. ACS Sustain. Chem. Eng. 2015, 3, 186–194. [Google Scholar] [CrossRef]
- Li, H.; Zhang, J.; Wu, P.; Xun, S.; Jiang, W.; Zhang, M.; Zhu, W.; Li, H. O2 Activation and Oxidative Dehydrogenation of Propane on Hexagonal Boron Nitride: Mechanism Revisited. J. Phys. Chem. C 2019, 123, 2256–2266. [Google Scholar] [CrossRef]
- Li, H.; Wang, C.; Xun, S.; He, J.; Jiang, W.; Zhang, M.; Zhu, W.; Li, H. An accurate empirical method to predict the adsorption strength for pi-orbital contained molecules on two dimensional materials. J. Mol. Graph. Model. 2018, 82, 93–100. [Google Scholar] [CrossRef]
- Hao, L.; Hurlock, M.J.; Ding, G.; Zhang, Q. Metal-Organic Frameworks towards Desulfurization of Fuels. Top. Curr. Chem. 2020, 378, 17. [Google Scholar] [CrossRef]
- Khan, N.A.; Hasan, Z.; Jhung, S.H. Ionic Liquids Supported on Metal-Organic Frameworks: Remarkable Adsorbents for Adsorptive Desulfurization. Chem. Eur. J. 2014, 20, 376–380. [Google Scholar] [CrossRef]
- Ganiyu, S.A.; Lateef, S.A. Review of adsorptive desulfurization process: Overview of the non-carbonaceous materials, mechanism and synthesis strategies. Fuel 2021, 294, 120273. [Google Scholar] [CrossRef]
- Yoosuk, B.; Silajan, A.; Prasassarakich, P. Deep adsorptive desulfurization over ion-exchanged zeolites: Individual and simultaneous effect of aromatic and nitrogen compounds. J. Cleaner Prod. 2020, 248, 119291. [Google Scholar] [CrossRef]
- Li, H.; Zhang, Y.; Lv, N.; Yin, J.; Zhang, J.; Ran, H.; Zhang, M.; Jiang, W.; Zhu, W.; Li, H. Unraveling the effects of O-doping into h-BN on the adsorptive desulfurization performance by DFT calculations. J. Environ. Chem. Eng. 2021, 9, 106463. [Google Scholar] [CrossRef]
- Li, H.; Ran, H.; Yujun, L.; Lv, N.; Yin, J.; Zhang, J.; Wang, C.; Jiang, W.; Zhu, W.; Li, H.; et al. Comparative Study of Halogen-Doped (X=Cl, Br, I) Hexagonal Boron Nitride: A Promising Strategy to Enhance the Capacity of Adsorptive Desulfurization. J. Environ. Chem. Eng. 2021, 9, 105886. [Google Scholar] [CrossRef]
- Li, Y.; Lv, N.; Wang, C.; Zhang, J.; Fu, W.; Yin, J.; Li, H.; Zhu, W.; Li, H. Theoretical prediction of F-doped hexagonal boron nitride: A promising strategy to enhance the capacity of adsorptive desulfurization. J. Mol. Graph. Model. 2020, 101, 107715. [Google Scholar] [CrossRef]
- Zhang, J.; Sun, R.; Ruan, D.; Zhang, M.; Li, Y.; Zhang, K.; Cheng, F.; Wang, Z.; Wang, Z.-M. Point defects in two-dimensional hexagonal boron nitride: A perspective. J. Appl. Phys. 2020, 128, 100902. [Google Scholar] [CrossRef]
- Lin, S.; Ye, X.; Johnson, R.S.; Guo, H. First-Principles Investigations of Metal (Cu, Ag, Au, Pt, Rh, Pd, Fe, Co, and Ir) Doped Hexagonal Boron Nitride Nanosheets: Stability and Catalysis of CO Oxidation. J. Phys. Chem. C 2013, 117, 17319–17326. [Google Scholar] [CrossRef]
- Lu, Z.; Lv, P.; Yang, Z.; Li, S.; Ma, D.; Wu, R. A promising single atom catalyst for CO oxidation: Ag on boron vacancies of h-BN sheets. Phys. Chem. Chem. Phys. 2017, 19, 16795–16805. [Google Scholar] [CrossRef] [Green Version]
- Esrafili, M.D.; Janebi, H.; Mousavian, P. Epoxidation of ethylene over an Ag atom embedded B-vacancy defective boron-nitride nanosheet via a trimolecular Langmuir-Hinshelwood mechanism: A DFT investigation. Mol. Catal. 2021, 514, 111843. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. Exploring the Limit of Accuracy of the Global Hybrid Meta Density Functional for Main-Group Thermochemistry, Kinetics, and Noncovalent Interactions. J. Chem. Theory Comput. 2008, 4, 1849–1868. [Google Scholar] [CrossRef]
- Grimme, S.; Hansen, A.; Brandenburg, J.G.; Bannwarth, C. Dispersion-Corrected Mean-Field Electronic Structure Methods. Chem. Rev. 2016, 116, 5105–5154. [Google Scholar] [CrossRef] [Green Version]
- Leininger, T.; Nicklass, A.; Stoll, H.; Dolg, M.; Schwerdtfeger, P. The accuracy of the pseudopotential approximation. II. A comparison of various core sizes for indium pseudopotentials in calculations for spectroscopic constants of InH, InF, and InCl. J. Chem. Phys. 1996, 105, 1052–1059. [Google Scholar] [CrossRef]
- Kwak, J.H.; Hu, J.; Mei, D.; Yi, C.-W.; Kim, D.H.; Peden, C.H.F.; Allard, L.F.; Szanyi, J. Coordinatively Unsaturated Al3+ Centers as Binding Sites for Active Catalyst Phases of Platinum on gamma-Al2O3. Science 2009, 325, 1670–1673. [Google Scholar] [CrossRef] [PubMed]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Li, H.; Zhu, S.; Zhang, M.; Wu, P.; Pang, J.; Zhu, W.; Jiang, W.; Li, H. Tuning the Chemical Hardness of Boron Nitride Nanosheets by Doping Carbon for Enhanced Adsorption Capacity. ACS Omega 2017, 2, 5385–5394. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chang, Y.; Zhu, W.; Wang, C. Theoretical evidence of charge transfer interaction between SO2 and deep eutectic solvents formed by choline chloride and glycerol. Phys. Chem. Chem. Phys. 2015, 17, 28729–28742. [Google Scholar] [CrossRef]
- Li, H.; Zhu, W.; Chang, Y.; Jiang, W.; Zhang, M.; Yin, S.; Xia, J.; Li, H. Theoretical investigation of the interaction between aromatic sulfur compounds and [BMIM]+[FeCl4]− ionic liquid in desulfurization: A novel charge transfer mechanism. J. Mol. Graph. Model. 2015, 59, 40–49. [Google Scholar] [CrossRef]
- Alvarez, S. A cartography of the van der Waals territories. Dalton Trans. 2013, 42, 8617–8636. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Zheng, Z.; Xia, S.; Yu, L. Synthesis, co-crystal structure, and DFT calculations of a multicomponent co-crystal constructed from 1H-benzotriazole and tetrafluoroterephthalic acid. J. Mol. Struct. 2020, 1219, 128480. [Google Scholar] [CrossRef]
- Yin, J.; Zhang, J.; Wang, C.; Lv, N.; Jiang, W.; Liu, H.; Li, H.; Zhu, W.; Li, H.; Ji, H. Theoretical insights into CO2/N2 selectivity of the porous ionic liquids constructed by ion-dipole interactions. J. Mol. Liq. 2021, 344, 117676. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. sigma-holes and -holes: Similarities and differences. J. Comput. Chem. 2018, 39, 464–471. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Quantitative analysis of molecular surface based on improved Marching Tetrahedra algorithm. J. Mol. Graph. Model. 2012, 38, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W.; Carroll, M.T.; Cheeseman, J.R.; Chang, C. Properties of Atoms in Molecules: Atomic Volumes. J. Am. Chem. Soc. 1987, 109, 7968–7979. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lu, T. Efficient evaluation of electrostatic potential with computerized optimized code. Phys. Chem. Chem. Phys. 2021, 23, 20323–20328. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Wang, F.; Chen, W.; Tang, S.; Zhang, W.; Li, W.; Sun, H.; Zhang, J.; Wang, R. Theoretical design and simulation of supramolecular polymer unit based on multiple hydrogen bonds. J. Mol. Graph. Model. 2015, 59, 31–39. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Fu, W.; Zhang, J.; Ran, H.; Lv, N.; Chao, Y.; Li, H.; Zhu, W.; Liu, H.; Li, H. Unraveling the mechanism of CO2 capture and separation by porous liquids. RSC Adv. 2020, 10, 42706–42717. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | |
|---|---|
| BN/Ag-B | |
| BN/Ag-B_ben | −20.6 |
| BN/Ag-B_nap | −24.4 |
| BN/Ag-B_flu | −25.3 |
| BN/Ag-B_BT | −23.3 |
| BN/Ag-B_DBT | −29.1 |
| BN/Ag-B_DMDBT | −33.9 |
| BN/Ag-N | |
| BN/Ag-N_ben | −17.9 |
| BN/Ag-N_nap | −20.3 |
| BN/Ag-N_flu | −22.5 |
| BN/Ag-N_BT | −20.4 |
| BN/Ag-N_DBT | −24.4 |
| BN/Ag-N_DMDBT | −29.1 |
| BN/Ag-BN | |
| BN/Ag-BN_ben | −25.5 |
| BN/Ag-BN_nap | −17.2 |
| BN/Ag-BN_flu | −32.4 |
| BN/Ag-BN_BT | −28.1 |
| BN/Ag-BN_DBT | −34.4 |
| BN/Ag-BN_DMDBT | −39.2 |
| Species | Δq (S) a | Δq (THs) b | Δq (Ag) c | Δq (Ads) d |
|---|---|---|---|---|
| h-BN | ||||
| BN_BT | 0.021 | 0.051 | - | −0.051 |
| BN_DBT | 0.024 | 0.059 | - | −0.059 |
| BN_DMDBT | 0.026 | 0.065 | - | −0.065 |
| BN/Ag-B | ||||
| BN/Ag-B_BT | 0.027 | 0.189 | −0.229 | −0.189 |
| BN/Ag-B_DBT | 0.035 | 0.198 | −0.465 | −0.198 |
| BN/Ag-B_DMDBT | 0.038 | 0.226 | −0.483 | −0.226 |
| BN/Ag-N | ||||
| BN/Ag-N_BT | 0.005 | 0.161 | 0.015 | −0.161 |
| BN/Ag-N_DBT | 0.022 | 0.164 | 0.005 | −0.164 |
| BN/Ag-N_DMDBT | 0.024 | 0.193 | 0.002 | −0.193 |
| BN/Ag-BN | ||||
| BN/Ag-BN_BT | 0.019 | 0.192 | −0.009 | −0.192 |
| BN/Ag-BN_DBT | 0.027 | 0.202 | −0.013 | −0.202 |
| BN/Ag-BN_DMDBT | 0.029 | 0.217 | −0.028 | −0.217 |
| Species | Bond Length | WBI |
|---|---|---|
| BN/Ag-B | ||
| BN/Ag-B_BT | 2.62 Å | 0.3031 |
| BN/Ag-B_DBT | 2.60 Å | 0.3108 |
| BN/Ag-B_DMDBT | 2.59 Å | 0.3255 |
| BN/Ag-N | ||
| BN/Ag-N_BT | 2.63 Å | 0.2200 |
| BN/Ag-N_DBT | 2.62 Å | 0.2353 |
| BN/Ag-N_DMDBT | 2.60 Å | 0.3093 |
| BN/Ag-BN | ||
| BN/Ag-BN_BT | 2.58 Å | 0.3115 |
| BN/Ag-BN_DBT | 2.59 Å | 0.2471 |
| BN/Ag-BN_DMDBT | 2.58 Å | 0.3118 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Yin, J.; Zhang, J.; Ran, H.; Lv, N.; Jiang, W.; Li, H.; Zhu, W.; Li, H. Ag Atom Anchored on Defective Hexagonal Boron Nitride Nanosheets As Single Atom Adsorbents for Enhanced Adsorptive Desulfurization via S-Ag Bonds. Nanomaterials 2022, 12, 2046. https://doi.org/10.3390/nano12122046
Liu H, Yin J, Zhang J, Ran H, Lv N, Jiang W, Li H, Zhu W, Li H. Ag Atom Anchored on Defective Hexagonal Boron Nitride Nanosheets As Single Atom Adsorbents for Enhanced Adsorptive Desulfurization via S-Ag Bonds. Nanomaterials. 2022; 12(12):2046. https://doi.org/10.3390/nano12122046
Chicago/Turabian StyleLiu, Hui, Jie Yin, Jinrui Zhang, Hongshun Ran, Naixia Lv, Wei Jiang, Hongping Li, Wenshuai Zhu, and Huaming Li. 2022. "Ag Atom Anchored on Defective Hexagonal Boron Nitride Nanosheets As Single Atom Adsorbents for Enhanced Adsorptive Desulfurization via S-Ag Bonds" Nanomaterials 12, no. 12: 2046. https://doi.org/10.3390/nano12122046