
γ-Valerolactone Production from Levulinic Acid Hydrogenation Using Ni Supported Nanoparticles: Influence of Tungsten Loading and pH of Synthesis

 , , , , and
, , , , and
Abstract
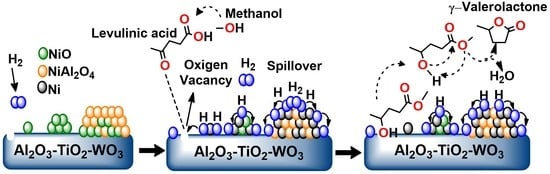
:
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Catalyst Support Synthesis
2.3. Ni Supported Catalysts Preparation
2.4. Characterization Techniques
2.4.1. Nitrogen Physisorption
2.4.2. X-ray Diffraction (XRD)
2.4.3. Diffuse Reflectance UV-Vis Spectroscopy (DRS UV-Vis)
2.4.4. SEM (Scanning Electron Microscopy)
2.4.5. X-ray Photoelectron Spectroscopy (XPS)
2.4.6. Pyridine FTIR Analysis
2.4.7. Temperature Programed Reduction of Hydrogen (TPR-H2)
2.4.8. Temperature Programed Desorption of Hydrogen (TPD-H2)
2.4.9. High Resolution Transmission Electron Microscopy (HRTEM)
2.5. Catalytic Tests
2.5.1. Analysis of the LA and GVL after Reaction Tests
Gas Chromatographic Analysis (GC)
Hydrogen Nuclear Magnetic Resonance 1H-NMR Analysis
3. Results and Discussions
3.1. Catalysts Characterization
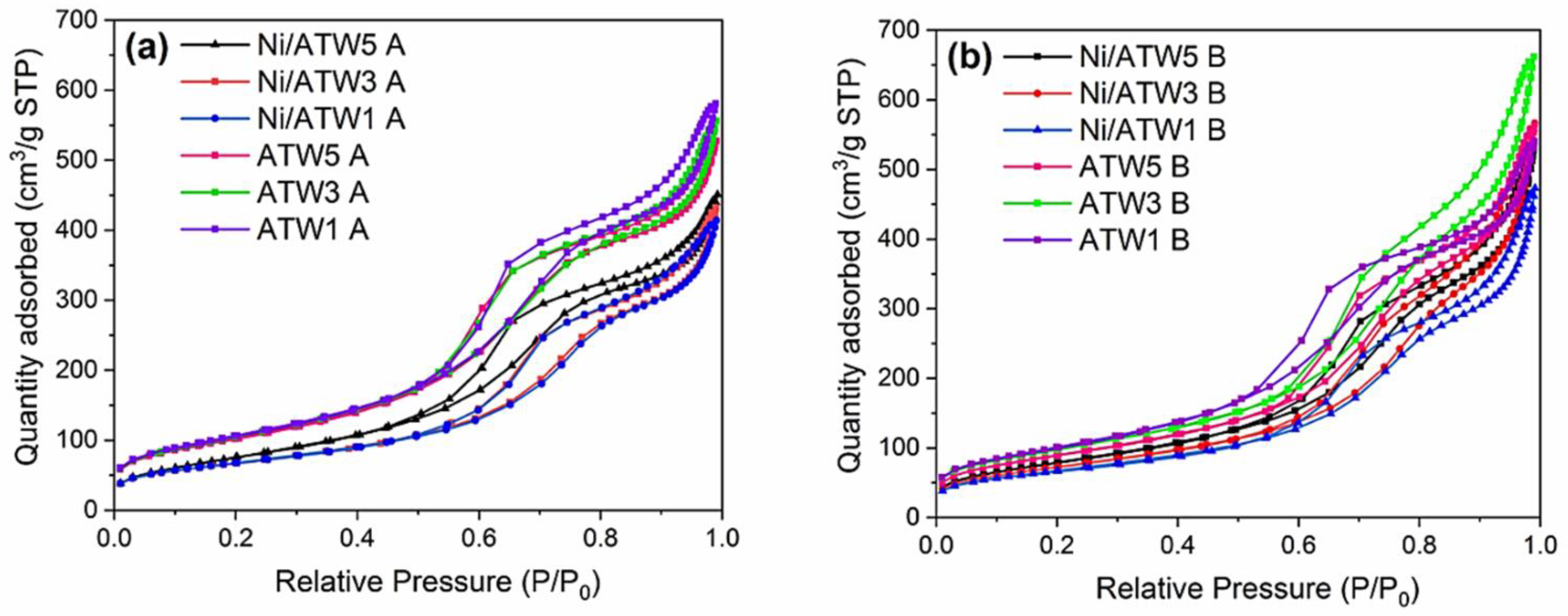
3.1.1. N2 Physisorption
3.1.2. X-ray Diffraction (XRD)
3.1.3. Diffuse Reflectance UV-Vis Spectroscopy (DRS UV-Vis)
3.1.4. TPR-H2
3.1.5. Temperature-Programmed Desorption of Hydrogen (TPD-H2)
3.1.6. Scanning Electron Microscopy (SEM)
3.1.7. HRTEM
3.1.8. FTIR-Pyridine
3.1.9. XPS
3.2. Conversion of Levulinic Acid (LA) to γ-Valerolactone (GVL)
3.2.1. Catalytic Activity of the ATW51 A
1H-NMR of the Crude after Reaction
3.3. Catalytic Tests of the Ni/ATW Catalysts
3.3.1. 1H-NMR Elucidation by Products
3.3.2. Effect of Lewis Acid Sites on the Yield to GVL
3.3.3. Effect of the Ni Particle Size on the Yield to GVL
3.3.4. Effect of de Ni0 Metallic Sites on the Yield to GVL
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Climent, M.J.; Corma, A.; Iborra, S. Conversion of biomass platform molecules into fuel additives and liquid hydrocarbon fuels. Green Chem. 2014, 16, 516–547. [Google Scholar] [CrossRef]
- Ragauskas, A.J.; Williams, C.K.; Davison, B.H.; Britovsek, G.; Cairney, J.; Eckert, C.A.; Frederick, W.J.; Hallett, J.P.; Leak, D.J.; Liotta, C.L.; et al. The Path Forward for Biofuels and Biomaterials. Science 2006, 311, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Mascal, M.; Dutta, S.; Wu, L. Preparation of Compounds from Levulinic Acid. Patent WO/2016/123459, 2020. [Google Scholar]
- Werpy, T.; Petersen, G. Top Value Added Chemicals from Biomass: Volume I—Results of Screening for Potential Candidates from Sugarsand Synthesis Gas; DOE/GO-102004-1992; TRN: US200427%%671 United States 10.2172/15008859 TRN: US200427%%671 NREL English; p. Medium: ED; National Renewable Energy Lab.: Golden, CO, USA, 2004; 76p. [Google Scholar]
- Tulchinsky, M.L.; Briggs, J.R. One-Pot Synthesis of Alkyl 4-Alkoxypentanoates by Esterification and Reductive Etherification of Levulinic Acid in Alcoholic Solutions. ACS Sustain. Chem. Eng. 2016, 4, 4089–4093. [Google Scholar] [CrossRef]
- Flores-Velázquez, V.; Córdova-Pérez, G.E.; Silahua-Pavón, A.A.; Torres-Torres, J.G.; Sierra, U.; Fernández, S.; Godavarthi, S.; Ortiz-Chi, F.; Espinosa-González, C.G. Cellulose obtained from banana plant waste for catalytic production of 5-HMF: Effect of grinding on the cellulose properties. Fuel 2020, 265, 116857. [Google Scholar] [CrossRef]
- Xu, R.; Liu, K.; Du, H.; Liu, H.; Cao, X.; Zhao, X.; Qu, G.; Li, X.; Li, B.; Si, C. Falling Leaves Return to Their Roots: A Review on the Preparation of γ-Valerolactone from Lignocellulose and Its Application in the Conversion of Lignocellulose. ChemSusChem 2020, 13, 6461–6476. [Google Scholar] [CrossRef]
- Hayes, D.; Fitzpatrick, S.; Hayes, M.; Ross, J. The Biofine Process@ Production of Levulinic Acid, Furfural, and Formic Acid from Lignocellulosic Feedstocks. Biorefin. Ind. Processes Prod. 2006, 1, 139–164. [Google Scholar]
- Morales, G.; Melero, J.A.; Iglesias, J.; Paniagua, M.; López-Aguado, C. From levulinic acid biorefineries to γ-valerolactone (GVL) using a bi-functional Zr-Al-Beta catalyst. React. Chem. Eng. 2019, 4, 1834–1843. [Google Scholar] [CrossRef]
- Alonso, D.M.; Wettstein, S.G.; Dumesic, J.A. Gamma-valerolactone, a sustainable platform molecule derived from lignocellulosic biomass. Green Chem. 2013, 15, 584–595. [Google Scholar] [CrossRef]
- Tang, X.; Sun, Y.; Zeng, X.; Lei, T.; Li, H.; Lin, L. Chapter 7—γ-Valerolactone—an excellent solvent and a promising building block. In Biomass, Biofuels, Biochemicals; Saravanamurugan, S., Pandey, A., Li, H., Riisager, A., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 199–226. [Google Scholar]
- Osatiashtiani, A.; Lee, A.F.; Wilson, K. Recent advances in the production of γ-valerolactone from biomass-derived feedstocks via heterogeneous catalytic transfer hydrogenation. J. Chem. Technol. Biotechnol. 2017, 92, 1125–1135. [Google Scholar] [CrossRef]
- Zhang, Z. Synthesis of γ-Valerolactone from Carbohydrates and its Applications. ChemSusChem 2016, 9, 156–171. [Google Scholar] [CrossRef]
- Yu, Z.; Lu, X.; Xiong, J.; Ji, N. Transformation of Levulinic Acid to Valeric Biofuels: A Review on Heterogeneous Bifunctional Catalytic Systems. ChemSusChem 2019, 12, 3915–3930. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Lu, X.; Bai, H.; Xiong, J.; Feng, W.; Ji, N. Effects of Solid Acid Supports on the Bifunctional Catalysis of Levulinic Acid to γ-Valerolactone: Catalytic Activity and Stability. Chem. Asian J. 2020, 15, 1182–1201. [Google Scholar] [CrossRef] [PubMed]
- Horváth, I.T.; Mehdi, H.; Fábos, V.; Boda, L.; Mika, L.T. γ-Valerolactone—A sustainable liquid for energy and carbon-based chemicals. Green Chem. 2008, 10, 238–242. [Google Scholar] [CrossRef]
- Bruno, T.J.; Wolk, A.; Naydich, A. Composition-Explicit Distillation Curves for Mixtures of Gasoline with Four-Carbon Alcohols (Butanols). Energy Fuels 2009, 23, 2295–2306. [Google Scholar] [CrossRef]
- Wang, R.; Chen, L.; Zhang, X.; Zhang, Q.; Li, Y.; Wang, C.; Ma, L. Conversion of levulinic acid to γ-valerolactone over Ru/Al2O3–TiO2 catalyst under mild conditions. RSC Adv. 2018, 8, 40989–40995. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, C.; Gong, W.; Song, J.; Zhang, H.; Zhang, Y.; Wang, G.; Zhao, H. Self-assembled Pd/CeO2 catalysts by a facile redox approach for high-efficiency hydrogenation of levulinic acid into gamma-valerolactone. Catal. Commun. 2017, 93, 10–14. [Google Scholar] [CrossRef]
- Mustafin, K.; Cárdenas-Lizana, F.; Keane, M.A. Continuous gas phase catalytic transformation of levulinic acid to γ-valerolactone over supported Au catalysts. J. Chem. Technol. Biotechnol. 2017, 92, 2221–2228. [Google Scholar] [CrossRef]
- Nemanashi, M.; Noh, J.-H.; Meijboom, R. Hydrogenation of biomass-derived levulinic acid to γ-valerolactone catalyzed by mesoporous supported dendrimer-derived Ru and Pt catalysts: An alternative method for the production of renewable biofuels. Appl. Catal. A Gen. 2018, 550, 77–89. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Y.; Tong, X.; Wang, Y.; Jin, G.; Guo, X. Highly active Ir/SiC catalyst for aqueous hydrogenation of levulinic acid to γ-valerolactone. Catal. Commun. 2020, 139, 105971. [Google Scholar] [CrossRef]
- Upare, P.P.; Lee, J.-M.; Hwang, D.W.; Halligudi, S.B.; Hwang, Y.K.; Chang, J.-S. Selective hydrogenation of levulinic acid to γ-valerolactone over carbon-supported noble metal catalysts. J. Ind. Eng. Chem. 2011, 17, 287–292. [Google Scholar] [CrossRef]
- Yan, Z.-P.; Lin, L.; Liu, S. Synthesis of γ-Valerolactone by Hydrogenation of Biomass-derived Levulinic Acid over Ru/C Catalyst. Energy Fuels 2009, 23, 3853–3858. [Google Scholar] [CrossRef]
- Lange, J.-P.; Price, R.; Ayoub, P.M.; Louis, J.; Petrus, L.; Clarke, L.; Gosselink, H. Valeric Biofuels: A Platform of Cellulosic Transportation Fuels. Angew. Chem. Int. Ed. 2010, 49, 4479–4483. [Google Scholar] [CrossRef] [PubMed]
- Mallesham, B.; Sudarsanam, P.; Venkata Shiva Reddy, B.; Govinda Rao, B.; Reddy, B.M. Nanostructured Nickel/Silica Catalysts for Continuous Flow Conversion of Levulinic Acid to γ-Valerolactone. ACS Omega 2018, 3, 16839–16849. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Sheng, D.; Lu, X. Hydrogenation of Levulinic Acid over Nickel Catalysts Supported on Aluminum Oxide to Prepare γ-Valerolactone. Catalysts 2016, 6, 6. [Google Scholar] [CrossRef]
- Stickney, J.A.; Sager, S.L.; Clarkson, J.R.; Smith, L.A.; Locey, B.J.; Bock, M.J.; Hartung, R.; Olp, S.F. An updated evaluation of the carcinogenic potential of 1,4-dioxane. Regul. Toxicol. Pharmacol. 2003, 38, 183–195. [Google Scholar] [CrossRef]
- Jiang, K.; Sheng, D.; Zhang, Z.; Fu, J.; Hou, Z.; Lu, X. Hydrogenation of levulinic acid to γ-valerolactone in dioxane over mixed MgO–Al2O3 supported Ni catalyst. Catal. Today 2016, 274, 55–59. [Google Scholar] [CrossRef]
- Lv, J.; Rong, Z.; Wang, Y.; Xiu, J.; Wang, Y.; Qu, J. Highly efficient conversion of biomass-derived levulinic acid into γ-valerolactone over Ni/MgO catalyst. RSC Adv. 2015, 5, 72037–72045. [Google Scholar] [CrossRef]
- Hengne, A.M.; Kadu, B.S.; Biradar, N.S.; Chikate, R.C.; Rode, C.V. Transfer hydrogenation of biomass-derived levulinic acid to γ-valerolactone over supported Ni catalysts. RSC Adv. 2016, 6, 59753–59761. [Google Scholar] [CrossRef]
- Hengne, A.M.; Rode, C.V. Cu–ZrO2 nanocomposite catalyst for selective hydrogenation of levulinic acid and its ester to γ-valerolactone. Green Chem. 2012, 14, 1064–1072. [Google Scholar] [CrossRef]
- Al-Shaal, M.G.; Wright, W.R.H.; Palkovits, R. Exploring the ruthenium catalysed synthesis of γ-valerolactone in alcohols and utilisation of mild solvent-free reaction conditions. Green Chem. 2012, 14, 1260–1263. [Google Scholar] [CrossRef]
- Enumula, S.S.; Gurram, V.R.B.; Chada, R.R.; Burri, D.R.; Kamaraju, S.R.R. Clean synthesis of alkyl levulinates from levulinic acid over one pot synthesized WO3-SBA-16 catalyst. J. Mol. Catal. A Chem. 2017, 426, 30–38. [Google Scholar] [CrossRef]
- Kumar, V.V.; Naresh, G.; Deepa, S.; Bhavani, P.G.; Nagaraju, M.; Sudhakar, M.; Chary, K.V.R.; Tardio, J.; Bhargava, S.K.; Venugopal, A. Influence of W on the reduction behaviour and Brønsted acidity of Ni/TiO2 catalyst in the hydrogenation of levulinic acid to valeric acid: Pyridine adsorbed DRIFTS study. Appl. Catal. A Gen. 2017, 531, 169–176. [Google Scholar] [CrossRef]
- Mafokoane, M.; Seguel, J.; García, R.; Díaz de León, J.N.; Sepúlveda, C.; Escalona, N. Conversion of levulinic acid using CuO/WO3(x)-Al2O3 catalysts. Catal. Today 2020, 367, 310–319. [Google Scholar] [CrossRef]
- Yuan, L.; Guo, S.; Li, Z.; Cui, H.; Dong, H.; Zhao, L.; Wang, J. Ring opening of decalin over bifunctional Ni–W carbide/Al2O3–USY catalysts and monofunctional acid Ni–W oxide/Al2O3–USY. RSC Adv. 2017, 7, 9446–9455. [Google Scholar] [CrossRef]
- Luo, W.; Deka, U.; Beale, A.M.; van Eck, E.R.H.; Bruijnincx, P.C.A.; Weckhuysen, B.M. Ruthenium-catalyzed hydrogenation of levulinic acid: Influence of the support and solvent on catalyst selectivity and stability. J. Catal. 2013, 301, 175–186. [Google Scholar] [CrossRef]
- Mohan, V.; Venkateshwarlu, V.; Pramod, C.V.; Raju, B.D.; Rao, K.S.R. Vapour phase hydrocyclisation of levulinic acid to γ-valerolactone over supported Ni catalysts. Catal. Sci. Technol. 2014, 4, 1253–1259. [Google Scholar] [CrossRef]
- Enumula, S.S.; Gurram, V.R.B.; Kondeboina, M.; Burri, D.R.; Kamaraju, S.R.R. ZrO2/SBA-15 as an efficient catalyst for the production of γ-valerolactone from biomass-derived levulinic acid in the vapour phase at atmospheric pressure. RSC Adv. 2016, 6, 20230–20239. [Google Scholar] [CrossRef]
- Shokrollahi Yancheshmeh, M.; Alizadeh Sahraei, O.; Aissaoui, M.; Iliuta, M.C. A novel synthesis of NiAl2O4 spinel from a Ni-Al mixed-metal alkoxide as a highly efficient catalyst for hydrogen production by glycerol steam reforming. Appl. Catal. B Environ. 2020, 265, 118535. [Google Scholar] [CrossRef]
- Zhang, Q.; Wu, T.; Zhang, P.; Qi, R.; Huang, R.; Song, X.; Gao, L. Facile synthesis of hollow hierarchical Ni/γ-Al2O3 nanocomposites for methane dry reforming catalysis. RSC Adv. 2014, 4, 51184–51193. [Google Scholar] [CrossRef]
- Hengst, K.; Ligthart, D.A.J.M.; Doronkin, D.E.; Walter, K.M.; Kleist, W.; Hensen, E.J.M.; Grunwaldt, J.-D. Continuous Synthesis of γ-Valerolactone in a Trickle-Bed Reactor over Supported Nickel Catalysts. Ind. Eng. Chem. Res. 2017, 56, 2680–2689. [Google Scholar] [CrossRef]
- Kondeboina, M.; Enumula, S.S.; Reddy, K.S.; Challa, P.; Burri, D.R.; Kamaraju, S.R.R. Bimetallic Ni-Co/γ-Al2O3 catalyst for vapour phase production of γ-valerolactone: Deactivation studies and feedstock selection. Fuel 2021, 285, 119094. [Google Scholar] [CrossRef]
- Primo, A.; Concepción, P.; Corma, A. Synergy between the metal nanoparticles and the support for the hydrogenation of functionalized carboxylic acids to diols on Ru/TiO2. Chem. Commun. 2011, 47, 3613–3615. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Yu, I.K.M.; Tsang, D.C.W.; Ng, Y.H.; Ok, Y.S.; Sherwood, J.; Clark, J.H. Green synthesis of gamma-valerolactone (GVL) through hydrogenation of biomass-derived levulinic acid using non-noble metal catalysts: A critical review. Chem. Eng. J. 2019, 372, 992–1006. [Google Scholar] [CrossRef]
- Hengst, K.; Schubert, M.; Carvalho, H.W.P.; Lu, C.; Kleist, W.; Grunwaldt, J.-D. Synthesis of γ-valerolactone by hydrogenation of levulinic acid over supported nickel catalysts. Appl. Catal. A Gen. 2015, 502, 18–26. [Google Scholar] [CrossRef]
- Sudhakar, M.; Kumar, V.V.; Naresh, G.; Kantam, M.L.; Bhargava, S.K.; Venugopal, A. Vapor phase hydrogenation of aqueous levulinic acid over hydroxyapatite supported metal (M = Pd, Pt, Ru, Cu, Ni) catalysts. Appl. Catal. B Environ. 2016, 180, 113–120. [Google Scholar] [CrossRef]
- Kumar, V.V.; Naresh, G.; Sudhakar, M.; Tardio, J.; Bhargava, S.K.; Venugopal, A. Role of Brønsted and Lewis acid sites on Ni/TiO2 catalyst for vapour phase hydrogenation of levulinic acid: Kinetic and mechanistic study. Appl. Catal. A Gen. 2015, 505, 217–223. [Google Scholar] [CrossRef]
- Singh, H.; Iyengar, N.; Yadav, R.; Rai, A.; Sinha, A.K. Facile conversion of levulinic acid to γ-valerolactone using a high surface area magnetically separable Ni/NiO catalyst. Sustain. Energy Fuels 2018, 2, 1699–1706. [Google Scholar] [CrossRef]
- Shimizu, K.-I.; Kanno, S.; Kon, K. Hydrogenation of levulinic acid to γ-valerolactone by Ni and MoOx co-loaded carbon catalysts. Green Chem. 2014, 16, 3899–3903. [Google Scholar] [CrossRef]
- Sun, D.; Ohkubo, A.; Asami, K.; Katori, T.; Yamada, Y.; Sato, S. Vapor-phase hydrogenation of levulinic acid and methyl levulinate to γ-valerolactone over non-noble metal-based catalysts. Mol. Catal. 2017, 437, 105–113. [Google Scholar] [CrossRef]
- Atanda, L.; Silahua, A.; Mukundan, S.; Shrotri, A.; Torres-Torres, G.; Beltramini, J. Catalytic behaviour of TiO2-ZrO2 binary oxide synthesized by sol-gel process for glucose conversion to 5-hydroxymethylfurfural. RSC Adv. 2015, 5, 80346–80352. [Google Scholar] [CrossRef]
- Silahua-Pavón, A.A.; Espinosa-González, C.G.; Ortiz-Chi, F.; Pacheco-Sosa, J.G.; Pérez-Vidal, H.; Arévalo-Pérez, J.C.; Godavarthi, S.; Torres-Torres, J.G. Production of 5-HMF from glucose using TiO2-ZrO2 catalysts: Effect of the sol-gel synthesis additive. Catal. Commun. 2019, 129, 105723. [Google Scholar] [CrossRef]
- Córdova-Pérez, G.E.; Torres-Torres, G.; Ortíz-Chi, F.; Godavarthi, S.; Silahua-Pavón, A.A.; Izquierdo-Colorado, A.; Da Costa, P.; Hernández-Como, N.; Aleman, M.; Espinosa-González, C.G. Effect of Acid-Basic Sites Ratio on the Catalytic Activity to Obtain 5-HMF from Glucose Using Al2O3-TiO2-W Catalysts. ChemistrySelect 2018, 3, 12854–12864. [Google Scholar] [CrossRef]
- Guo, J.; Zhu, S.; Cen, Y.; Qin, Z.; Wang, J.; Fan, W. Ordered mesoporous Nb–W oxides for the conversion of glucose to fructose, mannose and 5-hydroxymethylfurfural. Appl. Catal. B Environ. 2017, 200, 611–619. [Google Scholar] [CrossRef]
- Kwak, J.H.; Mei, D.; Peden, C.H.F.; Rousseau, R.; Szanyi, J. (100) facets of γ-Al2O3: The Active Surfaces for Alcohol Dehydration Reactions. Catal. Lett. 2011, 141, 649–655. [Google Scholar] [CrossRef]
- Hengne, A.; Biradar, N.; Rode, C. Surface species of supported ruthenium catalysts in selective hydrogenation of levulinic esters for bio-refinery application. Catal. Lett. 2012, 142, 779–787. [Google Scholar] [CrossRef]
- Cortez-Elizalde, J.; Cuauhtémoc-López, I.; Guerra-Que, Z.; Espinosa de los Monteros, A.E.; Lunagómez-Rocha, M.A.; Silahua-Pavón, A.A.; Arévalo-Pérez, J.C.; Cordero-García, A.; Cervantes-Uribe, A.; Torres-Torres, J.G. Chemical and Structural Changes by Gold Addition Using Recharge Method in NiW/Al2O3-CeO2-TiO2 Nanomaterials. Materials 2021, 14, 5470. [Google Scholar] [CrossRef]
- Tao, F.; Yang, S.; Yang, P.; Shi, Z.; Zhou, R. Effects of support property on the catalytic performance of CeO2-ZrO2-CrOx for 1,2-dichloroethane oxidation. J. Rare Earths 2016, 34, 381–389. [Google Scholar] [CrossRef]
- Rodríguez-Martínez, C.; García-Domínguez, Á.E.; Guerrero-Robles, F.; Saavedra-Díaz, R.O.; Torres-Torres, G.; Felipe, C.; Ojeda-López, R.; Silahua-Pavón, A.; Cervantes-Uribe, A. Synthesis of Supported Metal Nanoparticles (Au/TiO2) by the Suspension Impregnation Method. J. Compos. Sci. 2020, 4, 89. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Benoit, A.; Paramasivam, I.; Nah, Y.C.; Roy, P.; Schmuki, P. Decoration of TiO2 nanotube layers with WO3 nanocrystals for high-electrochromic activity. Electrochem. Commun. 2009, 11, 728–732. [Google Scholar] [CrossRef]
- Mohrhusen, L.; Al-Shamery, K. Conversion of methanol on rutile TiO2 (110) and tungsten oxide clusters: 1. population of defect-dependent thermal reaction pathways. Phys. Chem. Chem. Phys. 2021, 23, 12137–12147. [Google Scholar] [CrossRef]
- Kuo, I.J.; Suzuki, N.; Yamauchi, Y.; Wu, K.C.W. Cellulose-to-HMF conversion using crystalline mesoporous titania and zirconia nanocatalysts in ionic liquid systems. RSC Adv. 2013, 3, 2028–2034. [Google Scholar] [CrossRef]
- Zhao, F.; Yi, L.; Deng, R.; You, K.; Song, J.; Jian, J.; Liu, P.; Ai, Q.; Luo, H.A. Supported WO3/γ-Al2O3 as bifunctional catalyst for liquid-phase highly selective oxidation of cyclohexylamine to cyclohexanone oxime under solvent-free conditions. Mol. Catal. 2019, 475, 110494. [Google Scholar] [CrossRef]
- Wang, C.; Li, Y.; Qiu, P.; Duan, L.; Bi, W.; Chen, Y.; Guo, D.; Liu, Y.; Luo, W.; Deng, Y. Controllable synthesis of highly crystallized mesoporous TiO2/WO3 heterojunctions for acetone gas sensing. Chin. Chem. Lett. 2020, 31, 1119–1123. [Google Scholar] [CrossRef]
- Escobar, J.; De los Reyes, J.A.; Viveros, T. Influence of the Synthesis Additive on the Textural and Structural Characteristics of Sol−Gel Al2O3−TiO2. Ind. Eng. Chem. Res. 2000, 39, 666–672. [Google Scholar] [CrossRef]
- Li, Y.; Wu, L.; Wang, Y.; Ke, P.; Xu, J.; Guan, B. γ-Al2O3 doped with cerium to enhance electron transfer in catalytic ozonation of phenol. J. Water Process Eng. 2020, 36, 101313. [Google Scholar] [CrossRef]
- Zou, X.; Rui, Z.; Song, S.; Ji, H. Enhanced methane combustion performance over NiAl2O4-interface-promoted Pd/γ-Al2O3. J. Catal. 2016, 338, 192–201. [Google Scholar] [CrossRef]
- Calvo, M.; Jiménez González, C.; Rivas, B.; Gutiérrez-Ortiz, J.; Lopez-Fonseca, R. Effect of Ni/Al molar ratio on the performance of substoichiometric NiAl2O4 spinel-based catalysts for partial oxidation of methane. Appl. Catal. B Environ. 2017, 209, 128–138. [Google Scholar] [CrossRef]
- Ragupathi, C.; Vijaya, J.J.; Kennedy, L.J. Preparation, characterization and catalytic properties of nickel aluminate nanoparticles: A comparison between conventional and microwave method. J. Saudi Chem. Soc. 2017, 21, S231–S239. [Google Scholar] [CrossRef]
- Escobar, J.; De los Reyes, J.; Viveros-García, T. Nickel on TiO2-modified Al2O3 sol–gel oxides: Effect of synthesis parameters on the supported phase properties. Appl. Catal. A Gen. 2003, 253, 151–163. [Google Scholar] [CrossRef]
- Gullapelli, S.; Scurrell, M.S.; Valluri, D.K. Photocatalytic H2 production from glycerol–water mixtures over Ni/γ-Al2O3 and TiO2 composite systems. Int. J. Hydrogen Energy 2017, 42, 15031–15043. [Google Scholar] [CrossRef]
- Gholami, T.; Salavati-Niasari, M.; Varshoy, S. Electrochemical hydrogen storage capacity and optical properties of NiAl2O4/NiO nanocomposite synthesized by green method. Int. J. Hydrogen Energy 2017, 42, 5235–5245. [Google Scholar] [CrossRef]
- Jia, W.; Chen, Y.; Liu, M.; Liu, X.; Liu, X.; Yuan, J.; Lu, X.; Zhu, Z. Effect of calcination temperature and fluorination treatment on NiF2-AlF3 catalysts for dehydrofluorination of 1, 1, 1, 2-tetrafluoroethane to synthesize trifluoroethylene. Appl. Catal. A Gen. 2018, 571, 150–157. [Google Scholar] [CrossRef]
- Zurita-Mendez, N.N.; Carbajal-De la Torre, G.; Cadenas, E.; Liu, H.; Espinosa-Medina, M.A. Synthesis and characterization of nickel aluminate nanoparticles. Mater. Res. Express 2018, 6, 015036. [Google Scholar] [CrossRef]
- Scheffer, B.; Heijeinga, J.J.; Moulijn, J.A. An electron spectroscopy and x-ray diffraction study of nickel oxide/alumina and nickel-oxide-tungsten trioxide/alumina catalysts. J. Phys. Chem. 1987, 91, 4752–4759. [Google Scholar] [CrossRef]
- Prins, R. Hydrogen Spillover. Facts and Fiction. Chem. Rev. 2012, 112, 2714–2738. [Google Scholar] [CrossRef]
- Xu, Y.; Wu, S.; Wan, P.; Sun, J.; Hood, Z.D. Introducing Ti3+ defects based on lattice distortion for enhanced visible light photoreactivity in TiO2 microspheres. RSC Adv. 2017, 7, 32461–32467. [Google Scholar] [CrossRef]
- Ewbank, J.L.; Kovarik, L.; Diallo, F.Z.; Sievers, C. Effect of metal–support interactions in Ni/Al2O3 catalysts with low metal loading for methane dry reforming. Appl. Catal. A Gen. 2015, 494, 57–67. [Google Scholar] [CrossRef]
- Zieliński, J. Morphology of nickel/alumina catalysts. J. Catal. 1982, 76, 157–163. [Google Scholar] [CrossRef]
- Gao, J.; Jia, C.; Li, J.; Zhang, M.; Gu, F.; Xu, G.; Zhong, Z.; Su, F. Ni/Al2O3 catalysts for CO methanation: Effect of Al2O3 supports calcined at different temperatures. J. Energy Chem. 2013, 22, 919–927. [Google Scholar] [CrossRef]
- Kumar, V.V.; Naresh, G.; Sudhakar, M.; Anjaneyulu, C.; Bhargava, S.K.; Tardio, J.; Reddy, V.K.; Padmasri, A.H.; Venugopal, A. An investigation on the influence of support type for Ni catalysed vapour phase hydrogenation of aqueous levulinic acid to γ-valerolactone. RSC Adv. 2016, 6, 9872–9879. [Google Scholar] [CrossRef]
- Yang, R.; Du, X.; Zhang, X.; Xin, H.; Zhou, K.; Li, D.; Hu, C. Transformation of Jatropha Oil into High-Quality Biofuel over Ni–W Bimetallic Catalysts. ACS Omega 2019, 4, 10580–10592. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Cheng, H.; He, L.; Yu, Y.; Zhao, F. High performance of Ir-promoted Ni/TiO2 catalyst toward the selective hydrogenation of cinnamaldehyde. J. Catal. 2013, 303, 110–116. [Google Scholar] [CrossRef]
- Hinojosa-Reyes, M.; Rodríguez-González, V.; Zanella, R. Gold nanoparticles supported on TiO2–Ni as catalysts for hydrogen purification via water–Gas shift reaction. RSC Adv. 2014, 4, 4308–4316. [Google Scholar] [CrossRef]
- Mihet, M.; Lazar, M.D. Effect of Pd and Rh promotion on Ni/Al2O3 for NO reduction by hydrogen for stationary applications. Chem. Eng. J. 2014, 251, 310–318. [Google Scholar] [CrossRef]
- Zeng, Y.; Ma, H.; Zhang, H.; Ying, W.; Fang, D. Highly efficient NiAl2O4-free Ni/γ-Al2O3 catalysts prepared by solution combustion method for CO methanation. Fuel 2014, 137, 155–163. [Google Scholar] [CrossRef]
- Jordão, M.H.; Assaf, J.M.; Nascente, P.A.P. Surface Characterization of W/Ni/Al2O3 Catalysts. MRS Proc. 2011, 497, 243. [Google Scholar] [CrossRef]
- Horsley, J.A.; Wachs, I.E.; Brown, J.M.; Via, G.H.; Hardcastle, F.D. Structure of surface tungsten oxide species in the tungsten trioxide/alumina supported oxide system from x-ray absorption near-edge spectroscopy and Raman spectroscopy. J. Phys. Chem. 1987, 91, 4014–4020. [Google Scholar] [CrossRef]
- Yan, X.; Yuan, C.; Bao, J.; Li, S.; Qi, D.; Wang, Q.; Zhao, B.; Hu, T.; Fan, L.; Fan, B.; et al. A Ni-based catalyst with enhanced Ni–support interaction for highly efficient CO methanation. Catal. Sci. Technol. 2018, 8, 3474–3483. [Google Scholar] [CrossRef]
- Chary, K.V.R.; Ramana Rao, P.V.; Venkat Rao, V. Catalytic functionalities of nickel supported on different polymorphs of alumina. Catal. Commun. 2008, 9, 886–893. [Google Scholar] [CrossRef]
- Crisóstomo, C.A.B.; Almeida, T.S.S.; Soares, R.R. Towards triglycerides-based biorefineries: Hydrolysis-reforming-hydrogenation in one-pot over Ni/γ-Al2O3 based catalysts. Catal. Today 2020, 367, 124–136. [Google Scholar] [CrossRef]
- Díaz de León, J.N.; Picquart, M.; Villarroel, M.; Vrinat, M.; Gil Llambias, F.J.; Murrieta, F.; de los Reyes, J.A. Effect of gallium as an additive in hydrodesulfurization WS2/γ-Al2O3 catalysts. J. Mol. Catal. A Chem. 2010, 323, 1–6. [Google Scholar] [CrossRef]
- Liao, M.; Wang, C.; Bu, E.; Chen, Y.; Cheng, Z.; Luo, X.; Shu, R.; Wu, J. Efficient hydrogen production from partial oxidation of propane over SiC doped Ni/Al2O3 catalyst. Energy Procedia 2019, 158, 1772–1779. [Google Scholar] [CrossRef]
- Spanou, S.; Kontos, A.I.; Siokou, A.; Kontos, A.G.; Vaenas, N.; Falaras, P.; Pavlatou, E.A. Self cleaning behaviour of Ni/nano-TiO2 metal matrix composites. Electrochim. Acta 2013, 105, 324–332. [Google Scholar] [CrossRef]
- Billo, T.; Fu, F.-Y.; Raghunath, P.; Shown, I.; Chen, W.-F.; Lien, H.-T.; Shen, T.-H.; Lee, J.-F.; Chan, T.-S.; Huang, K.-Y.; et al. Ni-Nanocluster Modified Black TiO2 with Dual Active Sites for Selective Photocatalytic CO2 Reduction. Small 2018, 14, 1702928. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, Y.; Li, S.; Chen, J. Ni/NiO Nanocomposites with Rich Oxygen Vacancies as High-Performance Catalysts for Nitrophenol Hydrogenation. Catalysts 2019, 9, 944. [Google Scholar] [CrossRef]
- Garbarino, G.; Riani, P.; Infantes-Molina, A.; Rodríguez-Castellón, E.; Busca, G. On the detectability limits of nickel species on NiO/γ-Al2O3 catalytic materials. Appl. Catal. A Gen. 2016, 525, 180–189. [Google Scholar] [CrossRef]
- Panagiotou, G.D.; Petsi, T.; Bourikas, K.; Kordulis, C.; Lycourghiotis, A. The interfacial chemistry of the impregnation step involved in the preparation of tungsten(VI) supported titania catalysts. J. Catal. 2009, 262, 266–279. [Google Scholar] [CrossRef]
- Cruz-Perez, A.E.; Guevara-Lara, A.; Morales-Ceron, J.P.; Alvarez-Hernandez, A.; Reyes, J.A.D.L.; Massin, L.; Geantet, C.; Vrinat, M. Ni and W interactions in the oxide and sulfide states on an Al2O3–TiO2 support and their effects on dibenzothiophene hydrodesulfurization. Catal. Today 2011, 172, 203–208. [Google Scholar] [CrossRef]
- Parry, E.P. An infrared study of pyridine adsorbed on acidic solids. Characterization of surface acidity. J. Catal. 1963, 2, 371–379. [Google Scholar] [CrossRef]
- Zaki, M.I.; Hasan, M.A.; Al-Sagheer, F.A.; Pasupulety, L. In situ FTIR spectra of pyridine adsorbed on SiO2–Al2O3, TiO2, ZrO2 and CeO2: General considerations for the identification of acid sites on surfaces of finely divided metal oxides. Colloids Surf. A Physicochem. Eng. Asp. 2001, 190, 261–274. [Google Scholar] [CrossRef]
- Mohan, V.; Raghavendra, C.; Pramod, C.V.; Raju, B.D.; Rama Rao, K.S. Ni/H-ZSM-5 as a promising catalyst for vapour phase hydrogenation of levulinic acid at atmospheric pressure. RSC Adv. 2014, 4, 9660–9668. [Google Scholar] [CrossRef]
- Leal, G.F.; Lima, S.; Graça, I.; Carrer, H.; Barrett, D.H.; Teixeira-Neto, E.; Curvelo, A.A.S.; Rodella, C.B.; Rinaldi, R. Design of Nickel Supported on Water-Tolerant Nb2O5 Catalysts for the Hydrotreating of Lignin Streams Obtained from Lignin-First Biorefining. iScience 2019, 15, 467–488. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, K. (Ed.) Chapter 2—Determination of Acidic Properties on Solid Surfaces. In Solid Acids and Bases; Academic Press: London, UK, 1970; pp. 5–33. [Google Scholar]
- Tanabe, K. (Ed.) Chapter 4—Acid and Base Centres: Their Structure and Acid-Base Properties. In Solid Acids and Bases; Academic Press: London, UK, 1970; pp. 45–101. [Google Scholar]
- Busca, G.; Lorenzelli, V.; Escribano, V.S.; Guidetti, R. FT-113 study of the surface properties of the spinels NiAl2O4 and CoAl2O4 in relation to those of transitional aluminas. J. Catal. 1991, 131, 167–177. [Google Scholar] [CrossRef]
- Venezia, A.M.; Bertoncello, R.; Deganello, G. X-ray photoelectron spectroscopy investigation of pumice-supported nickel catalysts. Surf. Interface Anal. 1995, 23, 239–247. [Google Scholar] [CrossRef]
- Li, C.P.; Proctor, A.; Hercules, D.M. Curve Fitting Analysis of ESCA Ni 2p Spectra of Nickel-Oxygen Compounds and Ni/Al2O3 Catalysts. Appl. Spectrosc. 1984, 38, 880–886. [Google Scholar] [CrossRef]
- Ruan, L.; Zhang, H.; Zhou, M.; Zhu, L.; Pei, A.; Wang, J.; Yang, K.; Zhang, C.; Xiao, S.; Chen, B.H. A highly selective and efficient Pd/Ni/Ni(OH)2/C catalyst for furfural hydrogenation at low temperatures. Mol. Catal. 2020, 480, 110639. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, X.; Chen, C.; Zou, X.; Shang, X.; Ding, W.; Lu, X. Investigation of mesoporous NiAl2O4/MOx (M = La, Ce, Ca, Mg)–γ-Al2O3 nanocomposites for dry reforming of methane. RSC Adv. 2017, 7, 33143–33154. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, F.; Bai, J.; Liu, T.; Yu, Z.; Dai, M.; Zhou, L.; Wang, H.; Zhang, Y.; Suo, H.; et al. Direct growth of NiO films on Al2O3 ceramics by electrochemical deposition and its excellent H2S sensing properties. Sens. Actuators B Chem. 2019, 296, 126619. [Google Scholar] [CrossRef]
- Méndez-Mateos, D.; Barrio, V.L.; Requies, J.M.; Cambra, J.F. A study of deactivation by H2S and regeneration of a Ni catalyst supported on Al2O3, during methanation of CO2. Effect of the promoters Co, Cr, Fe and Mo. RSC Advances 2020, 10, 16551–16564. [Google Scholar] [CrossRef]
- Rodríguez, J.L.; Poznyak, T.; Valenzuela, M.A.; Tiznado, H.; Chairez, I. Surface interactions and mechanistic studies of 2,4-dichlorophenoxyacetic acid degradation by catalytic ozonation in presence of Ni/TiO2. Chem. Eng. J. 2013, 222, 426–434. [Google Scholar] [CrossRef]
- Reddy, B.M.; Chowdhury, B.; Reddy, E.P.; Fernández, A. An XPS study of dispersion and chemical state of MoO3 on Al2O3-TiO2 binary oxide support. Appl. Catal. A Gen. 2001, 213, 279–288. [Google Scholar] [CrossRef]
- Zhao, X.; Xie, W.; Deng, Z.; Wang, G.; Cao, A.; Chen, H.; Yang, B.; Wang, Z.; Su, X.; Yang, C. Salt templated synthesis of NiO/TiO2 supported carbon nanosheets for photocatalytic hydrogen production. Colloids Surf. A Physicochem. Eng. Asp. 2020, 587, 124365. [Google Scholar] [CrossRef]
- Riyapan, S.; Boonyongmaneerat, Y.; Mekasuwandumrong, O.; Praserthdam, P.; Panpranot, J. Effect of surface Ti3+ on the sol–gel derived TiO2 in the selective acetylene hydrogenation on Pd/TiO2 catalysts. Catal. Today 2015, 245, 134–138. [Google Scholar] [CrossRef]
- Xu, J.; Sun, K.; Zhang, L.; Ren, Y.; Xu, X. A highly efficient and selective catalyst for liquid phase hydrogenation of maleic anhydride to butyric acid. Catal. Commun. 2005, 6, 462–465. [Google Scholar] [CrossRef]
- Patra, N.; Karuturi, S.K.; Vasa, N.J.; Nakamura, D.; Higashihata, M.; Singh, V.; Palani, I. Influence of Ni, Ti and NiTi alloy nanoparticles on hydrothermally grown ZnO nanowires for photoluminescence enhancement. J. Alloys Compd. 2019, 770, 1119–1129. [Google Scholar] [CrossRef]
- Capel-Sanchez, M.C.; Campos-Martin, J.M.; Fierro, J.L.G.; de Frutos, M.P.; Polo, A.P. Effective alkene epoxidation with dilute hydrogen peroxide on amorphous silica-supported titanium catalysts. Chem. Commun. 2000, 855–856. [Google Scholar] [CrossRef]
- Reddy, B.M.; Chowdhury, B.; Smirniotis, P.G. An XPS study of the dispersion of MoO3 on TiO2–ZrO2, TiO2–SiO2, TiO2–Al2O3, SiO2–ZrO2, and SiO2–TiO2–ZrO2 mixed oxides. Appl. Catal. A Gen. 2001, 211, 19–30. [Google Scholar] [CrossRef]
- Reddy, Y.A.K.; Ajitha, B.; Sreedhar, A.; Varrla, E. Enhanced UV photodetector performance in bi-layer TiO2/WO3 sputtered films. Appl. Surf. Sci. 2019, 494, 575–582. [Google Scholar] [CrossRef]
- Chen, C.; Xu, M.; Zhang, K.; An, H.; Zhang, G.; Hong, B.; Li, J.; Lai, Y. Atomically ordered and epitaxially grown surface structure in core-shell NCA/NiAl2O4 enabling high voltage cyclic stability for cathode application. Electrochim. Acta 2019, 300, 437–444. [Google Scholar] [CrossRef]
- Yan, L.; Yao, Q.; Fu, Y. Conversion of levulinic acid and alkyl levulinates into biofuels and high-value chemicals. Green Chem. 2017, 19, 5527–5547. [Google Scholar] [CrossRef]
- Pham, T.N.; Shi, D.; Resasco, D.E. Reaction kinetics and mechanism of ketonization of aliphatic carboxylic acids with different carbon chain lengths over Ru/TiO2 catalyst. J. Catal. 2014, 314, 149–158. [Google Scholar] [CrossRef]
- Tukacs, J.M.; Sylvester, Á.; Kmecz, I.; Jones, R.V.; Óvári, M.; Mika, L.T. Continuous flow hydrogenation of methyl and ethyl levulinate: An alternative route to γ-valerolactone production. R. Soc. Open Sci. 2019, 6, 182233. [Google Scholar] [CrossRef] [PubMed]
- Abdelrahman, O.A.; Heyden, A.; Bond, J.Q. Analysis of Kinetics and Reaction Pathways in the Aqueous-Phase Hydrogenation of Levulinic Acid To Form γ-Valerolactone over Ru/C. ACS Catal. 2014, 4, 1171–1181. [Google Scholar] [CrossRef]
- Weng, R.; Yu, Z.; Xiong, J.; Lu, X. Effects of water in the heterogeneous catalytic valorization of levulinic acid into γ-valerolactone and its derivatives. Green Chem. 2020, 22, 3013–3027. [Google Scholar] [CrossRef]
- Sosa, L.F.; da Silva, V.T.; de Souza, P.M. Hydrogenation of levulinic acid to γ-valerolactone using carbon nanotubes supported nickel catalysts. Catal. Today 2020, 381, 86–95. [Google Scholar] [CrossRef]
- Song, S.; Yao, S.; Cao, J.; Di, L.; Wu, G.; Guan, N.; Li, L. Heterostructured Ni/NiO composite as a robust catalyst for the hydrogenation of levulinic acid to γ-valerolactone. Appl. Catal. B Environ. 2017, 217, 115–124. [Google Scholar] [CrossRef]
- Im, J.; Shin, H.; Jang, H.; Kim, H.; Choi, M. Maximizing the catalytic function of hydrogen spillover in platinum-encapsulated aluminosilicates with controlled nanostructures. Nat. Commun. 2014, 5, 3370. [Google Scholar] [CrossRef]
- Cheng, Z.X.; Zhao, X.G.; Li, J.L.; Zhu, Q.M. Role of support in CO2 reforming of CH4 over a Ni/γ-Al2O3 catalyst. Appl. Catal. A Gen. 2001, 205, 31–36. [Google Scholar] [CrossRef]
- Mieth, J.A.; Huang, Y.J.; Schwarz, J.A. Experimental procedures to evaluate dissolution, metal ion buffering, and catalytic precursor speciation during catalyst preparation. J. Colloid Interface Sci. 1988, 123, 366–379. [Google Scholar] [CrossRef]
- Huang, Y.J.; Schwarz, J.A.; Diehl, J.R.; Baltrus, J.P. Effect of Catalyst Preparation on Catalytic Activity: V. Chemical Structures on Nickel/Alumina Catalysts. Appl. Catal. 1988, 36, 163–175. [Google Scholar] [CrossRef]


























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Substrate | Solvent | H2 Source | T [°C] | Time [h] | GVL Yield (%) | LA Conversion (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| Ni/Al2O3 (40 wt.% Ni) | LA | H2O | 50 bar H2 | 200 | 4 | 57% | 57% | [47] |
| LA | Methanol | 10 bar H2 | 150 | 6 | 5% | 100% | ||
| Ni/HAP (5 wt.% Ni) | LA | Methanol | 10 bar H2 | 275 | 3 | 13% | 21% | [48] |
| Ni/TiO2 (5 wt.% Ni) | LA | H2O | - | 270 | 15 | 23% | 26% | [49] |
| Ni/TiO2 (10 wt.% Ni) | LA | H2O | - | 270 | 15 | 48% | 54% | [49] |
| Ni/Al2O3 (30 wt.% Ni) | LA | Methanol | 40 bar H2 | 120 | 4 | 80% | 89% | [50] |
| Ni/Al2O3 (10 wt.% Ni) | LA | - | 8 bar H2 | 140 | 5 | 11% | 18% | [51] |
| Ni/SiO2 (10 wt.% Ni) | LA | - | 8 bar H2 | 140 | 5 | 8% | 8% | [51] |
| Ni/CeO2 (10 wt.% Ni) | LA | - | 8 bar H2 | 140 | 5 | 5% | 18% | [51] |
| Ni/SiO2 | LA | - | 100 bar H2 | 250 | 5 | 100% | 89 | [52] |
| Supports | WO3 Loading (Wt%) | SBET (m2 g−1) | Pv (cm3 g−1) | Pd (nm) |
|---|---|---|---|---|
| ATW1 A a | 1 | 386.9 | 0.84 | 5.9 |
| ATW3 A a | 3 | 381.9 | 0.80 | 5.9 |
| ATW5 A a | 5 | 370.6 | 0.76 | 5.6 |
| ATW1 B b | 1 | 365.1 | 0.76 | 5.8 |
| ATW3 B b | 3 | 352.7 | 0.95 | 7.9 |
| ATW5 B b | 5 | 323.3 | 0.78 | 7.0 |
| Catalysts | WO3 Loading (Wt%) | SBET (m2 g−1) | Pv (cm3 g−1) | Pd (nm) |
|---|---|---|---|---|
| Ni/ATW1 A a | 1 | 245.95 | 0.59 | 6.89 |
| Ni/ATW3 A a | 3 | 245.98 | 0.59 | 6.88 |
| Ni/ATW5 A a | 5 | 321.59 | 0.64 | 5.74 |
| Ni/ATW1 B b | 1 | 244.31 | 0.62 | 7.60 |
| Ni/ATW3 B b | 3 | 282.36 | 0.77 | 8.39 |
| Ni/ATW5 B b | 5 | 317.18 | 0.73 | 7.20 |
| Catalyst | PSNi a (nm) | PSNi b (nm) | PSNi c (nm) | DNi d (%) | Lewis µmol/gcat | DensityLAS (µmol/m2) |
|---|---|---|---|---|---|---|
| Ni/ATW1 A | 9.4 | 9.3 | 8.7 | 11.5 | N.D | N.D |
| Ni/ATW3 A | 10.4 | N.D | N.D | N.D | 207.1 | 0.84 |
| Ni/ATW5 A | 9.8 | 11.9 | N.D | N.D | 428.9 | 1.74 |
| Ni/ATW1 B | 8.8 | 10.2 | N.D | N.D | 286.8 | 0.89 |
| Ni/ATW3 B | 8.3 | N.D | N.D | N.D | 458.9 | 1.87 |
| Ni/ATW5 B | 9.9 | 8.5 | 8.2 | 12.5 | 445.5 | 1.57 |
| Catalyst | Binding Energy Ni2p3/2 (Ni0) (eV) | Binding Energy Ni2p3/2 (Ni(OH)2 (eV) | Binding Energy Ni2p3/2 (NiAl2O4) (eV) |
|---|---|---|---|
| Ni/ATW1 A | 852.6 (9.2%) | 855.5 (25.2%) | 856.3 (65.6%) |
| Ni/ATW3 A | 852.4 (8.2%) | 855.4 (31.8%) | 856.2 (60%) |
| Ni/ATW5 A | 852.4 (7.3%) | 855.3 (46.9%) | 856.6 (45.8%) |
| Ni/ATW1 B | 852.3 (7.2%) | 855.3 (64.9%) | 856.7 (27.8%) |
| Ni/ATW5 B | 852.2 (10.1%) | 855.2 (61.3%) | 856.6 (28.6%) |
| Catalyst | Binding Energy Tip2/3 (Ti4+/Ti3+) (eV) | Binding Energy W4f7/2 (W6+) (eV) | Binding Energy Al2p (Al3+ AlO6/AlO4) (eV) | Binding Energy O1s (eV) |
|---|---|---|---|---|
| Ni/ATW1 A | 458.7/457.5 | 35.7 | 74.6/73.5 | 531.6, 530.5, 529.6 |
| Ni/ATW3 A | 458.7/457.5 | 35.8 | 74.6/73.5 | 531.5, 530.5, 529.6 |
| Ni/ATW5 A | 458.7/457.5 | 35.4 | 74.6/73.5 | 531.6, 530.5, 529.6 |
| Ni/ATW1 B | 458.0/457.1 | 35.3 | 74.4/73.3 | 531.1, 530.1, 529.4 |
| Ni/ATW5 B | 457.9/457.0 | 35.2 | 74.2/73.3 | 531.1, 530.0, 529.3 |
| Catalyst | LA Conversion (%) | GVL Yield (%) |
|---|---|---|
| ATW1 A | 100 | 6 a |
| Ni/ATW1 A | 100 | 59 |
| Ni/ATW3 A | 100 | 27 |
| Ni/ATW5 A | 97 | 18 |
| Ni/ATW1 B | 100 | 43 |
| Ni/ATW3 B | 100 | 65 |
| Ni/ATW5 B | 100 | 80 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Córdova-Pérez, G.E.; Cortez-Elizalde, J.; Silahua-Pavón, A.A.; Cervantes-Uribe, A.; Arévalo-Pérez, J.C.; Cordero-Garcia, A.; de los Monteros, A.E.E.; Espinosa-González, C.G.; Godavarthi, S.; Ortiz-Chi, F.; et al. γ-Valerolactone Production from Levulinic Acid Hydrogenation Using Ni Supported Nanoparticles: Influence of Tungsten Loading and pH of Synthesis. Nanomaterials 2022, 12, 2017. https://doi.org/10.3390/nano12122017
Córdova-Pérez GE, Cortez-Elizalde J, Silahua-Pavón AA, Cervantes-Uribe A, Arévalo-Pérez JC, Cordero-Garcia A, de los Monteros AEE, Espinosa-González CG, Godavarthi S, Ortiz-Chi F, et al. γ-Valerolactone Production from Levulinic Acid Hydrogenation Using Ni Supported Nanoparticles: Influence of Tungsten Loading and pH of Synthesis. Nanomaterials. 2022; 12(12):2017. https://doi.org/10.3390/nano12122017
Chicago/Turabian StyleCórdova-Pérez, Gerardo E., Jorge Cortez-Elizalde, Adib Abiu Silahua-Pavón, Adrián Cervantes-Uribe, Juan Carlos Arévalo-Pérez, Adrián Cordero-Garcia, Alejandra E. Espinosa de los Monteros, Claudia G. Espinosa-González, Srinivas Godavarthi, Filiberto Ortiz-Chi, and et al. 2022. "γ-Valerolactone Production from Levulinic Acid Hydrogenation Using Ni Supported Nanoparticles: Influence of Tungsten Loading and pH of Synthesis" Nanomaterials 12, no. 12: 2017. https://doi.org/10.3390/nano12122017