
Coupling between Polymer Conformations and Dynamics Near Amorphous Silica Surfaces: A Direct Insight from Atomistic Simulations

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Model and Simulation Details
- First, the slab was prepared as follows: An amorphous silica layer containing silicon and oxygen atoms was obtained from a bulk system prepared by following the protocol in [61]. The atoms inside the silica layer were connected according to the method reported in [62]; some of the unsaturated oxygen atoms on the surface were terminated by bonding hydrogen atoms to them.
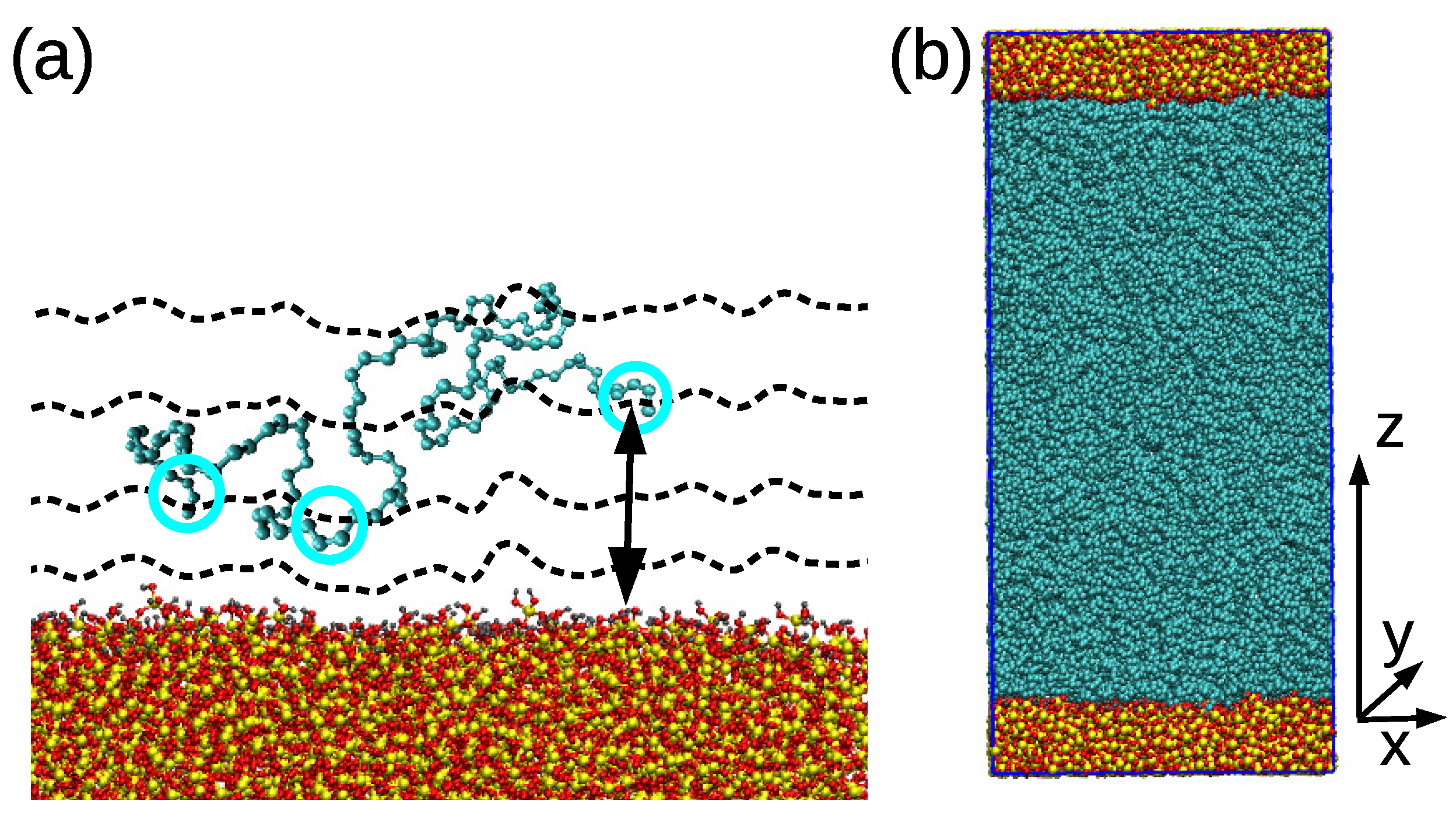
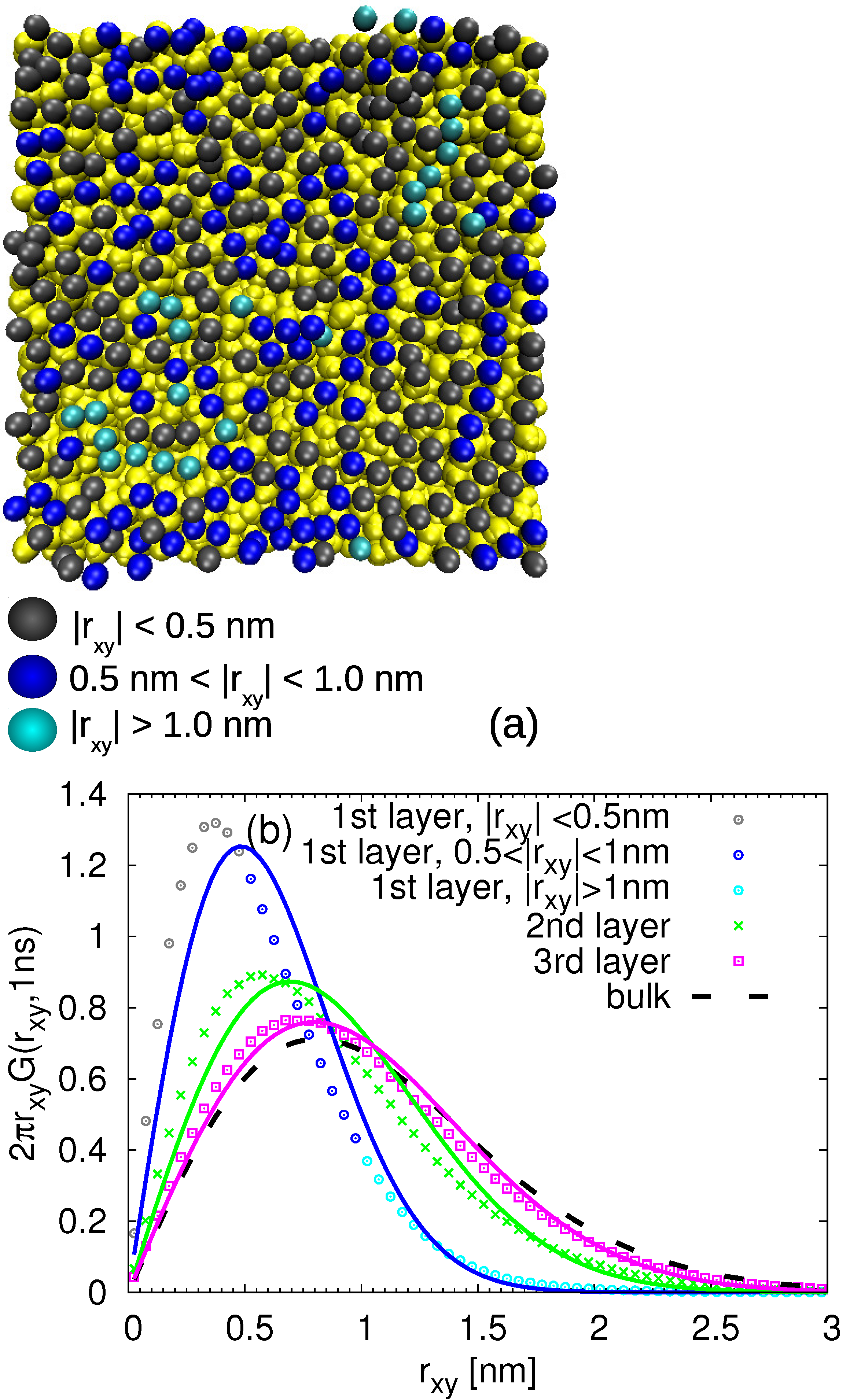
- Then, the polymer chains were added in the simulation box via a sequence of 30–60 runs, each of 1 ns with the time step of 0.5 fs. During those runs, the added chains were adsorbed to the surface and integrated into the polymer matrix; thus, the regions of heterogeneous density were eliminated. An example of a randomly selected PB chain adsorbed on the surface is shown in Figure 1a. The temperature was maintained by the stochastic velocity rescaling algorithm and the system was coupled to the Berendsen barostat.
- Once the film was prepared, the time step was increased to 1 fs and the film was equilibrated at the given temperature for about 50–75 ns. The same barostat and thermostat as in the step two were used.
- The final step was the pre-production run and this was conducted with the same settings as the production run (see below) for about 25 ns.
3. Results
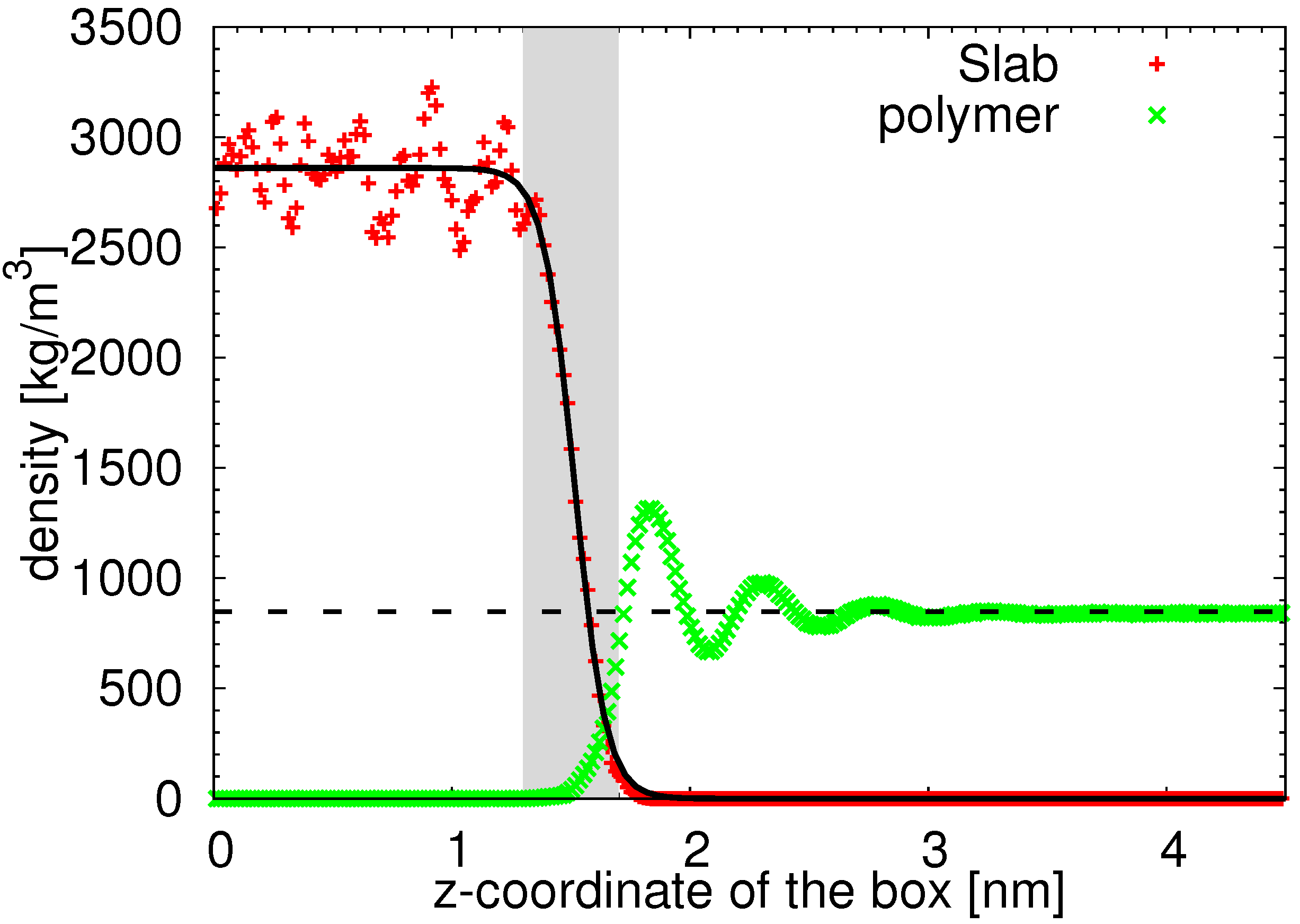
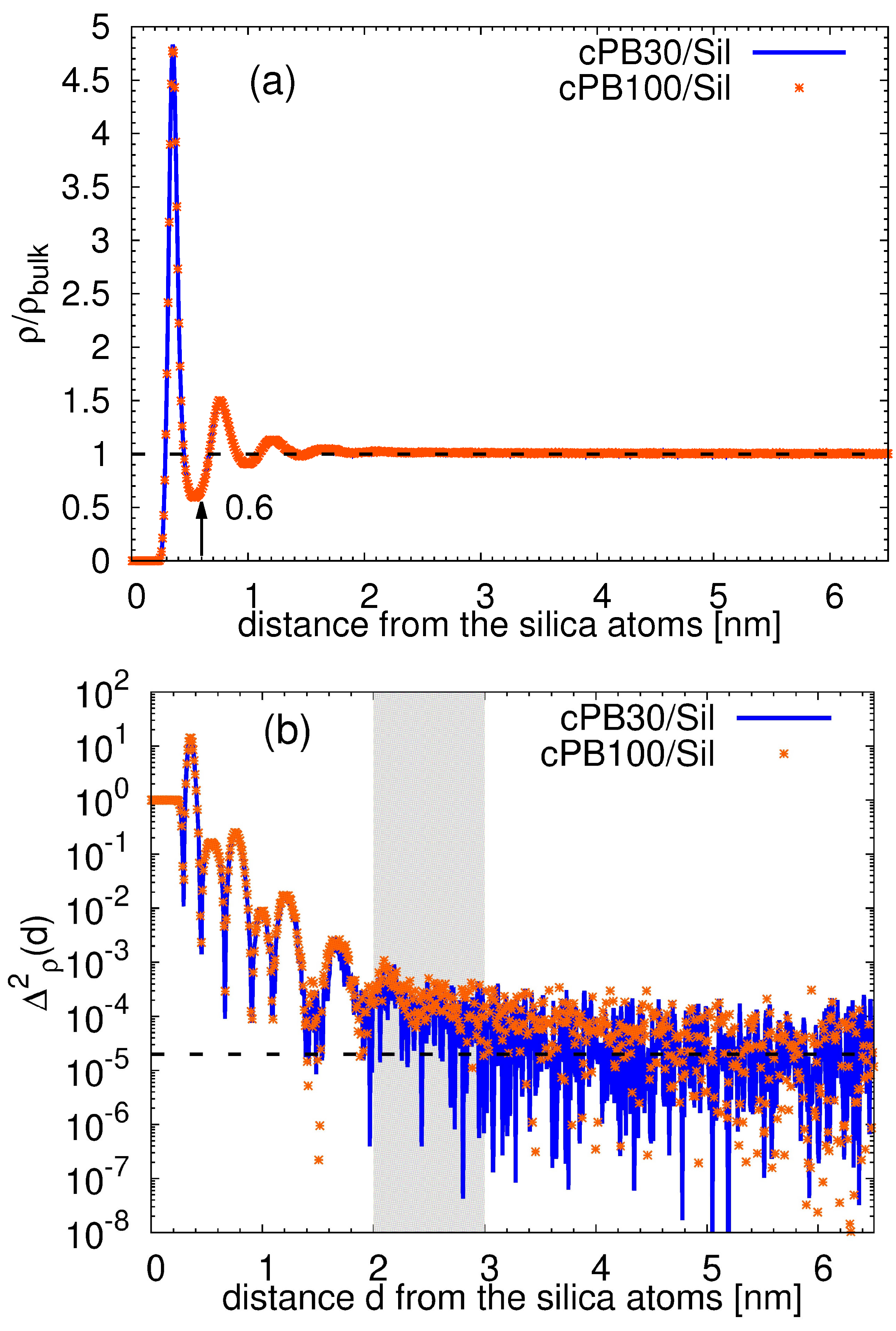
3.1. Structure of the Interphase
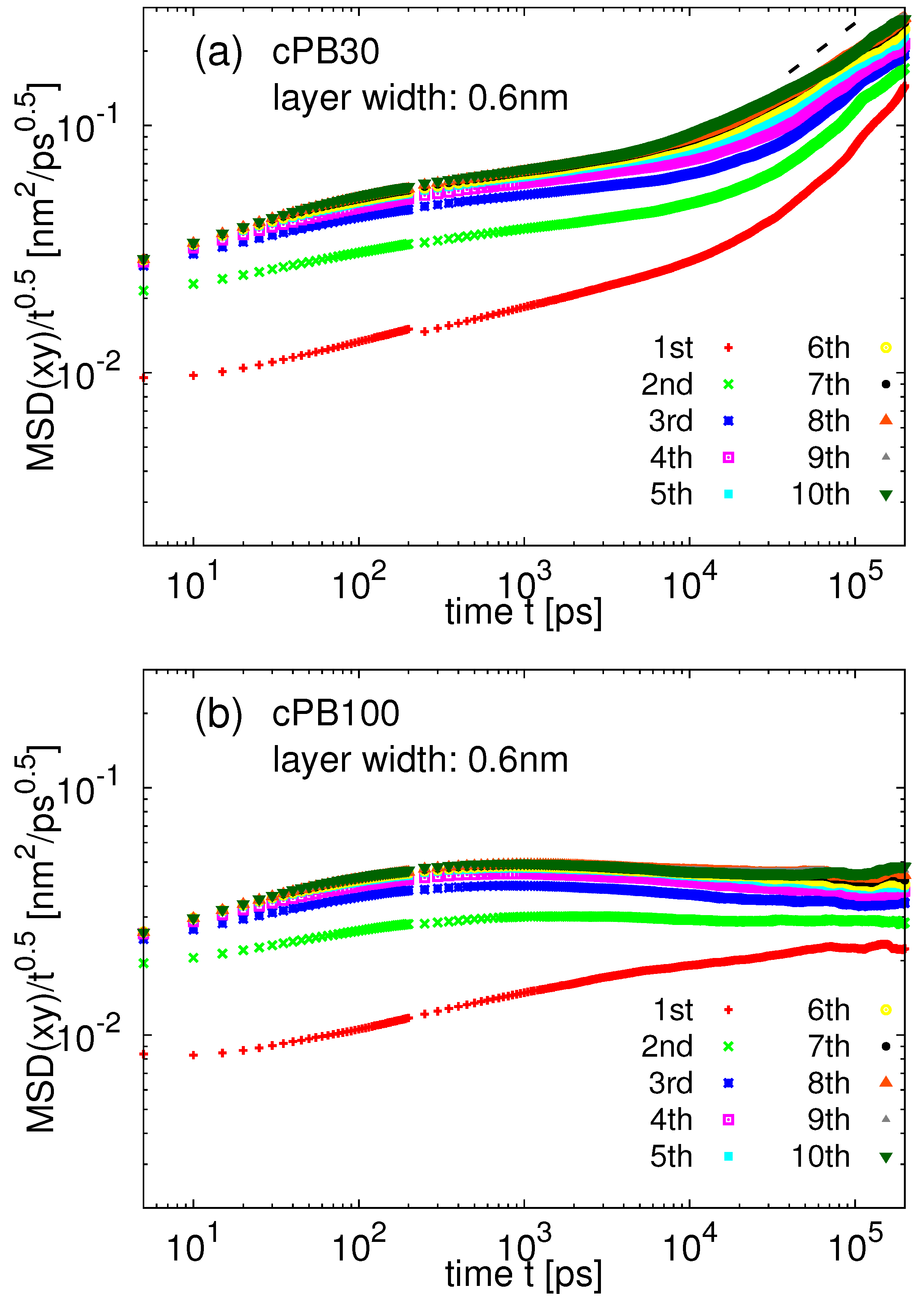
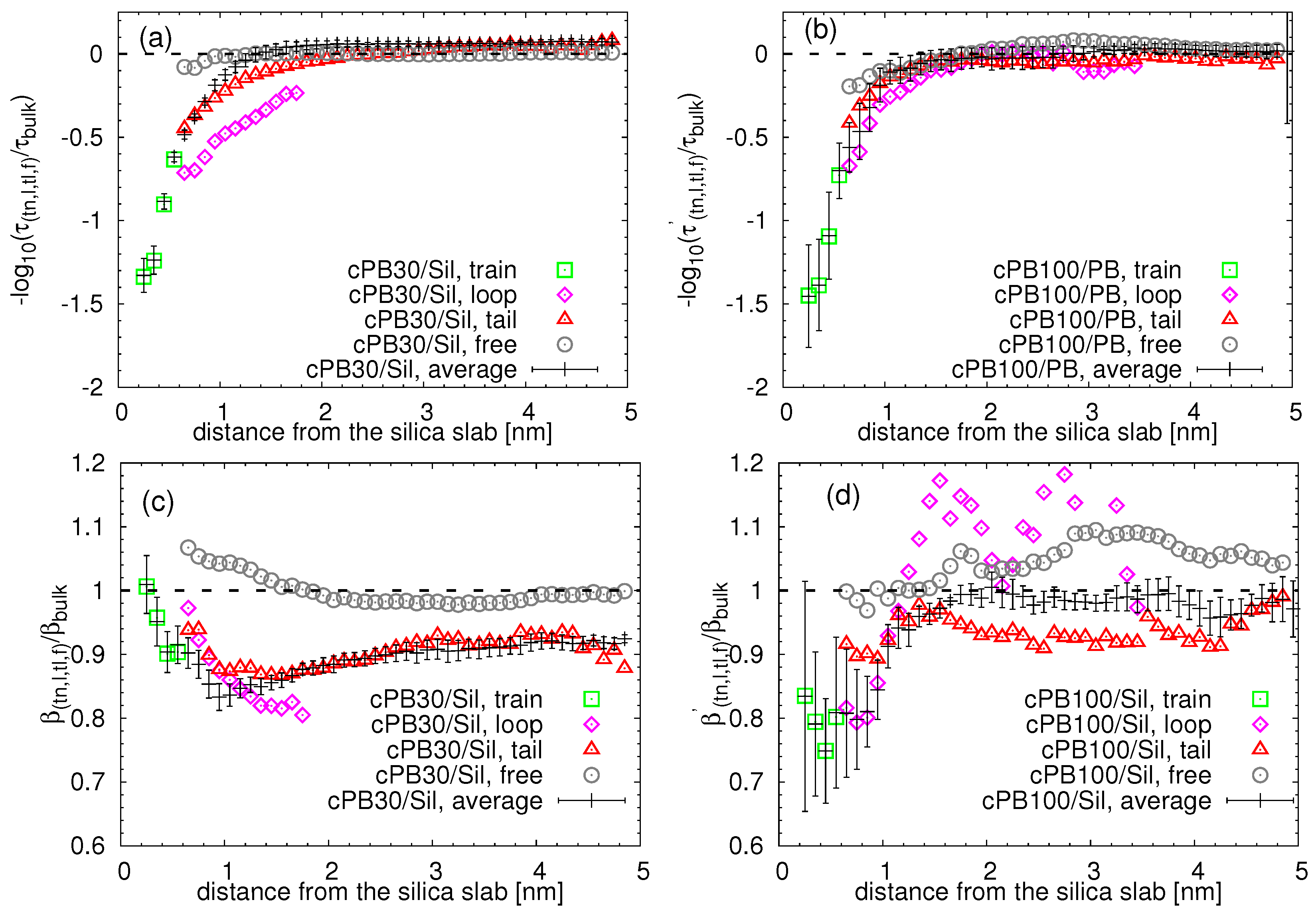
3.2. Dynamics at the Interface
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bailey, E.; Winey, K. Dynamics of Polymer Segments, Polymer Chains, and Nanoparticles in Polymer Nanocomposite Melts: A Review. Prog. Polym. Sci. 2020, 105, 101242. [Google Scholar] [CrossRef]
- Karatrantos, A.V.; Clarke, N. Polymer Dynamics in Polymer-Nanoparticle Interface. In Theory and Modeling of Polymer Nanocomposites; Ginzburg, V.V., Hall, L.M., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 81–100. [Google Scholar]
- Starr, F.W.; Schrøder, T.B.; Glotzer, S.C. Effects of a nanoscopic filler on the structure and dynamics of a simulated polymer melt and the relationship to ultrathin films. Phys. Rev. E 2001, 64, 021802. [Google Scholar] [CrossRef] [Green Version]
- Bansal, A.; Yang, H.; Li, C.; Cho, K.; Benicewicz, B.; Kumar, S.; Schadler, L. Quantitative equivalence between polymer nanocomposites and thin polymer films. Nat. Mater. 2005, 4, 693–698. [Google Scholar] [CrossRef]
- Hasegawa, R.; Doi, M. Adsorption Dynamics. Extension of Self-Consistent Field Theory to Dynamical Problems. Macromolecules 1997, 30, 3086–3089. [Google Scholar] [CrossRef]
- Semenov, A.; Joanny, J.F. Kinetics of Adsorption of Linear Homopolymers onto Flat Surfaces: Rouse Dynamics. J. Phys. II 1995, 5, 859–876. [Google Scholar] [CrossRef] [Green Version]
- Semenov, A.N.; Bonet-Avalos, J.; Johner, A.; Joanny, J.F. Adsorption of Polymer Solutions onto a Flat Surface. Macromolecules 1996, 29, 2179–2196. [Google Scholar] [CrossRef]
- Chandran, S.; Baschnagel, J.; Cangialosi, D.; Fukao, K.; Glynos, E.; Janssen, L.M.C.; Müller, M.; Muthukumar, M.; Steiner, U.; Xu, J.; et al. Processing Pathways Decide Polymer Properties at the Molecular Level. Macromolecules 2019, 52, 7146–7156. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.; Holt, A.; Wang, H.; Fan, F.; Bocharova, V.; Martin, H.; Etampawala, T.; White, B.; Saito, T.; Kang, N.G.; et al. Unexpected Molecular Weight Effect in Polymer Nanocomposites. Phys. Rev. Lett. 2016, 116, 038302. [Google Scholar] [CrossRef] [Green Version]
- Gin, P.; Jiang, N.; Liang, C.; Taniguchi, T.; Akgun, B.; Satija, S.K.; Endoh, M.K.; Koga, T. Revealed Architectures of Adsorbed Polymer Chains at Solid-Polymer Melt Interfaces. Phys. Rev. Lett. 2012, 109, 265501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panagopoulou, A.; Rodríguez-Tinoco, C.; White, R.P.; Lipson, J.E.G.; Napolitano, S. Substrate Roughness Speeds Up Segmental Dynamics of Thin Polymer Films. Phys. Rev. Lett. 2020, 124, 027802. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Thees, M.F.; Size, W.B.; Roth, C.B. Experimental study of substrate roughness on the local glass transition of polystyrene. J. Chem. Phys. 2020, 152, 244901. [Google Scholar] [CrossRef]
- Beena Unni, A.; Chat, K.; Duarte, D.M.; Wojtyniak, M.; Geppert-Rybczyńska, M.; Kubacki, J.; Wrzalik, R.; Richert, R.; Adrjanowicz, K. Experimental evidence on the effect of substrate roughness on segmental dynamics of confined polymer films. Polymer 2020, 199, 122501. [Google Scholar] [CrossRef]
- Tarnacka, M.; Wojtyniak, M.; Brzózka, A.; Talik, A.; Hachuła, B.; Kamińska, E.; Sulka, G.D.; Kaminski, K.; Paluch, M. Unique Behavior of Poly(propylene glycols) Confined within Alumina Templates Having a Nanostructured Interface. Nano Lett. 2020, 20, 5714–5719. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Ganesan, V.; Riggleman, R.A. Perspective: Outstanding theoretical questions in polymer-nanoparticle hybrids. J. Chem. Phys. 2017, 147, 020901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, Y.N.; Papakonstantopoulos, G.J.; Doxastakis, M. Polymer/Nanoparticle Interactions: Bridging the Gap. Macromolecules 2013, 46, 5097–5106. [Google Scholar] [CrossRef]
- Carrillo, J.M.Y.; Cheng, S.; Kumar, R.; Goswami, M.; Sokolov, A.P.; Sumpter, B.G. Untangling the Effects of Chain Rigidity on the Structure and Dynamics of Strongly Adsorbed Polymer Melts. Macromolecules 2015, 48, 4207–4219. [Google Scholar] [CrossRef]
- Baschnagel, J.; Meyer, H.; Wittmer, J.; Kulić, I.; Mohrbach, H.; Ziebert, F.; Nam, G.M.; Lee, N.K.; Johner, A. Semiflexible Chains at Surfaces: Worm-Like Chains and beyond. Polymers 2016, 8, 286. [Google Scholar] [CrossRef] [Green Version]
- Li, C.Y.; Luo, M.; Li, H.; Cao, W.P. Simulation study on the conformational properties of an adsorbed polymer on a nanoparticle. Colloid Polym. Sci. 2017, 295, 1–10. [Google Scholar] [CrossRef]
- Li, C.Y.; Huang, J.H.; Li, H.; Luo, M.B. Study on the interfacial properties of polymers around a nanoparticle. RSC Adv. 2020, 10, 28075–28082. [Google Scholar] [CrossRef]
- Kirk, J.; Ilg, P. Chain Dynamics in Polymer Melts at Flat Surfaces. Macromolecules 2017, 50, 3703–3718. [Google Scholar] [CrossRef]
- Kirk, J.; Wang, Z.; Ilg, P. Entanglement dynamics at flat surfaces: Investigations using multi-chain molecular dynamics and a single-chain slip-spring model. J. Chem. Phys. 2019, 150, 094906. [Google Scholar] [CrossRef] [PubMed]
- De Virgiliis, A.; Milchev, A.; Rostiashvili, V.; Vilgis, T. Structure and dynamics of a polymer melt at an attractive surface. Eur. Phys. J. Soft Matter 2012, 35, 9772. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, Q.H.; Fu, Y.; Wu, F.; Huang, J.H.; Luo, M.B. Study on the adsorption process of a semi-flexible polymer onto homogeneous attractive surfaces. Polymer 2019, 172, 83–90. [Google Scholar] [CrossRef]
- Yang, Q.H.; Qi, H.K.; Yang, X.; Luo, M.B. Simulation study on the critical adsorption and diffusion of polymer chains on heterogeneous surfaces with random adsorption sites. Soft Matter 2021, 17, 1000–1007. [Google Scholar] [CrossRef]
- Ziebarth, J.D.; Wang, Y.; Polotsky, A.; Luo, M. Dependence of the Critical Adsorption Point on Surface and Sequence Disorders for Self-Avoiding Walks Interacting with a Planar Surface. Macromolecules 2007, 40, 3498–3504. [Google Scholar] [CrossRef] [Green Version]
- Sarabadani, J.; Milchev, A.; Virgiliis, A.D.; Vilgis, T.A. Molecular Dynamic Study of the Structure and Dynamics of Polymer Melt at Solid Surfaces. Soft Mater. 2014, 12, S56–S70. [Google Scholar] [CrossRef]
- Li, J.; Lu, Y.; Hao, L.; Zhang, R.; Ding, M.; Shi, T. Dynamics Transition of Polymer Films Induced by Polymer–Obstacle Entanglements on Rough Surfaces. Macromolecules 2020, 53, 3873–3882. [Google Scholar] [CrossRef]
- Li, J.; Ding, M.; Zhang, R.; Shi, T. Effects of surface roughness on the self-diffusion dynamics of a single polymer. Soft Matter 2018, 14, 3550–3556. [Google Scholar] [CrossRef]
- Jeong, D.; Kim, J.; Sung, B.J. Effects of solvent and wall roughness on the dynamics and structure of a single polymer in a slit. Polymer 2016, 92, 256–263. [Google Scholar] [CrossRef]
- Hanakata, P.Z.; Douglas, J.F.; Starr, F.W. Interfacial mobility scale determines the scale of collective motion and relaxation rate in polymer films. Nat. Commun. 2014, 5, 1–8. [Google Scholar] [CrossRef]
- Raos, G.; Idé, J. Impact of Interaction Strength and Surface Heterogeneity on the Dynamics of Adsorbed Polymers. ACS Macro Lett. 2014, 3, 721–726. [Google Scholar] [CrossRef]
- Pastore, R.; David, A.; Casalegno, M.; Greco, F.; Raos, G. Influence of wall heterogeneity on nanoscopically confined polymers. Phys. Chem. Chem. Phys. 2019, 21, 772–779. [Google Scholar] [CrossRef] [Green Version]
- Pandey, Y.N.; Brayton, A.; Burkhart, C.; Papakonstantopoulos, G.J.; Doxastakis, M. Multiscale modeling of polyisoprene on graphite. J. Chem. Phys. 2014, 140, 054908. [Google Scholar] [CrossRef] [PubMed]
- Daoulas, K.C.; Harmandaris, V.A.; Mavrantzas, V.G. Detailed atomistic simulation of a polymer melt/solid interface: Structure, density, and conformation of a thin film of polyethylene melt adsorbed on graphite. Macromolecules 2005, 38, 5780–5795. [Google Scholar] [CrossRef]
- Harmandaris, V.A.; Daoulas, K.C.; Mavrantzas, V.G. Molecular dynamics simulation of a polymer melt/solid interface: Local dynamics and chain mobility in a thin film of polyethylene melt adsorbed on graphite. Macromolecules 2005, 38, 5796–5809. [Google Scholar] [CrossRef]
- Behbahani, A.F.; Vaez Allaei, S.M.; Motlagh, G.H.; Eslami, H.; Harmandaris, V.A. Structure, dynamics, and apparent glass transition of stereoregular poly (methyl methacrylate)/graphene interfaces through atomistic simulations. Macromolecules 2018, 51, 7518–7532. [Google Scholar] [CrossRef]
- Solar, M.; Mapesa, E.U.; Kremer, F.; Binder, K.; Paul, W. The dielectric α-relaxation in polymer films: A comparison between experiments and atomistic simulations. EPL (Europhys. Lett.) 2013, 104, 66004. [Google Scholar] [CrossRef]
- Solar, M.; Yelash, L.; Virnau, P.; Binder, K.; Paul, W. Polymer Dynamics in a Polymer-Solid Interphase: Molecular Dynamics Simulations of 1,4-Polybutadiene At a Graphite Surface. Soft Mater. 2014, 12, S80–S89. [Google Scholar] [CrossRef]
- Solar, M.; Binder, K.; Paul, W. Relaxation processes and glass transition of confined polymer melts: A molecular dynamics simulation of 1, 4-polybutadiene between graphite walls. J. Chem. Phys. 2017, 146, 203308. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh Sahraei, A.; Mokarizadeh, A.H.; George, D.; Rodrigue, D.; Baniassadi, M.; Foroutan, M. Insights into interphase thickness characterization for graphene/epoxy nanocomposites: A molecular dynamics simulation. Phys. Chem. Chem. Phys. 2019, 21, 19890–19903. [Google Scholar] [CrossRef]
- Kritikos, G.; Rissanou, A.N.; Harmandaris, V.; Karatasos, K. Bound Layer Polymer Behavior on Graphene and Graphene Oxide Nanosheets. Macromolecules 2020, 53, 6190–6203. [Google Scholar] [CrossRef]
- Behbahani, A.F.; Motlagh, G.H.; Vaez Allaei, S.M.; Harmandaris, V.A. Structure and Conformation of Stereoregular Poly(methyl methacrylate) Chains Adsorbed on Graphene Oxide and Reduced Graphene Oxide via Atomistic Simulations. Macromolecules 2019, 52, 3825–3838. [Google Scholar] [CrossRef]
- Behbahani, A.F.; Harmandaris, V. Gradient of Segmental Dynamics in Stereoregular Poly(Methyl Methacrylate) Melts Confined between Pristine or Oxidized Graphene Sheets. Polymers 2021, 13, 830. [Google Scholar] [CrossRef] [PubMed]
- Rissanou, A.N.; Harmandaris, V. Dynamics of various polymer-graphene interfacial systems through atomistic molecular dynamics simulations. Soft Matter 2014, 10, 2876–2888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rissanou, A.N.; Bačová, P.; Harmandaris, V. Properties of nanographene in polymer nanocomposites through all-atom simulations: Shape fluctuations and rippling. Comput. Mater. Sci. 2020, 172, 109330. [Google Scholar] [CrossRef]
- Pavlov, A.S.; Khalatur, P.G. Filler reinforcement in cross-linked elastomer nanocomposites: Insights from fully atomistic molecular dynamics simulation. Soft Matter 2016, 12, 5402–5419. [Google Scholar] [CrossRef]
- Pavlov, A.S.; Khalatur, P.G. Fully atomistic molecular dynamics simulation of nanosilica-filled crosslinked polybutadiene. Chem. Phys. Lett. 2016, 653, 90–95. [Google Scholar] [CrossRef]
- Kempfer, K.; Devemy, J.; Dequidt, A.; Couty, M.; Malfreyt, P. Atomistic descriptions of the cis-1, 4-polybutadiene/silica interfaces. ACS Appl. Polym. Mater. 2019, 1, 969–981. [Google Scholar] [CrossRef]
- Kempfer, K.; Devémy, J.; Dequidt, A.; Couty, M.; Malfreyt, P. Multi-scale modeling of the polymer–filler interaction. Soft Matter 2020, 16, 1538–1547. [Google Scholar] [CrossRef] [Green Version]
- Maurel, G.; Goujon, F.; Schnell, B.; Malfreyt, P. Multiscale modeling of the polymer-silica surface interaction: From atomistic to mesoscopic simulations. J. Phys. Chem. C 2015, 119, 4817–4826. [Google Scholar] [CrossRef]
- Guseva, D.; Komarov, P.; Lyulin, A.V. Molecular-dynamics simulations of thin polyisoprene films confined between amorphous silica substrates. J. Chem. Phys. 2014, 140, 114903. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Bačová, P.; Behbahani, A.F.; Burkhart, C.; Polińska, P.; Harmandaris, V.; Doxastakis, M. Tailoring Interfacial Properties in Polymer–Silica Nanocomposites via Surface Modification: An Atomistic Simulation Study. ACS Appl. Polym. Mater. 2021, 3, 2576–2587. [Google Scholar] [CrossRef]
- Behbahani, A.F.; Rissanou, A.; Kritikos, G.; Doxastakis, M.; Burkhart, C.; Polińska, P.; Harmandaris, V.A. Conformations and Dynamics of Polymer Chains in Cis and Trans Polybutadiene/Silica Nanocomposites through Atomistic Simulations: From the Unentangled to the Entangled Regime. Macromolecules 2020, 53, 6173–6189. [Google Scholar] [CrossRef]
- Johnston, K.; Harmandaris, V. Hierarchical simulations of hybrid polymer–solid materials. Soft Matter 2013, 9, 6696–6710. [Google Scholar] [CrossRef] [Green Version]
- Fetters, L.J.; Lohse, D.J.; Richter, D.; Witten, T.A.; Zirkel, A. Connection between Polymer Molecular Weight, Density, Chain Dimensions, and Melt Viscoelastic Properties. Macromolecules 1994, 27, 4639–4647. [Google Scholar] [CrossRef]
- Lopes, P.E.M.; Murashov, V.; Tazi, M.; Demchuk, E.; MacKerell, A.D. Development of an Empirical Force Field for Silica. Application to the Quartz-Water Interface. J. Phys. Chem. B 2006, 110, 2782–2792. [Google Scholar] [CrossRef] [Green Version]
- Ndoro, T.V.M.; Voyiatzis, E.; Ghanbari, A.; Theodorou, D.N.; Böhm, M.C.; Müller-Plathe, F. Interface of Grafted and Ungrafted Silica Nanoparticles with a Polystyrene Matrix: Atomistic Molecular Dynamics Simulations. Macromolecules 2011, 44, 2316–2327. [Google Scholar] [CrossRef]
- Pandey, Y.N.; Doxastakis, M. Detailed atomistic Monte Carlo simulations of a polymer melt on a solid surface and around a nanoparticle. J. Chem. Phys. 2012, 136, 094901. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Vollmayr, K.; Kob, W.; Binder, K. Cooling-rate effects in amorphous silica: A computer-simulation study. Phys. Rev. B 1996, 54, 15808–15827. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.; Marcadon, V.; Mélé, P.; Albérola, N.D. Effect of Filler Particle Size on the Properties of Model Nanocomposites. Macromolecules 2008, 41, 1499–1511. [Google Scholar] [CrossRef]
- Jacqmin, D. Calculation of Two-Phase Navier–Stokes Flows Using Phase-Field Modeling. J. Comput. Phys. 1999, 155, 96–127. [Google Scholar] [CrossRef]
- Zografos, K.; Afonso, A.M.; Poole, R.J.; Oliveira, M.S. A viscoelastic two-phase solver using a phase-field approach. J. Non-Newton. Fluid Mech. 2020, 284, 104364. [Google Scholar] [CrossRef]
- Tolan, M. X-ray Scattering from Soft-matter Thin Films: Materials Science and Basic Research; Springer Tracts in Modern Physics; Springer: Berlin/Heidelberg, Germany, 1999. [Google Scholar]
- Bačová, P.; Rissanou, A.N.; Harmandaris, V. Edge-Functionalized Graphene as a Nanofiller: Molecular Dynamics Simulation Study. Macromolecules 2015, 48, 9024–9038. [Google Scholar] [CrossRef]
- Koga, T.; Jiang, N.; Gin, P.; Endoh, M.K.; Narayanan, S.; Lurio, L.B.; Sinha, S.K. Impact of an Irreversibly Adsorbed Layer on Local Viscosity of Nanoconfined Polymer Melts. Phys. Rev. Lett. 2011, 107, 225901. [Google Scholar] [CrossRef] [PubMed]
- Likhtman, A.E.; Sukumaran, S.K.; Ramirez, J. Linear Viscoelasticity from Molecular Dynamics Simulation of Entangled Polymers. Macromolecules 2007, 40, 6748–6757. [Google Scholar] [CrossRef]
- Behbahani, A.F.; Schneider, L.; Rissanou, A.; Chazirakis, A.; Bačová, P.; Jana, P.K.; Li, W.; Doxastakis, M.; Polińska, P.; Burkhart, C.; et al. Dynamics and Rheology of Polymer Melts via Hierarchical Atomistic, Coarse-Grained, and Slip-Spring Simulations. Macromolecules 2021, 54, 2740–2762. [Google Scholar] [CrossRef]
- Ball, R.C.; Blunt, M.; Barford, W. Can surface bound states be induced by interfacial roughness? J. Phys. A Math. Gen. 1989, 22, 2587–2595. [Google Scholar] [CrossRef]
- Baumgärtner, A.; Muthukumar, M. Effects of surface roughness on adsorbed polymers. J. Chem. Phys. 1991, 94, 4062–4070. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Number of Chains | Length of the Chain (Number of Monomers) | (nm) | Film Thickness (nm) |
|---|---|---|---|---|
| cPB30/Sil | 261 | 30 | 1.434 ± 0.005 | 13.77 |
| cPB30 bulk | 100 | 30 | 1.439 ± 0.005 | - |
| cPB100/Sil | 90 | 100 | 2.70 ± 0.10 | 15.5 |
| cPB100 bulk | 100 | 100 | 2.79 ± 0.03 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bačová, P.; Li, W.; Behbahani, A.F.; Burkhart, C.; Polińska, P.; Doxastakis, M.; Harmandaris, V. Coupling between Polymer Conformations and Dynamics Near Amorphous Silica Surfaces: A Direct Insight from Atomistic Simulations. Nanomaterials 2021, 11, 2075. https://doi.org/10.3390/nano11082075
Bačová P, Li W, Behbahani AF, Burkhart C, Polińska P, Doxastakis M, Harmandaris V. Coupling between Polymer Conformations and Dynamics Near Amorphous Silica Surfaces: A Direct Insight from Atomistic Simulations. Nanomaterials. 2021; 11(8):2075. https://doi.org/10.3390/nano11082075
Chicago/Turabian StyleBačová, Petra, Wei Li, Alireza F. Behbahani, Craig Burkhart, Patrycja Polińska, Manolis Doxastakis, and Vagelis Harmandaris. 2021. "Coupling between Polymer Conformations and Dynamics Near Amorphous Silica Surfaces: A Direct Insight from Atomistic Simulations" Nanomaterials 11, no. 8: 2075. https://doi.org/10.3390/nano11082075