
Synthesis of Poly(Malic Acid) Derivatives End-Functionalized with Peptides and Preparation of Biocompatible Nanoparticles to Target Hepatoma Cells

 , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Apparatus
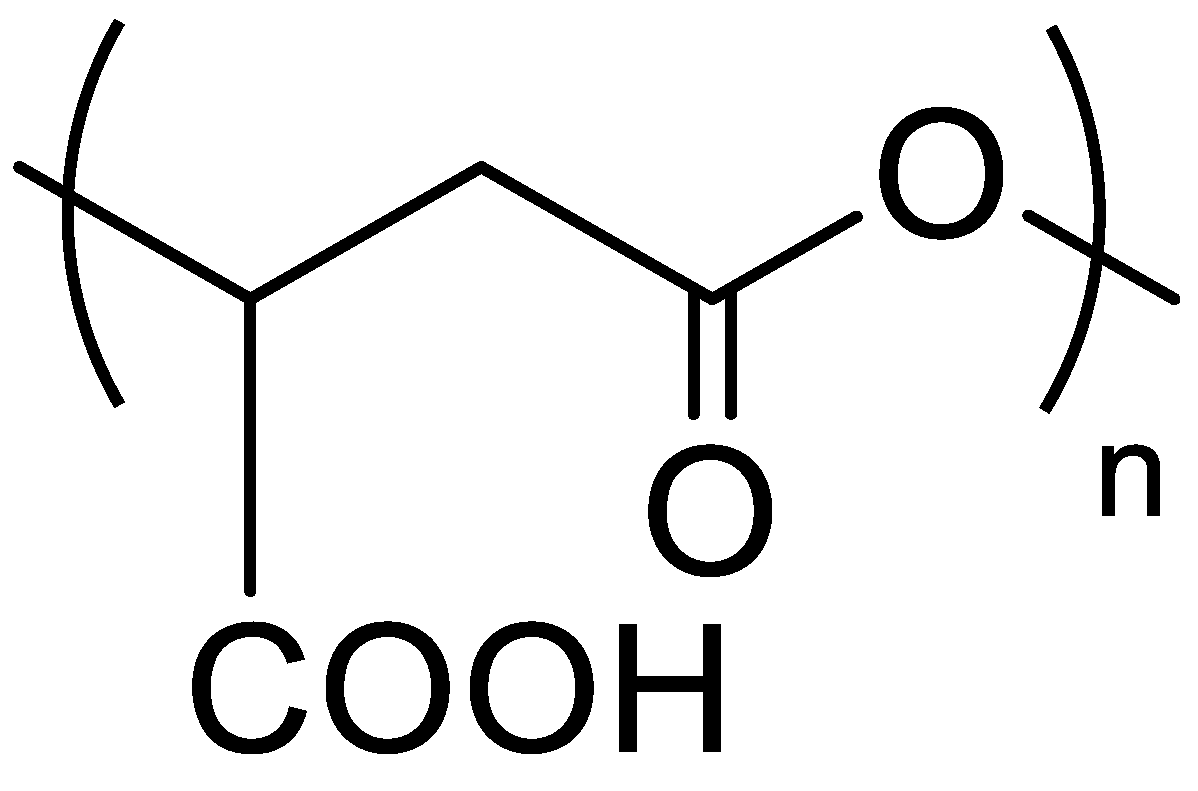
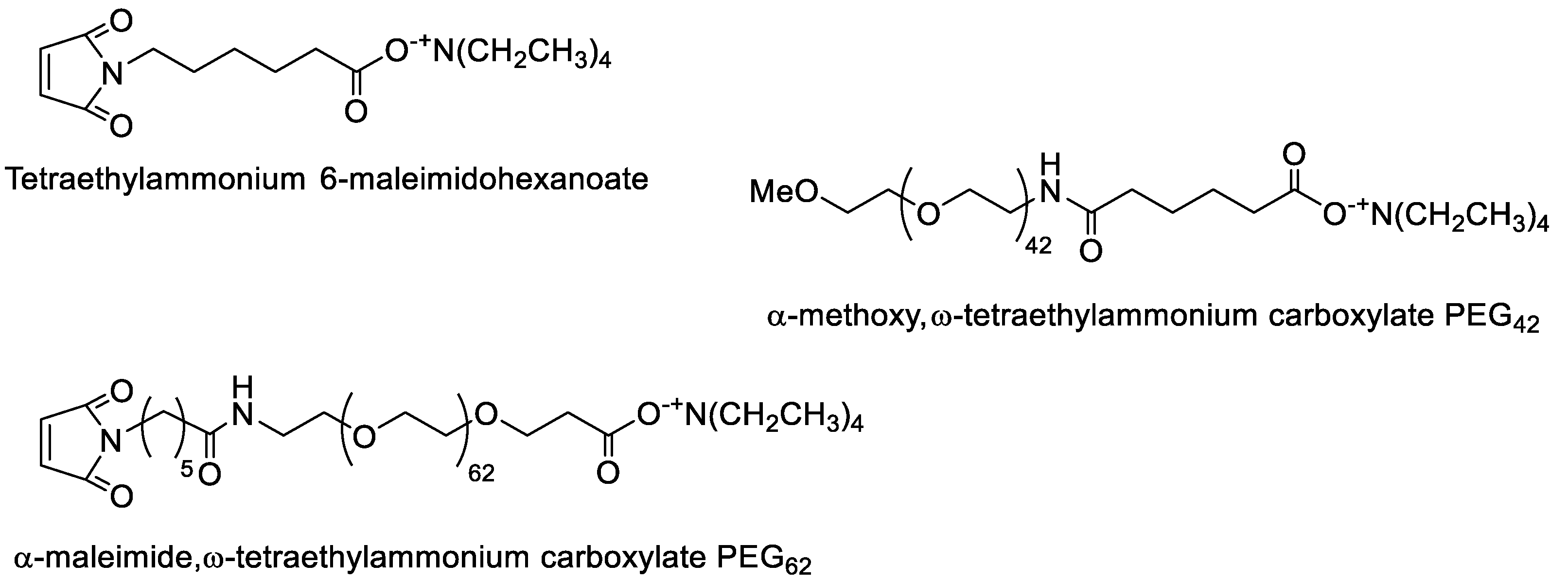
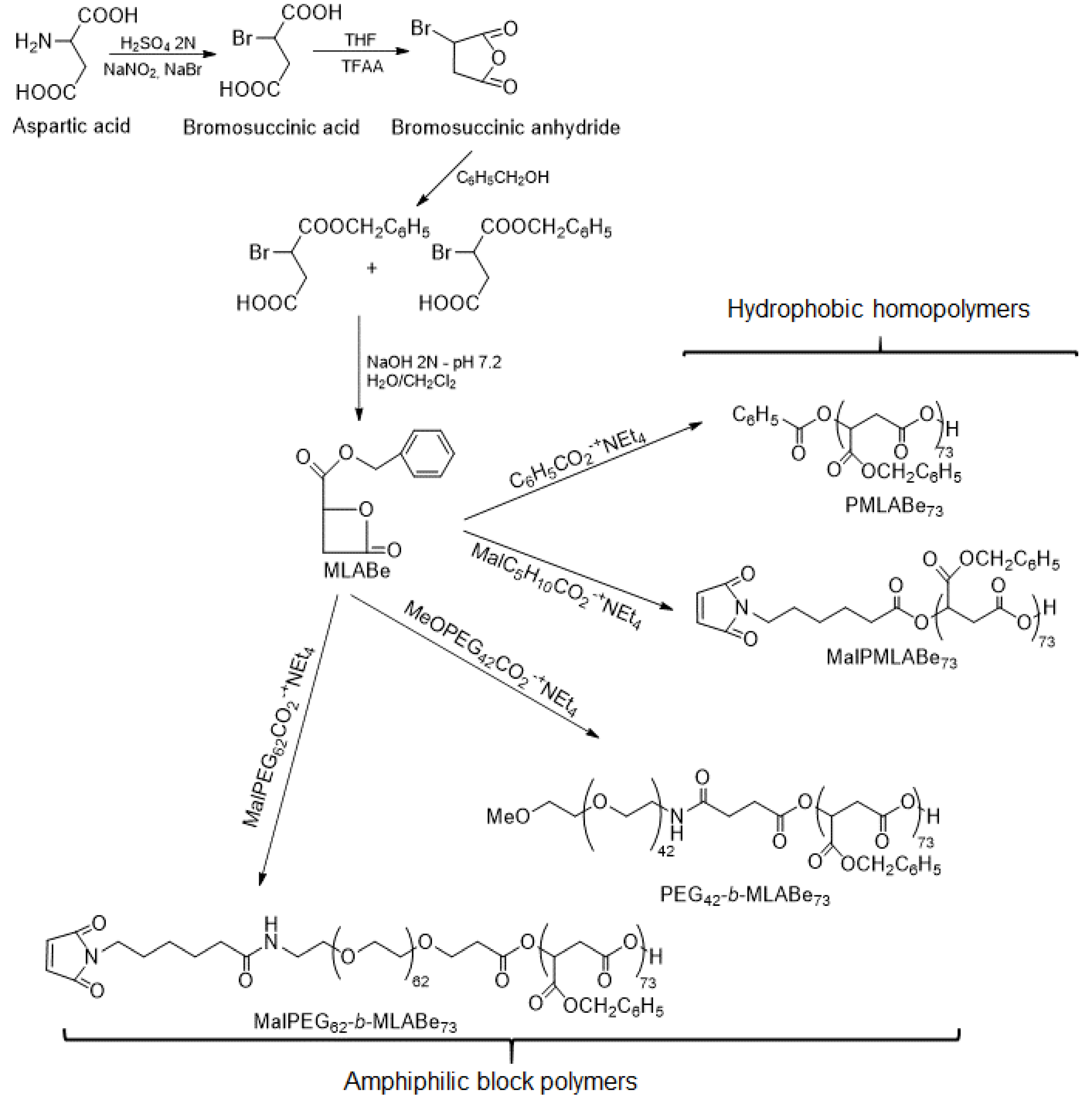
2.2. Synthesis of PMLABe Derivatives
2.3. Formulation of Nanoparticles
2.4. Cell Culture and In Vitro Uptake Assay of NPs
3. Results and Discussion
3.1. Synthesis and Characterization of Peptides End-Functionalized PMLABe Derivatives
3.1.1. Synthesis of PMLABe Derivatives
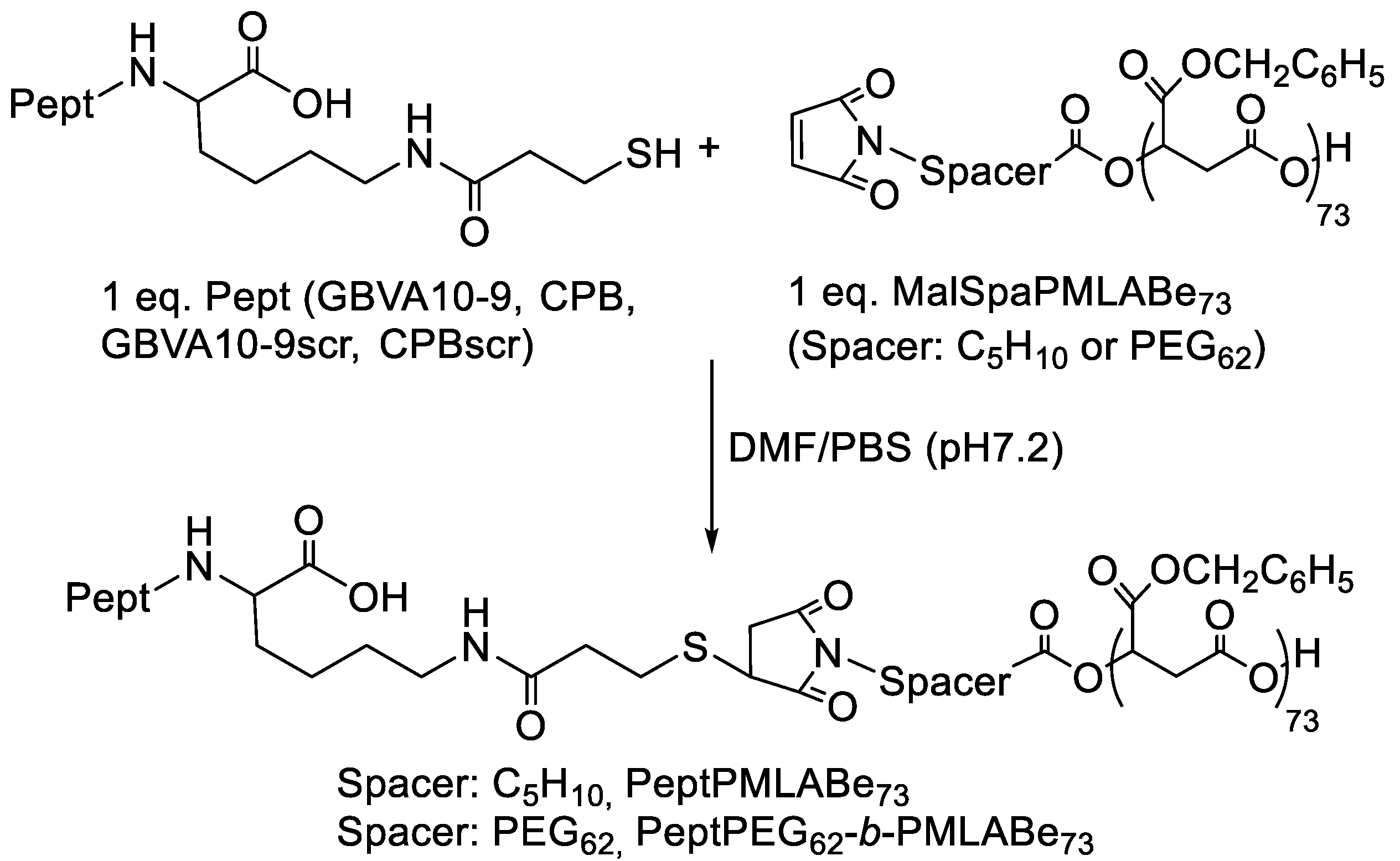
3.1.2. Peptide Grafting
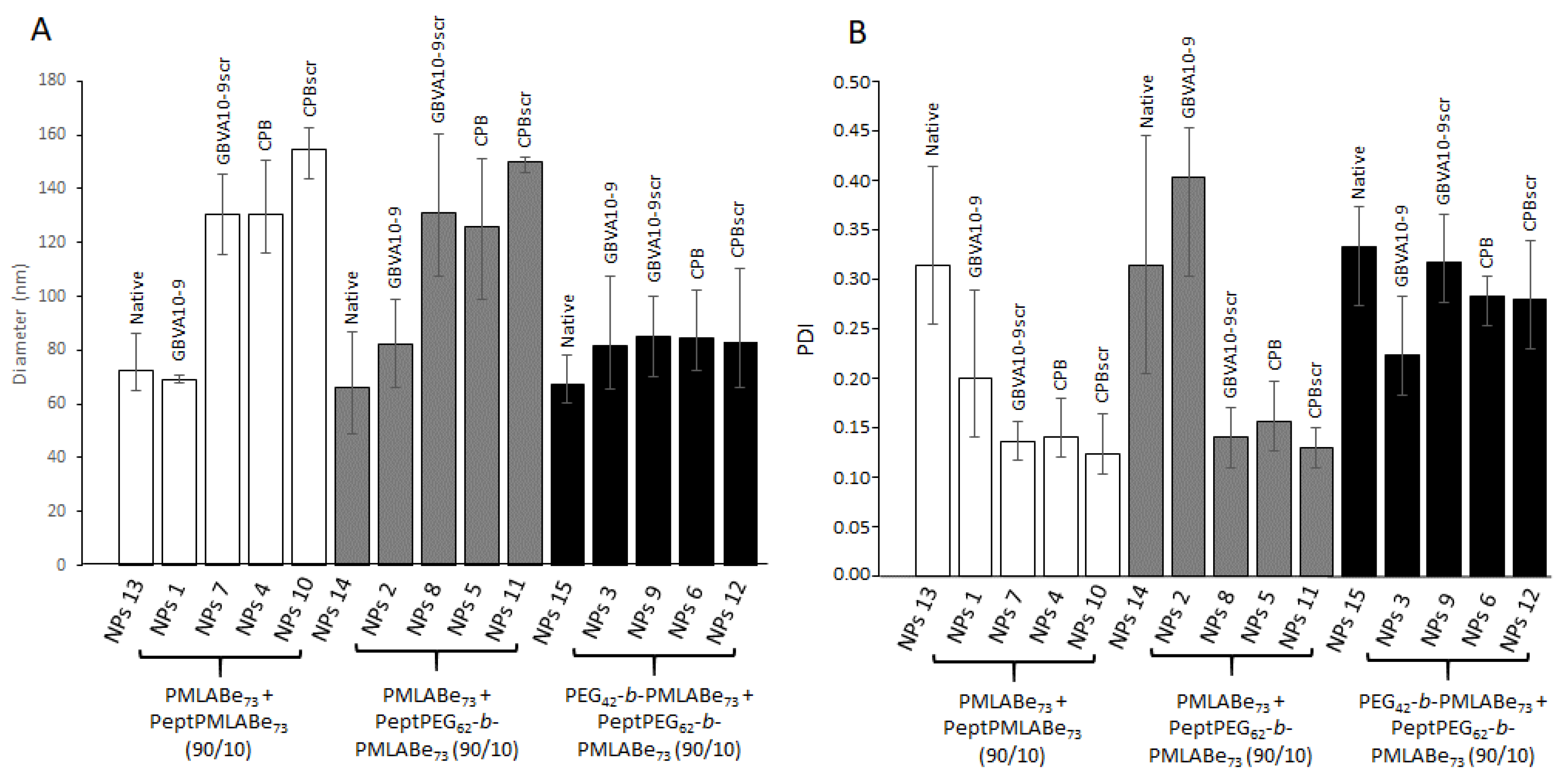
3.2. Preparation and Characterization of Peptides Functionalized Nanoparticles
3.3. In Vitro Internalization Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Winau, F.; Westphal, O.; Winau, R. Paul Ehrlich—In search of the magic bullet. Microbes Infect. 2004, 6, 786–789. [Google Scholar] [CrossRef]
- Toporkiewicz, M.; Meissner, J.; Matusewicz, L.; Czogalla, A.; Sikorski, A.F. Toward a magic or imaginary bullet? Ligands for drug targeting to cancer cells: Principles, hopes, and challenges. Int. J. Nanomed. 2015, 10, 1399–1414. [Google Scholar]
- Panowski, S.; Bhakta, S.; Raab, H.; Polakis, P.; Junutula, J.R. Site-specific antibody drug conjugates for cancer therapy. mAbs 2014, 6, 34–45. [Google Scholar] [CrossRef] [Green Version]
- Maso, K.; Grigolettob, A.; Vicenta, M.J.; Pasut, G. Molecular platforms for targeted drug delivery. Int. Rev. Cell. Mol. Biol. 2019, 346, 1–50. [Google Scholar] [PubMed]
- Russell-Jones, G.; McTavish, K.; McEwan, J.; Rice, J.; Nowotnik, D. Vitamin-mediated as a potential mechanism to increase drug uptake by tumor. J. Inorg. Biochem. 2004, 98, 1625–1633. [Google Scholar] [CrossRef]
- Tripodo, G.; Mandracchia, D.; Collina, S.; Rui, M.; Rossi, D. New Perspectives in Cancer Therapy: The Biotin-Antitumor Molecule Conjugates. Med. Chem. 2014, S1, 1–8. [Google Scholar]
- Saxera, A.; Kori, M.L. Preparation and characterization of paclitaxel-folic acid conjugates to target cancer cells. J. Adv. Sci. Res. 2019, 10, 362–369. [Google Scholar]
- Shukla, R.K.; Tiwari, A. Carbohydrate Molecules: An Expanding Horizon in Drug Delivery and Biomedicine. Crit. Rev. Therap. Drug Carr. Syst. 2011, 28, 255–292. [Google Scholar]
- Cadinoiu, A.N.; Rata, D.M.; Atanase, L.I.; Daraba, O.M.; Gherghel, D.; Vochita, G.; Popa, M. Aptamer-Functionalized Liposomes as a Potential Treatment for Basal Cell Carcinoma. Polymers 2019, 11, 1515. [Google Scholar] [CrossRef] [Green Version]
- Cesarini, V.; Scopa, C.; Silvestris, D.A.; Scafidi, A.; Petrera, V.; Del Baldo, G.; Gallo, A. Aptamer-Based In Vivo Therapeutic Targeting of Glioblastoma. Molecules 2020, 25, 4267. [Google Scholar] [CrossRef]
- Rata, D.M.; Cadinoiu, A.N.; Atanase, L.I.; Popa, M.; Mihai, C.T.; Solcan, C.; Ochiuz, L.; Vochita, G. Topical formulations containing aptamer-functionalized nanocapsules loaded with 5-fluorouracil—An innovative concept for the skin cancer therapy. Mater. Sci. Eng. C Mater. Biol. Appl. 2021, 119, 111591. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C. Peptide Receptors as Molecular Targets for Cancer Diagnosis and Therapy. Endocr. Rev. 2003, 24, 389–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roveri, M.; Bernasconi, M.; Leroux, J.M.; Luciani, P. Peptides for tumor-specific drug targeting: State of the art and beyond. J. Mat. Chem. B 2017, 5, 4348–4364. [Google Scholar] [CrossRef]
- Sun, H.; Dong, Y.; Jan, F.; Zhong, Z. Peptide-decorated polymeric nanomedicines for precision cancer therapy. J. Control. Release Off. J. Control. Release Soc. 2018, 290, 11–27. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Bae, C.; Kim, M.J.; Song, I.H.; Ryu, J.H.; Choi, J.H.; Lee, C.J.; Nam, J.S.; Kim, J.I. A novel nucleolin-binding peptide for Cancer Theranostics. Theranostics 2020, 10, 9153–9171. [Google Scholar] [CrossRef]
- Tang, S.Y.; Wei, H.; Yu, C.U. Peptide-functionalized delivery vehicles for enhanced cancer therapy. Int. J. Pharm. 2021, 593, 120141. [Google Scholar] [CrossRef]
- Goodman, M.; Cai, W.; Smith, N.D. The bold legacy of Emil Fisher. J. Pept. Sci. 2003, 9, 594–603. [Google Scholar] [CrossRef]
- Loffet, A. Peptides as drugs: Is there a market? J. Pept. Sci. 2002, 8, 1–7. [Google Scholar] [CrossRef]
- du Vigneaud, V. Hormones of the Posterior Pituitary Gland: Oxytocin and Vasopressin, The Harvey Lectures 1954–1955; Academic Press: New York, NY, USA, 1956; pp. 1–26. [Google Scholar]
- du Vigneaud, V.; Ressler, C.; Swan, C.J.M.; Roberts, C.W.; Katsoyannis, P.G.; Gordon, S. The synthesis of an octapeptide amide with the hormonal activity of oxytocin. J. Am. Chem. Soc. 1953, 75, 4879–4880. [Google Scholar] [CrossRef]
- Adessi, C.; Soto, C. Converting a peptide into a drug: Strategies to improve stability and bioavailability. Curr. Med. Chem. 2002, 9, 963–978. [Google Scholar] [CrossRef]
- McGregor, D.P. Discovering and improving novel peptide therapeutics. Curr. Opin. Pharmacol. 2008, 8, 616–619. [Google Scholar] [CrossRef]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef]
- Vans, B.J.; King, A.T.; Katsifis, A.; Matesic, L.; Jamie, J.F. Methods to enhance the metabolic stability of peptide-based PET radiopharmaceuticals. Molecules 2020, 25, 2314. [Google Scholar] [CrossRef]
- Stone, T.A.; Deber, C. Therapeutic design of peptide modulators of protein-protein interactions in membranes. Biochem. Biophys. Acta 2017, 1859, 577–585. [Google Scholar] [CrossRef]
- Eychenne, R.; Bouvry, C.; Bourgeois, M.; Loyer, P.; Benoist, E.; Lepareur, N. Overview of Radiolabeled Somatostatin Analogs for Cancer Imaging and Therapy. Molecules 2020, 25, 4012. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.O.; Pozderac, R.V.; Hinkle, G.; Hill, T.; O’Dosisio, T.M.; Schirmer, W.J.; Ellison, E.C.; O’Dorisio, M.S. Somatostatin receptor imaging of neuroendocrine tumors with Indium-111 pentetreotide (OctreoScan). Semin. Nucl. Med. 1995, XXV, 251–261. [Google Scholar] [CrossRef]
- Hennirch, U.; Kopka, K. Lutathera®: The first FDA- and EMA-approved radiopharmaceutical for peptide receptor radionuclide therapy. Pharmaceuticals 2019, 12, 114. [Google Scholar] [CrossRef] [Green Version]
- Faivre, S.; Rimassa, L.; Finn, R.S. Molecular therapies for HCC: Looking outside the box. J. Hepatol. 2020, 72, 342–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghavimi, S.; Apfel, T.; Azimi, H.; Persaud, A.; Pyrsopoulos, N.T. Management and Treatment of Hepatocellular Carcinoma with Immunotherapy: A Review of Current and Future Options. J. Clin. Transl. Hepatol. 2020, 8, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Akateh, C.; Black, S.M.; Conteh, L.; Miller, E.D.; Noonan, A.; Elliott, E.; Pawlik, T.M.; Tsung, A.; Cloyd, J.M. Neoadjuvant and adjuvant treatment strategies for hepatocellular carcinoma. World J. Gastroenterol. 2019, 25, 3704–3721. [Google Scholar] [CrossRef] [PubMed]
- Vène, E.; Jarnouen, K.; Ribault, C.; Vlach, M.; Verres, Y.; Bourgeois, M.; Lepareur, N.; Cammas-Marion, S.; Loyer, P. Hepatotropic circumsporozoite protein of Plasmodium berghei- and GB Virus A-derived peptides strongly enhance cell uptake of bioconjugates and functionalized polymeric nanoparticles by human hepatoma cells: Influence of the amino acid sequence and opsonization. Biomaterials 2021. under review. [Google Scholar]
- Fessi, H.; Puisieux, F.; Devissaguet, J.P.; Ammoury, N.; Benita, S. Nanocapsule formation by interfacial deposition following solvent displacement. Int. J. Pharm. 1989, 55, R1–R4. [Google Scholar] [CrossRef]
- Huang, Z.W.; Laurent, V.; Chetouani, G.; Ljubimova, J.Y.; Holler, E.; Benvegnu, T.; Loyer, P.; Cammas-Marion, S. New functional degradable and bio-compatible nanoparticles based on poly(malic acid) derivatives for site-specific anti-cancer drug delivery. Int. J. Pharm. 2012, 423, 84–92. [Google Scholar] [CrossRef] [Green Version]
- Su, S.; Kang, P.M. Recent advances in nanocarrier-assisted therapeutics delivery systems. Pharmaceutics 2020, 12, 837. [Google Scholar] [CrossRef]
- Siddique, S.; Chow, J.C.L. Application of nanomaterials in biomedical imaging and cancer therapy. Nanomaterials 2020, 10, 1700. [Google Scholar] [CrossRef]
- Indoria, S.; Singh, V.; Hsieh, M.F. Recent advances in theranostic polymeric nanoparticles for cancer treatment: A review. Int. J. Pharm. 2020, 582, 119314. [Google Scholar] [CrossRef]
- Hwang, D.; Ramsey, J.D.; Kabanov, A.V. Polymeric micelles for the delivery of poorly soluble drugs: From nanoformulation to clinical approval. Adv. Drug Del. Rev. 2020, 156, 80–118. [Google Scholar] [CrossRef]
- Cammas-Marion, S.; Loyer, P. Natural and synthetic poly(malic acid)-based derivates: A family of versatile biopolymers for the design of drug nanocarriers. J. Drug Targ. 2014, 22, 556–575. [Google Scholar]
- Loyer, P.; Bedhouche, W.; Huang, Z.W.; Cammas-Marion, S. Degradable and Biocompatible Nanoparticles Decorated with Cyclic RGD Peptide for Efficient Drug Delivery to Hepatoma Cells In Vitro. Int. J. Pharm. 2013, 454, 727–737. [Google Scholar] [CrossRef] [Green Version]
- Vene, E.; Jarnouen, K.; Huang, Z.W.; Bedhouche, W.; Montier, T.; Cammas-Marion, S.; Loyer, P. In vitro Toxicity Evaluation and in vivo Biodistribution of Polymeric Micelles Derived from Poly(ethylene glycol)-b-poly(benzyl malate) Copolymers. Pharm. Nanotech. 2016, 4, 24–37. [Google Scholar] [CrossRef]
- Venkatraj, N.; Nanjan, M.J.; Loyer, P.; Chandrashekar, M.J.N.; Cammas-Marion, S. Poly(malic acid) bearing Doxorubicin and N-Acetyl Galactosamine as a site-specific prodrugs for targeting HepatoCellular Carcinoma. J. Biomat. Sci. Polym. Ed. 2017, 28, 1140–1157. [Google Scholar] [CrossRef] [PubMed]
- Pounder, R.J.; Stanford, M.J.; Books, P.; Richards, S.P.; Dove, A.P. Metal free thiol-maleimide ‘click’ reaction as a mild functionalization strategy for degradable polymers. Chem. Commun. 2008, 5158–5160. [Google Scholar] [CrossRef]
- Liu, Z.; Dong, C.; Wang, X.; Wang, H.; Li, W.; Tan, J.; Chang, J. Self-assembled biodegradable protein-polymer vesicle as a tumor-targeted nanocarrier. ACS Appl. Mater. Interfaces 2014, 6, 2393–2400. [Google Scholar] [CrossRef]
- Petrelli, A.; Borsali, R.; Fort, S.; Halila, S. Oligosaccharide-based block copolymers: Metal-free thiol-maleimide click conjugation and self-assembly into nanoparticles. Carbohydr. Polym. 2015, 124, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Gunnoo, S.B.; Madder, A. Chemical protein modification through cysteine. ChemBioChem 2016, 17, 529–553. [Google Scholar] [CrossRef] [Green Version]
- Nawroth, J.F.; McDaniel, J.R.; Chilkoti, A.; Jordan, R.; Luxenhofer, R. Maleimide-functionalized poly(2-oxazoline)s and their conjugation to elastin-like polypeptides. Macromol. Biosci. 2016, 16, 322–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patil, N.A.; Karas, J.A.; Turner, B.J.; Shabanpoor, F. Thiol-cyanobenzothiazole ligation for the efficient preparation of peptide-PNA conjugates. Bioconjugate Chem. 2019, 30, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Donahue, N.D.; Mao, A.S.; Karim, A.; Komarneni, M.; Thomas, E.E.; Francek, E.R.; Yang, W.; Wilhem, S. Exploring maleimide-based nanoparticle surface engineering to control cellular interactions. ACS Appl. Nano. Mater. 2020, 3, 2421–2429. [Google Scholar] [CrossRef]
- Vauthier, C.; Bouchemal, K. Methods for the Preparation and Manufacture of Polymeric Nanoparticles. Pharm. Res. 2009, 26, 1025–1058. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.R.; Agrawal, Y.K. Nanosuspension: An approach to enhance solubility of drugs. J. Adv. Pharm. Technol. Res. 2011, 2, 81–87. [Google Scholar] [PubMed]
- Pearce, A.K.; O’Reilly, R.K. Insight into active targeting of nanoparticles in drug delivery: Advances in clinical studies and design considerations for cancer nanomedicine. Bioconjugate Chem. 2019, 30, 2300–2311. [Google Scholar] [CrossRef]
- Elsabahy, M.; Wooley, K.L. Design of polymeric nanoparticles for biomedical delivery applications. Chem. Soc. Rev. 2012, 41, 2545–2561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tornesello, A.L.; Buonaguro, L.; Tornesello, M.L.; Buonaguro, F.M. New Insights in the Design of Bioactive Peptides and Chelating Agents for Imaging and Therapy in Oncology. Molecules 2017, 22, 1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benesova, M.; Schafer, M.; Bauder-Wust, U.; Afshar-Oromieh, A.; Kratochwil, C.; Mier, W.; Haberkorn, U.; Kopka, K.; Eder, M. Preclinical Evaluation of a Tailor-Made DOTA-Conjugated PSMA Inhibitor with Optimized Linker Moiety for Imaging and Endoradiotherapy of Prostate Cancer. J. Nucl. Med. 2015, 56, 914–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S. Bifunctional coupling agents for radiolabeling of biomolecules and target-specific delivery of metallic radionuclides. Adv. Drug Deliv. Rev. 2008, 60, 1347–1370. [Google Scholar] [CrossRef] [Green Version]
- Sahay, G.; Alakhova, D.Y.; Kabanov, A.V. Endocytosis of nanomedicines. J. Control. Release 2010, 145, 182–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iversen, T.G.; Skotland, T.; Sandvig, K. Endocytosis and intracellular transport of nanoparticles: Present knowledge and need for future studies. Nano Today 2011, 6, 176–185. [Google Scholar] [CrossRef]
- Ho, L.W.C.; Yung, W.Y.; Sy, K.H.S.; Li, H.Y.; Choi, C.K.K.; Leung, K.C.F.; Lee, T.W.Y.; Choi, C.H.J. Effect of alkylation on the cellular uptake of polyethylene glycol-coated gold nanoparticles. ACS Nano 2017, 11, 6058–6101. [Google Scholar] [CrossRef]
- Jiang, Z.; He, H.; Liu, H.; Thayumanavan, S. Cellular uptake evaluation of amphiphilic polymer assemblies: Importance of interplay between pharmacological and genetic approaches. Biomacromolecules 2019, 20, 4407–4418. [Google Scholar] [CrossRef]
- Vene, E.; Barouti, G.; Jarnouen, K.; Gicquel, T.; Rauch, C.; Ribault, C.; Guillaume, S.M.; Cammas-Marion, S.; Loyer, P. Opsonisation of nanoparticles prepared from poly(β-hydroxybutyrate) and poly(trimethylene carbonate)-b-poly(malic acid) amphiphilic diblock copolymers: Impact on the in vitro cell uptake by primary human macrophages and HepaRG hepatoma cells. Int. J. Pharm. 2016, 513, 438–452. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Code | Composition |
|---|---|---|
| GBVA10-9 | NPs 1 | 10 wt% GBVA10-9PMLABe73 + 90 wt% PMLABe73 |
| NPs 2 | 10 wt% GBVA10-9PEG62-b-PMLABe73 + 90 wt% PMLABe73 | |
| NPs 3 | 10 wt% GBVA10-9PEG62-b-PMLABe73 + 90 wt% PEG42-b-PMLABe73 | |
| CPB | NPs 4 | 10 wt% CPBPMLABe73 + 90 wt% PMLABe73 |
| NPs 5 | 10 wt% CPBPEG62-b-PMLABe73 + 90 wt% PMLABe73 | |
| NPs 6 | 10 wt% CPBPEG62-b-PMLABe73 + 90 wt% PEG42-b-PMLABe73 | |
| GBVA10-9scr | NPs 7 | 10 wt% GBVA10-9scrPMLABe73 + 90 wt% PMLABe73 |
| NPs 8 | 10 wt% GBVA10-9scrPEG62-b-PMLABe73 + 90 wt% PMLABe73 | |
| NPs 9 | 10 wt% GBVA10-9scrPEG62-b-PMLABe73 + 90 wt% PEG42-b-PMLABe73 | |
| CPBscr | NPs 10 | 10 wt% CPBscrPMLABe73 + 90 wt% PMLABe73 |
| NPs 11 | 10 wt% CPBscrPEG62-b-PMLABe73 + 90 wt% PMLABe73 | |
| NPs 12 | 10 wt% CPBscrPEG62-b-PMLABe73 + 90 wt% PEG42-b-PMLABe73 |
| Code | Composition |
|---|---|
| NPs 13 | PMLABe73 |
| NPs 14 | 10 wt% PEG42-b-PMLABe73 + 90 wt% PMLABe73 |
| NPs 15 | PEG42-b-PMLABe73 |
| Mtheo PMLABe a (g/mol) | MNMR PMLABe b (g/mol) | Mw c (g/mol) | Ð c | |
|---|---|---|---|---|
| PMLABe73 | 15,000 | nd d | 7280 | 1.46 |
| MalPMLABe73 | 15,000 | nd d | 5730 | 1.48 |
| PEG42-b-PMLABe73 | 15,000 | 12,980 | 6460 | 1.45 |
| MalPEG62-b-PMLABe73 | 15,000 | 13,800 | 8470 | 1.59 |
| (Co)Polymer | Conjugation Percentage (%) |
|---|---|
| GBVA10-9PMLABe73 | 90 |
| GBVA10-9PEG62-b-PMLABe73 | 81 |
| CPBPMLABe73 | 95 |
| CPBPEG62-b-PMLABe73 | 95 |
| GBVA10-9scrPMLABe73 | 95 |
| GBVA10-9scrPEG62-b-PMLABe73 | 77 |
| CPBscrPMLABe73 | 83 |
| CPBscrPEG62-b-PMLABe73 | 77 |
| Code | Composition (wt%) | Dh a (nm) | PDI a | ζ b (mV) | E.E. c (%) |
|---|---|---|---|---|---|
| NPs 1 | GBVA10-9PMLABe73/PMLABe73 (10/90) | 108 ± 60 | 0.18 | −48 ± 7 | 76 |
| NPs 2 | GBVA10-9PEG62-b-PMLABe73/PMLABe73 (10/90) | 66 ± 30 | 0.21 | −46 ± 8 | 68 |
| NPs 3 | GBVA10-9PEG62-b-PMLABe73/PEG42-b-PMLABe73 (10/90) | 71 ± 50 | 0.28 | −37 ± 5 | 80 |
| NPs 4 | CPBPMLABe73/PMLABe73 (10/90) | 139 ± 60 | 0.12 | −54 ± 10 | 55 |
| NPs 5 | CPBPEG62-b-PMLABe73/PMLABe73 (10/90) | 128 ± 60 | 0.13 | −50 ± 6 | 93 |
| NPs 6 | CPBPEG62-b-PMLABe73/PEG42-b-PMLABe73 (10/90) | 102 ± 60 | 0.30 | −43 ± 6 | 81 |
| NPs 7 | GBVA10-9scrPMLABe73/PMLABe73 (10/90) | 115 ± 50 | 0.16 | −58 ± 8 | 66 |
| NPs 8 | GBVA10-9scrPEG62-b-PMLABe73/PMLABe73 (10/90) | 106 ± 60 | 0.17 | −56 ± 7 | 95 |
| NPs 9 | GBVA10-9scrPEG62-b-PMLABe73/PEG42-b-PMLABe73 (10/90) | 70 ± 50 | 0.28 | −51 ± 8 | 71 |
| NPs 10 | CPBscrPMLABe73/PMLABe73 (10/90) | 163 ± 60 | 0.10 | −53 ± 8 | 76 |
| NPs 11 | CPBscrPEG62-b-PMLABe73/PMLABe73 (10/90) | 146 ± 70 | 0.13 | −52 ± 6 | 95 |
| NPs 12 | CPBscrPEG62-b-PMLABe73/PEG42-b-PMLABe73 (10/90) | 74 ± 50 | 0.23 | −47 ± 10 | 77 |
| NPs 13 | PMLABe73 | 71 ± 40 | 0.21 | −62 ± 8 | 74 |
| NPs 14 | PEG42-b-PMLABe73/PMLABe73 (10/90) | 62 ± 40 | 0.22 | −59 ± 9 | 49 |
| NPs 15 | PEG42-b-PMLABe73 | 64 ± 50 | 0.27 | −55 ± 7 | 68 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brossard, C.; Vlach, M.; Vène, E.; Ribault, C.; Dorcet, V.; Noiret, N.; Loyer, P.; Lepareur, N.; Cammas-Marion, S. Synthesis of Poly(Malic Acid) Derivatives End-Functionalized with Peptides and Preparation of Biocompatible Nanoparticles to Target Hepatoma Cells. Nanomaterials 2021, 11, 958. https://doi.org/10.3390/nano11040958
Brossard C, Vlach M, Vène E, Ribault C, Dorcet V, Noiret N, Loyer P, Lepareur N, Cammas-Marion S. Synthesis of Poly(Malic Acid) Derivatives End-Functionalized with Peptides and Preparation of Biocompatible Nanoparticles to Target Hepatoma Cells. Nanomaterials. 2021; 11(4):958. https://doi.org/10.3390/nano11040958
Chicago/Turabian StyleBrossard, Clarisse, Manuel Vlach, Elise Vène, Catherine Ribault, Vincent Dorcet, Nicolas Noiret, Pascal Loyer, Nicolas Lepareur, and Sandrine Cammas-Marion. 2021. "Synthesis of Poly(Malic Acid) Derivatives End-Functionalized with Peptides and Preparation of Biocompatible Nanoparticles to Target Hepatoma Cells" Nanomaterials 11, no. 4: 958. https://doi.org/10.3390/nano11040958