
Spin-Orbit Coupling Electronic Structures of Organic-Group Functionalized Sb and Bi Topological Monolayers

Abstract
:1. Introduction
2. Atomic Model and Calculation Schemes
3. Results and Discussion
3.1. Atomic Structure
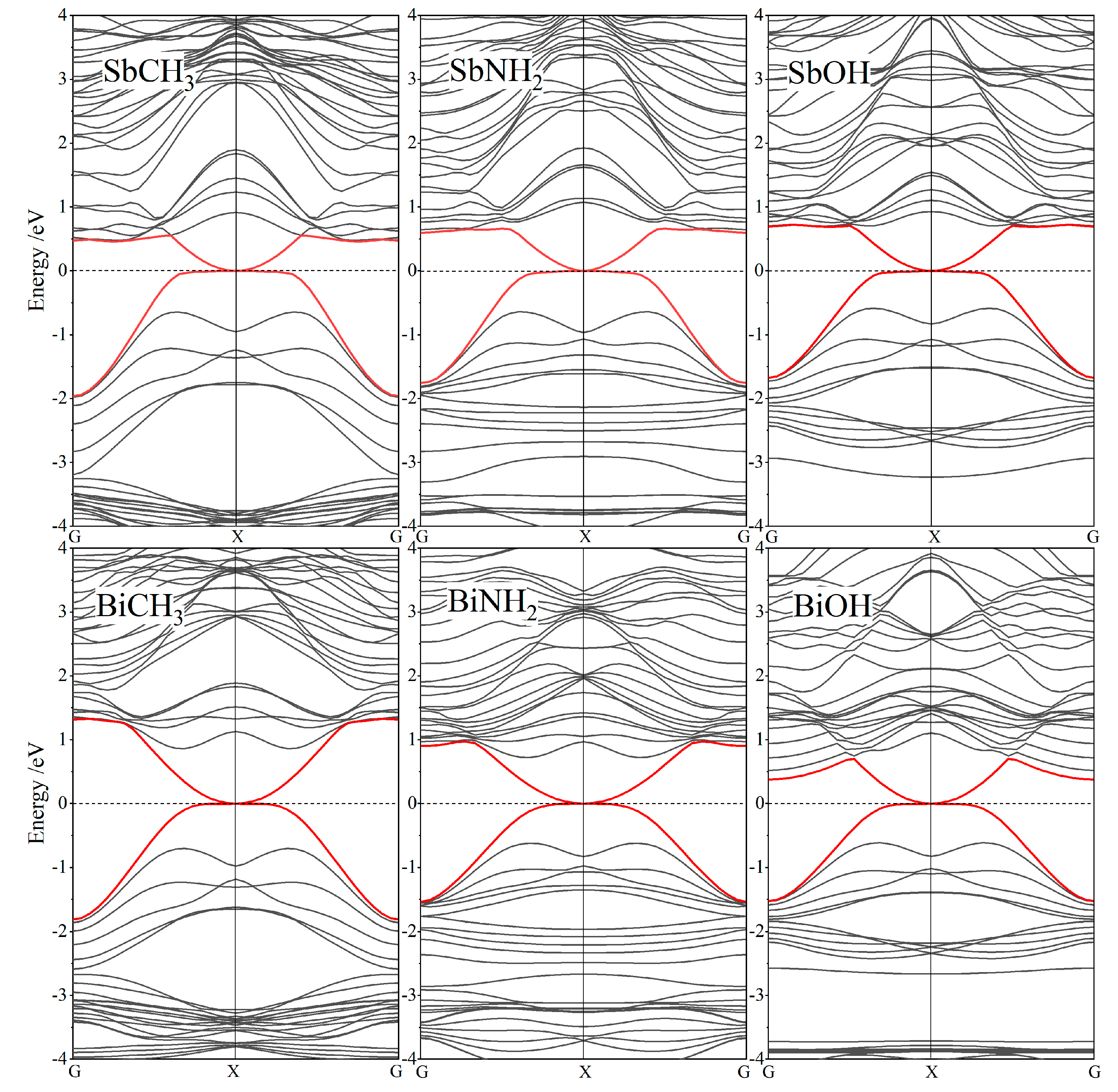
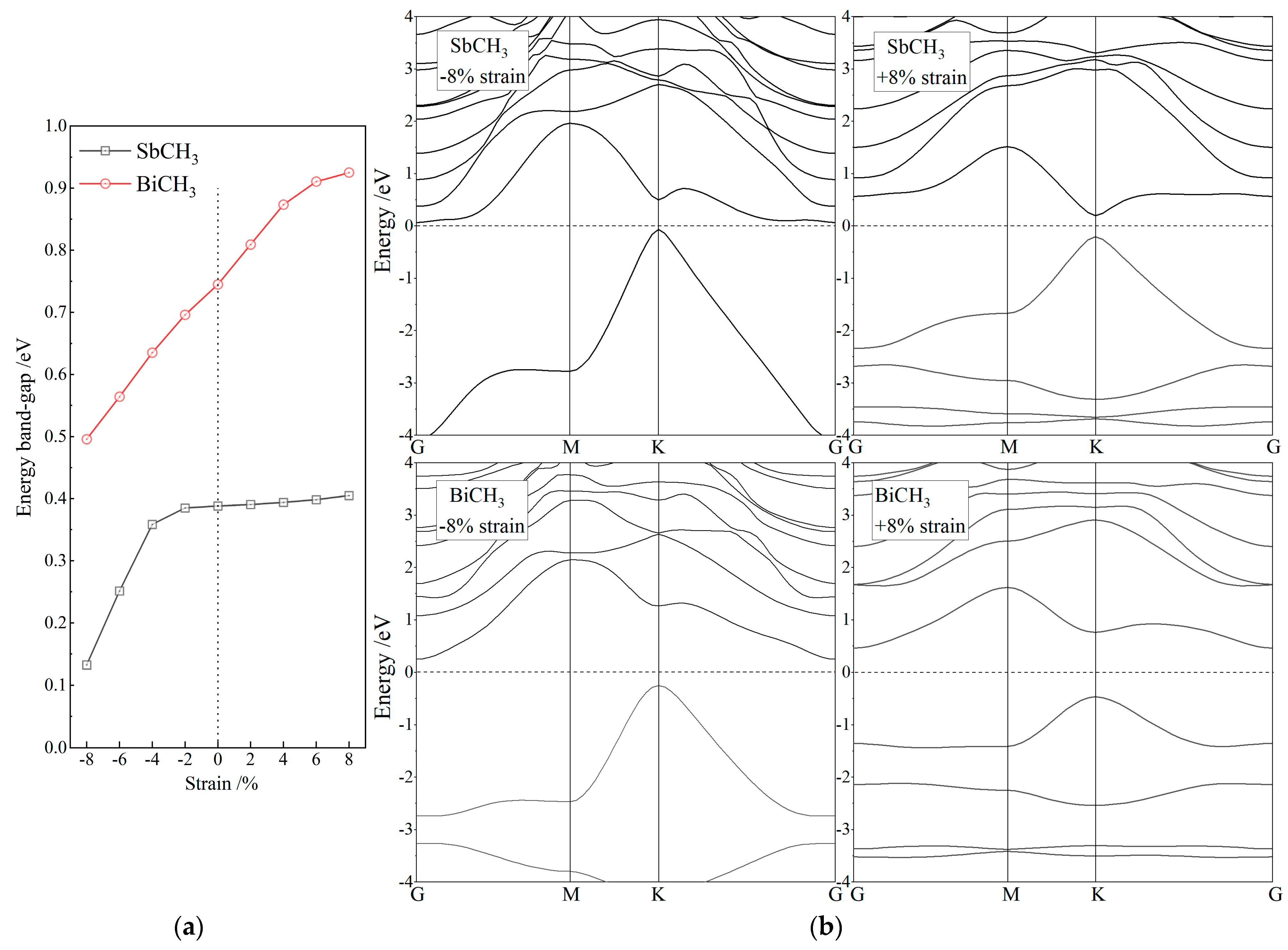
3.2. Band Structure and Topological Property
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bernevig, B.A.; Hughes, T.L.; Zhang, S.C. Quantum spin Hall effect and topological phase transition in HgTe quantum wells. Science 2006, 314, 1757–1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- König, M.; Wiedmann, S.; Brüne, C.; Roth, A.; Buhmann, H.; Molenkamp, L.W.; Qi, X.L.; Zhang, S.C. Quantum spin Hall insulator state in HgTe quantum wells. Science 2007, 318, 766–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasche, B.; Isaeva, A.; Ruck, M.; Borisenko, S.; Zabolotnyy, V.; Büchner, B.; Koepernik, K.; Ortix, C.; Richtern, M.; van den Brink, J. Stacked topological insulator built from bismuth-based graphene sheet analogues. Nat. Mater. 2013, 12, 422–425. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.E. The birth of topological insulators. Nature 2013, 464, 194–198. [Google Scholar] [CrossRef]
- Bernevig, B.A.; Zhang, S.C. Quantum spin Hall effect. Phys. Rev. Lett. 2006, 96, 106802. [Google Scholar] [CrossRef] [Green Version]
- Kane, C.L.; Mele, E.J. Quantum spin Hall effect in graphene. Phys. Rev. Lett. 2005, 95, 226801. [Google Scholar] [CrossRef] [Green Version]
- Chuang, F.C.; Hsu, C.H.; Chen, C.Y.; Huang, Z.Q.; Ozolins, V.; Lin, H.; Bansil, A. Tunable topological electronic structures in Sb (111) bilayers: A first-principles study. Appl. Phys. Lett. 2013, 102, 22424. [Google Scholar] [CrossRef]
- Xu, Y.; Yan, B.H.; Zhang, H.J.; Wang, J.; Xu, G.; Tang, P.; Duan, W.H.; Zhang, S.C. Large-gap quantum spin Hall insulators in tin films. Phys. Rev. Lett. 2013, 111, 136804. [Google Scholar] [CrossRef]
- Weng, H.M.; Dai, X.; Fang, Z. Transition-metal pentatelluride ZrTe5 and HfTe5: A paradigm for large-gap quantum spin Hall insulators. Phys. Rev. X 2014, 4, 11002. [Google Scholar]
- Si, C.; Liu, J.W.; Xu, Y.; Wu, J.; Gu, B.L.; Duan, W.H. Functionalized germanene as a prototype of large-gap two-dimensional topological insulators. Phys. Rev. B 2014, 89, 115429. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.F.; Liu, Z.; Duan, W.; Liu, F.; Wu, J. Topological and electronic transitions in a Sb(111) nanofilm: The interplay between quantum confinement and surface effect. Phys. Rev. B 2013, 85, 201410. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.Q.; Chuang, F.C.; Hsu, C.H.; Liu, Y.T.; Chang, H.R.; Lin, H.; Bansil, A. Nontrivial topological electronic structures in a single Bi(111) bilayer on different substrates: A first-principles study. Phys. Rev. B 2013, 88, 165301. [Google Scholar] [CrossRef]
- Wada, M.; Murakami, S.; Freimuth, F.; Bihlmayer, G. Localized edge states in two-dimensional topological insulators: Ultrathin Bi films. Phys. Rev. B 2011, 83, 121310. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Liu, C.X.; Wu, Y.S.; Duan, W.H.; Liu, F.; Wu, J. Stable nontrivial Z2 topology in ultrathin Bi(111) films: A first-principles study. Phys. Rev. Lett. 2011, 107, 136805. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Miao, L.; Wang, Z.F.; Yao, M.Y.; Zhu, F.; Song, Y.R.; Wang, M.X.; Xu, J.P.; Fedorov, A.V.; Sun, Z.; et al. Spatial and energy distribution of topological edge states in single Bi(111) bilayer. Phys. Rev. Lett. 2012, 109, 16801. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.F.; Yao, M.Y.; Ming, W.; Miao, L.; Zhu, F.; Liu, C.; Gao, C.L.; Qian, D.; Jia, J.F.; Liu, F. Creation of helical Dirac fermions by interfacing two gapped systems of ordinary fermions. Nat. Commun. 2013, 4, 1384. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Xie, L.M.; Li, Y.; Wang, H.; Ouyang, Y.; Guo, J.; Dai, H.G. Controlled chlorine plasma reaction for noninvasive graphene doping. J. Am. Chem. Soc. 2011, 133, 19668–19671. [Google Scholar] [CrossRef]
- Jiang, S.S.; Butler, S.; Bianco, E.; Restrepo, O.D.; Windl, W.; Goldberger, J.E. Improving the stability and optical properties of germanane via one-step covalent methyl-termination. Nat. Commun. 2014, 5, 3389. [Google Scholar] [CrossRef]
- Freitas, R.R.Q.; Rivelino, R.; de Brito Mota, F.; de Castilho, M.C. Topological insulating phases in two-dimensional bismuth containing single layers preserved by hydrogenation. J. Phys. Chem. C 2015, 119, 23599–23606. [Google Scholar] [CrossRef] [Green Version]
- Becerril, H.A.; Mao, J.; Liu, Z.; Stoltenberg, R.M.; Bao, Z.; Chen, Y. Evaluation of solution-processed reduced graphene oxide films as transparent conductors. ACS Nano 2008, 2, 463–470. [Google Scholar] [CrossRef]
- Knapp, D.; Brunschwig, B.S.; Lewis, N.S. Chemical, electronic, and electrical properties of alkylated Ge(111) surfaces. J. Phys. Chem. C 2010, 114, 12300–12307. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.D.; Dai, Y.; Kou, L.Z.; Frauenheim, T.; Heine, T. Robust two-dimensional topological insulators in methylfunctionalized bismuth, antimony, and lead bilayer films. Nano Lett. 2015, 15, 1083–1089. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Burke, K. Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.S.; Qteish, A.; Payne, M.C.; Heine, V. Optimized and transferable nonlocal separable ab initio pseudopotentials. Phys. Rev. B 1993, 47, 4174–4180. [Google Scholar] [CrossRef]
- Milman, V.; Lee, M.H.; Payne, M.C. Ground-state properties of CoSi2 determined by a total-energy pseudopotential method. Phys. Rev. B 1994, 49, 16300–16308. [Google Scholar] [CrossRef]
- Payne, M.C.; Teter, M.P.; Allan, D.C.; Arias, T.A.; Joannopoulos, J.D. Iterative minimization techniques for ab initio total energy calculations: Molecular dynamics and conjugate gradients. Rev. Mod. Phys. 1992, 64, 1045–1097. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations—A reply. Phys. Rev. B 1977, 16, 1748. [Google Scholar]
- Packwood, D.; Kermode, J.; Mones, L.; Bernstein, N.; Woolley, J.; Gould, N.; Ortner, C.; Csanyi, G. A universal pre-conditioner for simulating condensed phase materials. J. Chem. Phys. 2016, 144, 164109. [Google Scholar] [CrossRef] [Green Version]
- Baroni, S.; de Gironcoli, S.; dal Corso, A.; Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 2001, 73, 515–562. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Cao, D.; Wang, J.; Liang, P.; Chen, X.; Shu, H. Semiconducting edges and flake-shape evolution of monolayer GaSe: Role of edge reconstructions. Nanoscale 2018, 10, 12133–12140. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Zhou, S.; Bai, Y.; Zhao, J. Enhanced piezoelectric effect in Janus group-III chalcogenide monolayers. Appl. Phys. Lett. 2017, 110, 163102. [Google Scholar] [CrossRef]
- Xu, X.D.; Sun, W.F. First-principles investigation of GaInSe2 monolayer as a janus material. J. Chin. Ceram. Soc. 2020, 48, 507–513. [Google Scholar]
- Borlido, P.; Schmidt, J.; Huran, A.W.; Tran, F.; Marques, M.A.L.; Botti, S. Exchange-correlation functionals for band gaps of solids: Benchmark, reparametrization and machine learning. npj Comput. Mater. 2020, 6, 96. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Monolayers | Space Symmetry | a/Å | dMM/Å | dMX/Å | hm/Å | ht/Å | Ecoh/(eV·unitcell−1) |
|---|---|---|---|---|---|---|---|
| Sb | P6/MMM | 4.744 | 2.739 | – | – | – | 8.728 |
| SbCH3 | P-3M1 | 5.020 | 2.898 | 2.099 | 0.058 | 5.005 | 14.367 |
| SbNH2 | C2/M | 4.965 | 2.894 | 1.955 | 0.017 | 4.984 | 15.293 |
| SbOH | P-3M1 | 5.016 | 2.896 | 1.769 | 0.044 | 5.485 | 16.232 |
| Bi | P6/MMM | 4.960 | 2.864 | – | – | – | 7.998 |
| BiCH3 | P-3M1 | 5.309 | 3.066 | 2.225 | 0.067 | 5.244 | 12.943 |
| BiNH2 | C2/M | 5.455 | 3.184 | 2.134 | 0.479 | 5.547 | 13.520 |
| BiOH | P-3M1 | 5.289 | 3.059 | 1.917 | 0.173 | 5.928 | 14.501 |
| Monolayers | ED (K) | Eg (G-K) | Monolayers | ED K) | Eg (G-K) |
|---|---|---|---|---|---|
| SbCH3 | 0.388 | - | BiCH3 | 1.312 | 0.745 |
| SbNH2 | 0.370 | - | BiNH2 | 1.396 | 0.498 |
| SbOH | 0.311 | - | BiOH | 1.348 | 0.309 |
| TRIP | Parity δ | P | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| G | +1 | +1 | +1 | −1 | −1 | −1 | +1 | +1 | −1 | +1 | −1 | +1 | −1 |
| 3 M | +1 | −1 | −1 | +1 | +1 | −1 | −1 | +1 | +1 | −1 | +1 | −1 | +1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, Q.; Zhang, G. Spin-Orbit Coupling Electronic Structures of Organic-Group Functionalized Sb and Bi Topological Monolayers. Nanomaterials 2022, 12, 2041. https://doi.org/10.3390/nano12122041
Gong Q, Zhang G. Spin-Orbit Coupling Electronic Structures of Organic-Group Functionalized Sb and Bi Topological Monolayers. Nanomaterials. 2022; 12(12):2041. https://doi.org/10.3390/nano12122041
Chicago/Turabian StyleGong, Qi, and Guiling Zhang. 2022. "Spin-Orbit Coupling Electronic Structures of Organic-Group Functionalized Sb and Bi Topological Monolayers" Nanomaterials 12, no. 12: 2041. https://doi.org/10.3390/nano12122041