
Nanoplatforms for Targeted Stimuli-Responsive Drug Delivery: A Review of Platform Materials and Stimuli-Responsive Release and Targeting Mechanisms



Abstract
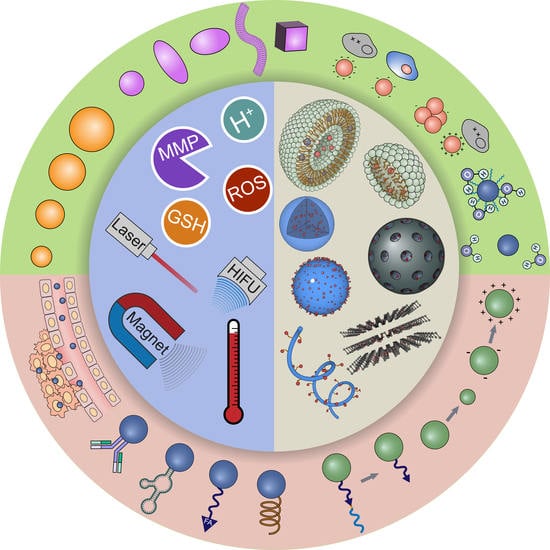
:
1. Introduction
2. Nanocarrier Platforms and Binding Strategies
2.1. Polymeric Micelles
2.2. Liposomes
2.3. Polymeric Nanoparticles
2.4. Porous Inorganic Nanocarriers
2.5. Systems Not Based on Encapsulation
2.6. Targeting and Physiochemical Properties

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Targeting Method | Optimization Method | References |
|---|---|---|
| Passive targeting optimization | Nanocarrier size | [273,274] |
| Protective polymeric layer (density and polymer length) | [275,276] | |
| Novel non-PEG protective layer | [117,277,278,279] | |
| Reduction in interstitial fluid pressure | [280,281] | |
| Degradation of physical barrier in ECM | [281,282,283] | |
| Normalization of tumor ECM | [284] | |
| Active ligand targeting | Optimization of ligand-receptor interaction | [285,286] |
| Ligand density | [287,288,289,290,291,292,293,294,295,296] | |
| Ligand orientation | [297] | |
| Ligand clustering | [298] | |
| Tether of ligands | [287,296,299] |
2.6.1. Passive Targeting
2.6.2. Active Ligand Targeting
2.6.3. Related Physicochemical Properties
3. Stimuli
3.1. pH-Responsive DDSs
| Mechanism | Chemistry | References |
|---|---|---|
| Covalent bond cleavage |  | [445] a,b,c |
 | [41] b,c,** | |
 | [40] b,c,*** [173] b,d [441] a,b,c,*** | |
 | [42] b,c,* [155] b,d,*** [156] b,c,** [443] [449] b,c,** | |
 | [27] b,c,* [202] b,c,*** [272] c,** [325],a,b,c,*** [450] a,b,c,*** | |
 | [26] b,c,** [270] b,c,*** [451] b,c,*** | |
 | [166] b,c,** [203] a,b,c,*** [264] b,c,** [333] b,c,** [452] a,b,c,* | |
 | [119] b,d,* [442] a,b,d [453] b,c,*** | |
 | [241] a,b,c,*** | |
 | [454] b,c,** [455] b,c,*** | |
| Coordination bond cleavage |  | [223] a,b,c,** [224] c [225] b,c,*** [236] a,b [456] a,b,c,*** |
| Inorganic chemical degradation |  | [179] a,b,d,*** [457] c,** [458] b,c,** |
 | [459] b,c,*** [460] c,* | |
 | [461] b,c,*** | |
| Membrane fusion |  | [138] b,c (peptide) |
| [119] b,d,* [123] b,c,*** [442] a,b,d [452] a,b,c,* [462] a,b,c (lipid) | ||
| Protonation |  | [70] b,c,*** [71] b,c,*** [121,122], [181] c,*** [462] a,b,c [463] b,c,* [464] c,** [465] c,* |
 | [51] a,b,c,** [123] b,c,*** [237] a,b,c,** [238] a,b,c,** [462] a,b,c [464] c,** [466] b,c,*** [467] c,* | |
 | [50] a,b [206] b,c,*** | |
 | [468] a,b,c,*** | |
 | [157] a,b,c,*** [166] b,c,** [174] a,b,c,* [175] a,b [469] b,c,** [470] a,b,c,*** | |
 | [65] b,c,** [174] a,b,c,* [175] a,b |
3.1.1. pH-Responsive Micelles
3.1.2. pH-Responsive Liposomes
3.1.3. pH-Responsive Polymeric Nanoparticles
3.1.4. pH-Responsive Porous Inorganic Nanoparticles
3.1.5. pH-Responsive Non-Encapsulated DDDs
3.1.6. pH-Responsive DDSs: Limitations and Remaining Issues
3.2. Redox-Responsive DDSs
| Mechanism | Stimuli-Induced Response | Reference |
|---|---|---|
| Oxidation enhanced polarity |  | [49] b,c,*** [277] a,b,c,** [486] a,b,c,** [493] b,c,*** [494] a,b,c |
 | [484] c [494] a,b,c [495] c | |
 | [485] a,b,c,*** | |
 | [490] c,*** [491] b,c,*** [492] b,c [496] b,c,*** | |
| Redox-induced bond cleavage |    | [32] a,b,c,** [44] b,c,*** [45] b,c,*** [46] c,*** [62] b,c,* [117] a,b,d [163] b,d [165] b,c,*** [166] b,c,** [180] a,b,c,*** [191] a,b,c,*** [197] a,b,c,** [200] b,c,*** [201] b,c,*** [207] b,c,*** [208] b,c,*** [210] c,*** [227] b,c,*** [228] a,b,c,** [229] a,b,c,* [240] b,c,** [241] a,b,c,*** [332] a,b,c,* [326] b,c,*** [327] b,c,*** [328] a,b,c,*** [494] a,b,c [496] b,c,*** [497] b,d [498] b,c,*** [499] c,*** [500] b,c,** (disulfide) |
| [494] a,b,c [501] c [502] b,c,** (diselenide) | ||
| [503] a,b,c,** (ditelluride) | ||
 | [167] b,c,** [494] a,b,c [501] c [502] b,c,** (diselenide) | |
 | [164] a,b,d (thioketal) | |
 | [43] a,b,c,* | |
 | [504] b,d | |
 | [505] a,b,c,*** | |
| Other responses |  | [441] a,b,c,*** |
 | [486] a,b,c,** [487] a,b,c,*** | |
 | [277] a,b,c,** [486] a,b,c,** [487] a,b,c,*** |
3.2.1. Redox-Responsive Micelles
3.2.2. Redox-Responsive Liposomes
3.2.3. Redox-Responsive Polymeric Nanoparticles
3.2.4. Redox-Responsive Porous Inorganic Nanoparticles
3.2.5. Redox-Responsive Non-Encapsulated DDDs
3.2.6. Redox-Responsive DDSs: Limitations and Remaining Issues
3.3. Enzyme-Responsive DDSs
| Mechanism | Enzyme and Substrate | Reference |
|---|---|---|
| Cleavage by hydrolases |  | [124] c |
 | [120] c,*** | |
 | [534] c,** | |
 | [535] b,c,** | |
 | [160] b,c,*** [329] b,c,** [536] b,d [537] a,b,c,** | |
 | [157] a,b,c,*** | |
 | [261] a,b,c [242] c [243] a,b,c [265] a,b,c,*** [330] b,c,*** [538] b,c,*** [539] a,c,*** [540] a,b,c,*** [541] a,b,c | |
 | [525] a,b,c,* | |
 | [19] a,b,c,* [212] b,c,*** [542] a,b [543] a,b,c,* [544] b,c,** | |
 | [205] b,c,*** [530] a,b,c,*** [531] a,b,c,*** | |
| Cleavage by reductases |  | [545] c |
 | [529] a,b,c,*** | |
| Cleavage by other enzymes |  | [190] b |
 | [182] c,*** | |
 | [47] [328] a,b,c,*** [532] b,c,** [533] |
3.3.1. Enzyme-Responsive Micelles
3.3.2. Enzyme-Responsive Liposome
3.3.3. Enzyme-Responsive Polymeric Nanoparticles
3.3.4. Enzyme-Responsive Porous Inorganic Nanoparticles
3.3.5. Enzyme-Responsive Non-Encapsulated DDDs
3.3.6. Enzyme-Responsive DDSs: Limitations and Remaining Issues
3.4. Thermo-Responsive DDSs
| Mechanism | Stimuli-Induced Response | Reference |
|---|---|---|
| LCST Polymers |  | [21] b,c,* [204] c,*** [464] c,*** [468] a,b,c,*** [561] c,** [562] a,b,c,** [563] c,*** [564] c,** [565] c,* (PNIPAM) |
| [52] b,c,** (Pluronics) | ||
| [54] b,c,** [566] a,c [567] a,b,c,*** (OEG-grafted copolymer) | ||
| [568] b,c,* (PNVCL) | ||
| UCST polymers |  | [569] b,c,** [570] a,b,c,* [571] b,c,*** [572] a,b,c,** acrylamide-based copolymer) |
| [279] b,c,** (PNAGA) | ||
| [573] c (ureido-grafted copolymer) | ||
| Thermo-responsive peptides |  | [574] c,* |
 | [575] a,b,c,** | |
 | [18] a,b,c,* [576] c,*** | |
| Lipid-related transitions |  | [105] c,** [279] b,c,** [577] [578] a,b,c,*** [579] [580] c |
 | [105] c,** [106] a,c,* [581] c,* [582] c,** [583] a,c | |
| Gas disruption |  | [116] b,c,** [584] b,c,** [585] a,c,** |
| Thermo-sensitive binding |  | [586] c,*** [587] b,c,*** [588] b,c,** |
 | [311] a,b,c,** [589] a,b,c,*** [590] b,c,*** | |
 | [71] b,c,*** | |
| Thermo-weakened adsorption |  | [238] a,b,c,** [591] b,c,** [237] a,b,c,** [252] a,b,c,** |
3.4.1. Thermo-Responsive Micelles
3.4.2. Thermo-Responsive Liposomes
3.4.3. Thermo-Responsive Polymeric Nanoparticles
3.4.4. Thermo-Responsive Porous Inorganic Nanoparticles
3.4.5. Thermo-Responsive Non-Encapsulated DDDs
3.4.6. Thermo-Responsive DDSs: Limitations and Remaining Issues
3.5. Magneto-Responsive DDSs
| Mechanism | Stimuli-Induced Response | Reference |
|---|---|---|
| Alternating magnetic field |  | [111] c [213] b,c [590] b,c,*** [614] b,c,** [616] a,b,c,*** [617] c,*** [618] c,*** [619] c,*** [620] b,d,** [621] b,c,** [622] b,c,** [623] c,*** [624] b,c,*** [625] b,c,*** [626], [627] b,c,*** [628] b,c,*** [629] b,c,* [630] a,c [631] c [632] c,* (high frequency AMF) |
| [222] c,* (low-frequency AMF) | ||
| Static magnetic field |  | [67] c,*** (static magnetic field stimulated release) |
| [633] b,c [634] a,b,c [635] a,b,c [636] b,d [637] b,d [638] b,d (static magnetic field guidance delivery) |
3.5.1. Magnetic Field-Guided Drug Delivery
3.5.2. Magneto-Responsive Micelles
3.5.3. Magneto-Responsive Liposomes
3.5.4. Magneto-Responsive Polymeric Nanoparticles
3.5.5. Magneto-Responsive Porous Inorganic Nanoparticles
3.5.6. Magneto-Responsive Non-Encapsulated DDDs
3.5.7. Magneto-Responsive DDSs: Limitations and Remaining Issues
3.6. Photo-Responsive DDSs
| Mechanism | Stimuli-Induced Response | Reference |
|---|---|---|
| Photo-induced cleavage |  | [159] c,*** [169] c,*** [170] c,*** [171] c,*** [268] b,d [269] c [246] b,c,d,*** [650] c,*** [652] c,** [653] c,*** |
 | [172] c,*** [654] a,b,c,*** | |
 | [244] a,b,c,*** [655] b,c,*** | |
 | [656] b,c,*** | |
| Photo-isomerization |  | [178] d,* [454] b,c,** [455] b,c,*** [657] a,b,c,** [658] b,c |
 | [221] c,*** [230] b,c,*** [659] c,** [660] c,* [661] d,* [662] b,c,*** [663] b,c | |
| Photo-induced bonding |  | [118] c,*** |
| Photo-induced rearrangement |  | [48] b,c |
| Photo-dynamic effect |  | [167] b,c,** [664] b,c,* [665] b,d [666] a,b,c [667] b,c,*** [668] c [669] c [670] c,*** [671] b,c,*** |
 | [485] a,b,c,*** [672] a,c,*** [673] a,c,*** [674] a,b,c,*** | |
 | [675] a,b,c,** | |
| Photo-thermal effect |  | [21] b,c,* [237] a,b,c,** [252] a,b,c,** [279] b,c,** [456] a,b,c,*** [467] c,* [572] a,b,c,** [578] a,b,c,*** [588] b,c,** [589] a,b,c,*** [676] c,*** [677] b,c,*** [678] a,b,c,** [679] a,b,c,* [680] c,*** [681] a,b,c,*** |
| Photo-acoustic effect |  | [682] c [683] b,c,*** |
3.6.1. Photo-Responsive Micelles
3.6.2. Photo-Responsive Liposomes
3.6.3. Photo-Responsive Polymeric Nanoparticle
3.6.4. Photo-Responsive Porous Inorganic Nanoparticle
3.6.5. Photo-Responsive Non-Encapsulated DDDs
3.6.6. Photo-Responsive DDSs: Limitations and Remaining Issues
3.7. Ultrasound-Responsive DDSs
| Mechanism | Stimuli-Induced Response | Reference |
|---|---|---|
| Cavitation |  | [57] c [58] a,b,c,*** [107] c [108] c,** [720] b,c [721] a,c [722] c [723] c [724] c,*** [725] b,c,** |
 | [55] a,b,c [113] b,c,*** [114] b,d [500] b,c,** [726] b,c | |
 | [109] d,* [458] b,c,** [727] c,** [728] b,c,* | |
| Thermal effect |  | [575] a,b,c,** [729] c,*** [730] a,b,c,*** [731] c,* |
| Acoustic streaming |  | [732] c |
| Bond cleavage |  | [46] c,*** [59] c [61] c [60] c [214] b,c,** [733] c,*** |
 | [734] c,*** |
3.7.1. Ultrasound-Responsive Micelles
3.7.2. Ultrasound-Responsive Liposomes
3.7.3. Other Ultrasound-Responsive Systems
3.7.4. Ultrasound-Responsive DDSs: Limitations and Remaining Issues
3.8. Stimuli-Induced Targeting
| Strategy | Stimuli-Induced Response | References |
|---|---|---|
| Exposure of target ligands |  | [180,208,331,774,775] |
 | [776,777,778,779] | |
| Change in size of nanocarriers |  | [158,441,780,781,782,783,784] |
 | [774,785,786] | |
 | [772,773,787,788] | |
| Change in surface property of nanocarriers |  | [771,784,789,790] |
3.8.1. Stimuli-Induced Targeting Ligand Recovery
3.8.2. Stimuli-Induced Size Change
3.8.3. Stimuli-Induced Surface Property Changes
4. Perspective and Outlook
| Stimuli | Advantages | Limitations |
|---|---|---|
| pH |
|
|
| Redox |
|
|
| Enzyme |
|
|
| Thermo |
|
|
| Magneto |
|
|
| Photo |
|
|
| US |
|
|
| Stimuli | Strategy | Testing | References |
|---|---|---|---|
| pH + gelatinase | pH-responsive polyelectrolyte layer protects gelatin-based nanocarrier from gelatinase present in the liver and is dissociated in the weakly acid condition of tumor ECM. | In vivo: S180 tumor | [157] |
| pH + hyperthermia | LCST polymer-based nanogel maintains structure until significant shrinkage occurring in the presence of both protonation and temperature elevation. | [464] | |
| pH + photoirradiation | pH-responsive gatekeepers and photo-responsive gatekeepers are decorated on the pores of MSN sequentially. Payloads can be released only when both stimuli present. | [791] | |
| GSH + azoreductase | The micelle-based system contains both a disulfide-bridged amphiphilic prodrug and azo bond-bridged amphiphiles. The conjugated drug can be released only when both GSH and azoreductase are present to induce dissociation of the micelle. | in vitro: HepG2 and EC in vivo: H22 tumor on mice | [328] |
| GSH + hyaluronidase | A liposome composed of a disulfide-containing lipid is coated with a HA layer. The payload is released only when the HA is degraded by hyaluronidase, and the liposome is dissociated due to GSH cleavage of the disulfide bond. | in vitro: A549 in vivo: LLC tumor on mice | [117] |
| Protease + photoirradiation | A photo-cleavable group protects crosslinks based on a protease substrate. The polymeric nanoparticles can not be degraded by protease until the system is activated by photoirradiation to remove the protective group. | in vitro: HeLa cell | [159] |
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Scott, A.W.; Tyler, B.M.; Masi, B.C.; Upadhyay, U.M.; Patta, Y.R.; Grossman, R.; Basaldella, L.; Langer, R.S.; Brem, H.; Cima, M.J. Intracranial microcapsule drug delivery device for the treatment of an experimental gliosarcoma model. Biomaterials 2011, 32, 2532–2539. [Google Scholar] [CrossRef]
- Chasin, M.; Hollenbeck, G.; Brem, H.; Grossman, S.; Colvin, M.; Langer, R. Interstitial drug therapy for brain tumors: A case study. Drug Dev. Ind. Pharm. 1990, 16, 2579–2594. [Google Scholar] [CrossRef]
- Langer, R. Polymer implants for drug delivery in the brain. J. Control. Release 1991, 16, 53–59. [Google Scholar] [CrossRef]
- Shapira-Furman, T.; Serra, R.; Gorelick, N.; Doglioli, M.; Tagliaferri, V.; Cecia, A.; Peters, M.; Kumar, A.; Rottenberg, Y.; Langer, R. Biodegradable wafers releasing temozolomide and carmustine for the treatment of brain cancer. J. Control. Release 2019, 295, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Zhou, Z.; Qiu, N.; Shen, Y. Rational design of cancer nanomedicine: Nanoproperty integration and synchronization. Adv. Mater. 2017, 29, 1606628. [Google Scholar] [CrossRef]
- Sun, T.; Zhang, Y.S.; Pang, B.; Hyun, D.C.; Yang, M.; Xia, Y. Engineered nanoparticles for drug delivery in cancer therapy. Angew. Chem. Int. Ed. 2014, 53, 12320–12364. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Xu, L.; Song, G.; Liu, Z. Emerging nanomedicine approaches fighting tumor metastasis: Animal models, metastasis-targeted drug delivery, phototherapy, and immunotherapy. Chem. Soc. Rev. 2016, 45, 6250–6269. [Google Scholar] [CrossRef]
- Chiriva-Internati, M.; Grizzi, F.; Wachtel, M.S.; Jenkins, M.; Ferrari, R.; Cobos, E.; Frezza, E.E. Biological treatment for liver tumor and new potential biomarkers. Dig. Dis. Sci. 2008, 53, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Baxevanis, C.N.; Perez, S.A.; Papamichail, M. Combinatorial treatments including vaccines, chemotherapy and monoclonal antibodies for cancer therapy. Cancer Immunol. Immunother. 2009, 58, 317–324. [Google Scholar] [CrossRef]
- Postow, M.A.; Sidlow, R.; Hellmann, M.D. Immune-related adverse events associated with immune checkpoint blockade. N. Engl. J. Med. 2018, 378, 158–168. [Google Scholar] [CrossRef]
- Johnson, J.A. Biologics and Biosimilars: Background and Key Issues; Congressional Research Service: Washington, DC, USA, 2016. [Google Scholar]
- Clarkson, B.R.; Schön, A.; Freire, E. Conformational stability and self-association equilibrium in biologics. Drug Discov. Today 2016, 21, 342–347. [Google Scholar] [CrossRef] [Green Version]
- Zhong, H.; Chan, G.; Hu, Y.; Hu, H.; Ouyang, D. A comprehensive map of FDA-approved pharmaceutical products. Pharmaceutics 2018, 10, 263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Ahmady, Z.; Kostarelos, K. Chemical components for the design of temperature-responsive vesicles as cancer therapeutics. Chem. Rev. 2016, 116, 3883–3918. [Google Scholar] [CrossRef] [PubMed]
- Cassano, D.; Pocoví-Martínez, S.; Voliani, V. Ultrasmall-in-nano approach: Enabling the translation of metal nanomaterials to clinics. Bioconjugate Chem. 2018, 29, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Li, L.; Xing, J.; Jalde, S.; Li, Y.; Cai, J.; Chen, J.; Liu, P.; Gu, N.; Ji, M. Redox responsive liposomal nanohybrid cerasomes for intracellular drug delivery. Colloids Surf. B Biointerfaces 2016, 148, 518–525. [Google Scholar] [CrossRef]
- Al-Ahmady, Z.S.; Al-Jamal, W.T.; Bossche, J.V.; Bui, T.T.; Drake, A.F.; Mason, A.J.; Kostarelos, K. Lipid–peptide vesicle nanoscale hybrids for triggered drug release by mild hyperthermia in vitro and in vivo. ACS Nano 2012, 6, 9335–9346. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Wang, X.; Yao, X.; Zhang, Y.; Wu, W.; Jiang, X. Hyaluronic acid nanogels with enzyme-sensitive cross-linking group for drug delivery. J. Control. Release 2015, 205, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Z.-Y.; Zhang, R.; Du, F.-S.; Liang, D.-H.; Li, Z.-C. Multi-responsive nanogels containing motifs of ortho ester, oligo(ethylene glycol) and disulfide linkage as carriers of hydrophobic anti-cancer drugs. J. Control. Release 2011, 152, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.; Tsai, W.-B. Fabrication of photothermo-responsive drug-loaded nanogel for synergetic cancer therapy. Polymers 2018, 10, 1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.; Cheng, X.; Li, N.; Wang, H.; Chen, H. Nanocarriers and their loading strategies. Adv. Healthc. Mater. 2019, 8, 1801002. [Google Scholar] [CrossRef]
- Kamaly, N.; Xiao, Z.; Valencia, P.M.; Radovic-Moreno, A.F.; Farokhzad, O.C. Targeted polymeric therapeutic nanoparticles: Design, development and clinical translation. Chem. Soc. Rev. 2012, 41, 2971–3010. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Wang, X.; Zhang, S.; Liu, X. Applications of polymeric micelles with tumor targeted in chemotherapy. J. Nanoparticle Res. 2012, 14, 1254. [Google Scholar] [CrossRef]
- Milovanovic, M.; Arsenijevic, A.; Milovanovic, J.; Kanjevac, T.; Arsenijevic, N. Chapter 14—Nanoparticles in antiviral therapy. In Antimicrobial Nanoarchitectonics; Grumezescu, A.M., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 383–410. [Google Scholar]
- Wang, H.; Wang, Y.; Chen, Y.; Jin, Q.; Ji, J. A biomimic pH-sensitive polymeric prodrug based on polycarbonate for intracellular drug delivery. Polym. Chem. 2014, 5, 854–861. [Google Scholar] [CrossRef]
- Yang, B.; Lv, Y.; Zhu, J.-Y.; Han, Y.-T.; Jia, H.-Z.; Chen, W.-H.; Feng, J.; Zhang, X.-Z.; Zhuo, R.-X. A pH-responsive drug nanovehicle constructed by reversible attachment of cholesterol to PEGylated poly(l-lysine) via catechol–boronic acid ester formation. Acta Biomater. 2014, 10, 3686–3695. [Google Scholar] [CrossRef] [PubMed]
- Van Domeselaar, G.H.; Kwon, G.S.; Andrew, L.C.; Wishart, D.S. Application of solid phase peptide synthesis to engineering PEO–peptide block copolymers for drug delivery. Colloids Surf. B Biointerfaces 2003, 30, 323–334. [Google Scholar] [CrossRef]
- Adams, M.L.; Kwon, G.S. The effects of acyl chain length on the micelle properties of poly(ethylene oxide)-block-poly(N-hexylL-aspartamide)-acyl conjugates. J. Biomater. Sci. Polym. Ed. 2002, 13, 991–1006. [Google Scholar] [CrossRef] [PubMed]
- Shan, X.; Mao, J.; Long, M.; Ahmed, K.S.; Sun, C.; Qiu, L.; Chen, J. Influence of polyethylene glycol molecular weight on the anticancer drug delivery of pH-sensitive polymeric micelle. J. Appl. Polym. Sci. 2019, 136, 47854. [Google Scholar] [CrossRef]
- Fernandez-Villamarin, M.; Sousa-Herves, A.; Porto, S.; Guldris, N.; Martínez-Costas, J.; Riguera, R.; Fernandez-Megia, E. A dendrimer–hydrophobic interaction synergy improves the stability of polyion complex micelles. Polym. Chem. 2017, 8, 2528–2537. [Google Scholar] [CrossRef]
- Li, Y.; Xiao, K.; Luo, J.; Xiao, W.; Lee, J.S.; Gonik, A.M.; Kato, J.; Dong, T.A.; Lam, K.S. Well-defined, reversible disulfide cross-linked micelles for on-demand paclitaxel delivery. Biomaterials 2011, 32, 6633–6645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; van der Meel, R.; Theek, B.; Oude Blenke, E.; Pieters, E.H.E.; Fens, M.H.A.M.; Ehling, J.; Schiffelers, R.M.; Storm, G.; van Nostrum, C.F.; et al. Complete regression of xenograft tumors upon targeted delivery of paclitaxel via Π–Π stacking stabilized polymeric micelles. ACS Nano 2015, 9, 3740–3752. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Jiang, M. Polymeric self-assembly into micelles and hollow spheres with multiscale cavities driven by inclusion complexation. J. Am. Chem. Soc. 2006, 128, 3703–3708. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; van Steenbergen, M.J.; Teunissen, E.A.; Novo, L.s.; Gradmann, S.; Baldus, M.; van Nostrum, C.F.; Hennink, W.E. Π–Π stacking increases the stability and loading capacity of thermosensitive polymeric micelles for chemotherapeutic drugs. Biomacromolecules 2013, 14, 1826–1837. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Tan, J.P.K.; Nederberg, F.; Fukushima, K.; Colson, J.; Yang, C.; Nelson, A.; Yang, Y.-Y.; Hedrick, J.L. Hydrogen bonding-enhanced micelle assemblies for drug delivery. Biomaterials 2010, 31, 8063–8071. [Google Scholar] [CrossRef]
- Dong, X.; Guo, X.; Liu, G.; Fan, A.; Wang, Z.; Zhao, Y. When self-assembly meets topology: An enhanced micelle stability. Chem. Commun. 2017, 53, 3822–3825. [Google Scholar] [CrossRef] [PubMed]
- Bronich, T.K.; Keifer, P.A.; Shlyakhtenko, L.S.; Kabanov, A.V. Polymer micelle with cross-linked ionic core. J. Am. Chem. Soc. 2005, 127, 8236–8237. [Google Scholar] [CrossRef]
- Chen, J.; Ouyang, J.; Kong, J.; Zhong, W.; Xing, M.M. Photo-cross-linked and pH-sensitive biodegradable micelles for doxorubicin delivery. ACS Appl. Mater. Interfaces 2013, 5, 3108–3117. [Google Scholar] [CrossRef]
- Deng, H.; Liu, J.; Zhao, X.; Zhang, Y.; Liu, J.; Xu, S.; Deng, L.; Dong, A.; Zhang, J. PEG-b-PCL copolymer micelles with the ability of pH-controlled negative-to-positive charge reversal for intracellular delivery of doxorubicin. Biomacromolecules 2014, 15, 4281–4292. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Song, L.; Su, Y.; Zhu, L.; Pang, Y.; Qiu, F.; Tong, G.; Yan, D.; Zhu, B.; Zhu, X. Oxime linkage: A robust tool for the design of pH-sensitive polymeric drug carriers. Biomacromolecules 2011, 12, 3460–3468. [Google Scholar] [CrossRef]
- Hu, J.; He, J.; Zhang, M.; Ni, P. Precise modular synthesis and a structure–property study of acid-cleavable star-block copolymers for pH-triggered drug delivery. Polym. Chem. 2015, 6, 1553–1566. [Google Scholar] [CrossRef]
- Su, Z.; Chen, M.; Xiao, Y.; Sun, M.; Zong, L.; Asghar, S.; Dong, M.; Li, H.; Ping, Q.; Zhang, C. ROS-triggered and regenerating anticancer nanosystem: An effective strategy to subdue tumor’s multidrug resistance. J. Control. Release 2014, 196, 370–383. [Google Scholar] [CrossRef]
- Sun, H.; Guo, B.; Cheng, R.; Meng, F.; Liu, H.; Zhong, Z. Biodegradable micelles with sheddable poly(ethylene glycol) shells for triggered intracellular release of doxorubicin. Biomaterials 2009, 30, 6358–6366. [Google Scholar] [CrossRef]
- Zhu, W.; Wang, Y.; Cai, X.; Zha, G.; Luo, Q.; Sun, R.; Li, X.; Shen, Z. Reduction-triggered release of paclitaxel from in situ formed biodegradable core-cross-linked micelles. J. Mater. Chem. B 2015, 3, 3024–3031. [Google Scholar] [CrossRef]
- Tong, R.; Lu, X.; Xia, H. A facile mechanophore functionalization of an amphiphilic block copolymer towards remote ultrasound and redox dual stimulus responsiveness. Chem. Commun. 2014, 50, 3575–3578. [Google Scholar] [CrossRef]
- Rao, J.; Khan, A. Enzyme sensitive synthetic polymer micelles based on the azobenzene motif. J. Am. Chem. Soc. 2013, 135, 14056–14059. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.-Y.; Chen, C.-J.; Li, D.-D.; Wang, S.-S.; Ji, J. Near-infrared light-sensitive micelles for enhanced intracellular drug delivery. J. Mater. Chem. 2012, 22, 16865–16871. [Google Scholar] [CrossRef]
- Xiao, C.; Ding, J.; Ma, L.; Yang, C.; Zhuang, X.; Chen, X. Synthesis of thermal and oxidation dual responsive polymers for reactive oxygen species (ROS)-triggered drug release. Polym. Chem. 2015, 6, 738–747. [Google Scholar] [CrossRef]
- Ling, D.; Park, W.; Park, S.-j.; Lu, Y.; Kim, K.S.; Hackett, M.J.; Kim, B.H.; Yim, H.; Jeon, Y.S.; Na, K.; et al. Multifunctional tumor pH-sensitive self-assembled nanoparticles for bimodal imaging and treatment of resistant heterogeneous tumors. J. Am. Chem. Soc. 2014, 136, 5647–5655. [Google Scholar] [CrossRef] [PubMed]
- Quadir, M.A.; Morton, S.W.; Deng, Z.J.; Shopsowitz, K.E.; Murphy, R.P.; Epps, T.H.; Hammond, P.T. PEG–polypeptide block copolymers as pH-responsive endosome-solubilizing drug nanocarriers. Mol. Pharm. 2014, 11, 2420–2430. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Li, D.; Yang, G.; Shi, C.; Tang, Z.; Wang, J.; Zhou, S. Thermo-triggered drug release from actively targeting polymer micelles. ACS Appl. Mater. Interfaces 2014, 6, 8549–8559. [Google Scholar] [CrossRef]
- Chen, S.; Li, Y.; Guo, C.; Wang, J.; Ma, J.; Liang, X.; Yang, L.-R.; Liu, H.-Z. Temperature-responsive magnetite/PEO−PPO−PEO block copolymer nanoparticles for controlled drug targeting delivery. Langmuir 2007, 23, 12669–12676. [Google Scholar] [CrossRef]
- Cheng, Y.; Hao, J.; Lee, L.A.; Biewer, M.C.; Wang, Q.; Stefan, M.C. Thermally controlled release of anticancer drug from self-assembled γ-substituted amphiphilic poly(ε-caprolactone) micellar nanoparticles. Biomacromolecules 2012, 13, 2163–2173. [Google Scholar] [CrossRef]
- Rapoport, N.; Nam, K.-H.; Gupta, R.; Gao, Z.; Mohan, P.; Payne, A.; Todd, N.; Liu, X.; Kim, T.; Shea, J.; et al. Ultrasound-mediated tumor imaging and nanotherapy using drug loaded, block copolymer stabilized perfluorocarbon nanoemulsions. J. Control. Release 2011, 153, 4–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Z.; Fain, H.D.; Rapoport, N. Ultrasound-enhanced tumor targeting of polymeric micellar drug carriers. Mol. Pharm. 2004, 1, 317–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husseini, G.A.; Myrup, G.D.; Pitt, W.G.; Christensen, D.A.; Rapoport, N.Y. Factors affecting acoustically triggered release of drugs from polymeric micelles. J. Control. Release 2000, 69, 43–52. [Google Scholar] [CrossRef]
- Wu, P.; Jia, Y.; Qu, F.; Sun, Y.; Wang, P.; Zhang, K.; Xu, C.; Liu, Q.; Wang, X. Ultrasound-responsive polymeric micelles for sonoporation-assisted site-specific therapeutic action. ACS Appl. Mater. Interfaces 2017, 9, 25706–25716. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xia, H.; Wang, J.; Li, Y. High intensity focused ultrasound-responsive release behavior of PLA-b-PEG copolymer micelles. J. Control. Release 2009, 139, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Pelletier, M.; Zhang, H.; Xia, H.; Zhao, Y. High-frequency ultrasound-responsive block copolymer micelle. Langmuir 2009, 25, 13201–13205. [Google Scholar] [CrossRef] [PubMed]
- Xuan, J.; Boissière, O.; Zhao, Y.; Yan, B.; Tremblay, L.; Lacelle, S.; Xia, H.; Zhao, Y. Ultrasound-responsive block copolymer micelles based on a new amplification mechanism. Langmuir 2012, 28, 16463–16468. [Google Scholar] [CrossRef]
- Zhou, Z.; Tang, J.; Sun, Q.; Murdoch, W.J.; Shen, Y. A multifunctional PEG–PLL drug conjugate forming redox-responsive nanoparticles for intracellular drug delivery. J. Mater. Chem. B 2015, 3, 7594–7603. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, Z.; Zhang, X. Amphiphilic building blocks for self-assembly: From amphiphiles to supra-amphiphiles. Acc. Chem. Res. 2012, 45, 608–618. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yu, G.; Huang, F. Supramolecular chemotherapy based on host–guest molecular recognition: A novel strategy in the battle against cancer with a bright future. Chem. Soc. Rev. 2017, 46, 7021–7053. [Google Scholar] [CrossRef]
- Xu, X.; Li, Y.; Li, H.; Liu, R.; Sheng, M.; He, B.; Gu, Z. Smart nanovehicles based on pH-triggered disassembly of supramolecular peptide-amphiphiles for efficient intracellular drug delivery. Small 2014, 10, 1133–1140. [Google Scholar] [CrossRef]
- Song, N.; Lou, X.-Y.; Ma, L.; Gao, H.; Yang, Y.-W. Supramolecular nanotheranostics based on pillarenes. Theranostics 2019, 9, 3075–3093. [Google Scholar] [CrossRef]
- Zhou, J.; Chen, M.; Diao, G. Magnetic-responsive supramolecular vesicles from self-organization of amphiphilic pillar[5]arene and application in controlled release. ACS Appl. Mater. Interfaces 2014, 6, 18538–18542. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Zou, X.; Xiong, S.; Li, Y.; Shen, Y.; Hu, X.; Wang, L. Supramolecular prodrug micelles constructed by drug-drug conjugate with water soluble pillar[6]arene for controllable and rapid drug release. Chin. J. Chem. 2015, 33, 329–334. [Google Scholar] [CrossRef]
- Zhou, Y.; Jie, K.; Huang, F. A redox-responsive selenium-containing pillar[5]arene-based macrocyclic amphiphile: Synthesis, controllable self-assembly in water, and application in controlled release. Chem. Commun. 2017, 53, 8364–8367. [Google Scholar] [CrossRef]
- Huang, X.; Du, X. Pillar[6]arene-valved mesoporous silica nanovehicles for multiresponsive controlled release. ACS Appl. Mater. Interfaces 2014, 6, 20430–20436. [Google Scholar] [CrossRef]
- Tan, L.-L.; Song, N.; Zhang, S.X.-A.; Li, H.; Wang, B.; Yang, Y.-W. Ca2+, pH and thermo triple-responsive mechanized Zr-based MOFs for on-command drug release in bone diseases. J. Mater. Chem. B 2016, 4, 135–140. [Google Scholar] [CrossRef]
- Bangham, A.D.; De Gier, J.; Greville, G.D. Osmotic properties and water permeability of phospholipid liquid crystals. Chem. Phys. Lipids 1967, 1, 225–246. [Google Scholar] [CrossRef]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otake, K.; Shimomura, T.; Goto, T.; Imura, T.; Furuya, T.; Yoda, S.; Takebayashi, Y.; Sakai, H.; Abe, M. Preparation of liposomes using an improved supercritical reverse phase evaporation method. Langmuir 2006, 22, 2543–2550. [Google Scholar] [CrossRef]
- Corace, G.; Angeloni, C.; Malaguti, M.; Hrelia, S.; Stein, P.C.; Brandl, M.; Gotti, R.; Luppi, B. Multifunctional liposomes for nasal delivery of the anti-Alzheimer drug tacrine hydrochloride. J. Liposome Res. 2014, 24, 323–335. [Google Scholar] [CrossRef]
- Dos Santos, N.; Cox, K.A.; McKenzie, C.A.; van Baarda, F.; Gallagher, R.C.; Karlsson, G.; Edwards, K.; Mayer, L.D.; Allen, C.; Bally, M.B. pH gradient loading of anthracyclines into cholesterol-free liposomes: Enhancing drug loading rates through use of ethanol. Biochim. Biophys. Acta-Biomembr. 2004, 1661, 47–60. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.; Maitani, Y.; Qi, X.-R.; Takayama, K.; Nagai, T. Remote loading of diclofenac, insulin and fluorescein isothiocyanate labeled insulin into liposomes by pH and acetate gradient methods. Int. J. Pharm. 1999, 179, 85–95. [Google Scholar] [CrossRef]
- Han, H.D.; Lee, A.; Song, C.K.; Hwang, T.; Seong, H.; Lee, C.O.; Shin, B.C. In vivo distribution and antitumor activity of heparin-stabilized doxorubicin-loaded liposomes. Int. J. Pharm. 2006, 313, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Abraham, S.A.; Edwards, K.; Karlsson, G.; Hudon, N.; Mayer, L.D.; Bally, M.B. An evaluation of transmembrane ion gradient-mediated encapsulation of topotecan within liposomes. J. Control. Release 2004, 96, 449–461. [Google Scholar] [CrossRef]
- Sioud, M.; Sørensen, D.R. Cationic liposome-mediated delivery of siRNAs in adult mice. Biochem. Biophys. Res. Commun. 2003, 312, 1220–1225. [Google Scholar] [CrossRef]
- Ruozi, B.; Battini, R.; Montanari, M.; Mucci, A.; Tosi, G.; Forni, F.; Vandelli, M.A. DOTAP/UDCA vesicles: Novel approach in oligonucleotide delivery. Nanomed. Nanotechnol. Biol. Med. 2007, 3, 1–13. [Google Scholar] [CrossRef]
- Engudar, G.; Schaarup-Jensen, H.; Fliedner, F.P.; Hansen, A.E.; Kempen, P.; Jølck, R.I.; Kjaer, A.; Andresen, T.L.; Clausen, M.H.; Jensen, A.I.; et al. Remote loading of liposomes with a 124I-radioiodinated compound and their in vivo evaluation by PET/CT in a murine tumor model. Theranostics 2018, 8, 5828–5841. [Google Scholar] [CrossRef]
- Sur, S.; Fries, A.C.; Kinzler, K.W.; Zhou, S.; Vogelstein, B. Remote loading of preencapsulated drugs into stealth liposomes. Proc. Natl. Acad. Sci. USA 2014, 111, 2283–2288. [Google Scholar] [CrossRef] [Green Version]
- Vemuri, S.; Rhodes, C.T. Development and characterization of a liposome preparation by a pH-gradient method. J. Pharm. Pharmacol. 1994, 46, 778–783. [Google Scholar] [CrossRef]
- Zucker, D.; Marcus, D.; Barenholz, Y.; Goldblum, A. Liposome drugs’ loading efficiency: A working model based on loading conditions and drug’s physicochemical properties. J. Control. Release 2009, 139, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Fritze, A.; Hens, F.; Kimpfler, A.; Schubert, R.; Peschka-Süss, R. Remote loading of doxorubicin into liposomes driven by a transmembrane phosphate gradient. Biochim. Biophys. Acta - Biomembr. 2006, 1758, 1633–1640. [Google Scholar] [CrossRef] [Green Version]
- Zununi Vahed, S.; Salehi, R.; Davaran, S.; Sharifi, S. Liposome-based drug co-delivery systems in cancer cells. Mater. Sci. Eng. C 2017, 71, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Crowe, L.M.; Crowe, J.H.; Rudolph, A.; Womersley, C.; Appel, L. Preservation of freeze-dried liposomes by trehalose. Arch. Biochem. Biophys. 1985, 242, 240–247. [Google Scholar] [CrossRef]
- Pietzyk, B.; Henschke, K. Degradation of phosphatidylcholine in liposomes containing carboplatin in dependence on composition and storage conditions. Int. J. Pharm. 2000, 196, 215–218. [Google Scholar] [CrossRef]
- Rudolphi-Skórska, E.; Filek, M.; Zembala, M. The effects of the structure and composition of the hydrophobic parts of phosphatidylcholine-containing systems on phosphatidylcholine oxidation by ozone. J. Membr. Biol. 2017, 250, 493–505. [Google Scholar] [CrossRef] [Green Version]
- Alhajlan, M.; Alhariri, M.; Omri, A. Efficacy and safety of liposomal clarithromycin and its effect on Pseudomonas aeruginosa virulence factors. Antimicrob. Agents Chemother. 2013, 57, 2694–2704. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zhang, Y.; Tang, C.; Tian, C.; Sun, Q.; Su, Z.; Xue, L.; Yin, Y.; Ju, C.; Zhang, C. Co-delivery of paclitaxel and anti-survivin siRNA via redox-sensitive oligopeptide liposomes for the synergistic treatment of breast cancer and metastasis. Int. J. Pharm. 2017, 529, 102–115. [Google Scholar] [CrossRef]
- El-Nesr, O.H.; Yahiya, S.A.; El-Gazayerly, O.N. Effect of formulation design and freeze-drying on properties of fluconazole multilamellar liposomes. Saudi Pharm. J. 2010, 18, 217–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semple, S.C.; Chonn, A.; Cullis, P.R. Influence of cholesterol on the association of plasma proteins with liposomes. Biochemistry 1996, 35, 2521–2525. [Google Scholar] [CrossRef] [PubMed]
- Gaber, M.H.; Hong, K.; Huang, S.K.; Papahadjopoulos, D. Thermosensitive sterically stabilized liposomes: Formulation and in vitro studies on mechanism of doxorubicin release by bovine serum and human plasma. Pharm. Res. 1995, 12, 1407–1416. [Google Scholar] [CrossRef] [PubMed]
- Riaz, K.M.; Riaz, A.M.; Zhang, X.; Lin, C.; Wong, H.K.; Chen, X.; Zhang, G.; Lu, A.; Yang, Z. Surface functionalization and targeting strategies of liposomes in solid tumor therapy: A review. Int. J. Mol. Sci. 2018, 19, 195. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.H.; Lee, S.G.; Kang, M.J.; Lee, S.; Choi, Y.W. Surface modification of lipid-based nanocarriers for cancer cell-specific drug targeting. J. Pharm. Investig. 2017, 47, 203–227. [Google Scholar] [CrossRef]
- Nag, O.K.; Yadav, V.R.; Hedrick, A.; Awasthi, V. Post-modification of preformed liposomes with novel non-phospholipid poly(ethylene glycol)-conjugated hexadecylcarbamoylmethyl hexadecanoic acid for enhanced circulation persistence in vivo. Int. J. Pharm. 2013, 446, 119–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uster, P.S.; Allen, T.M.; Daniel, B.E.; Mendez, C.J.; Newman, M.S.; Zhu, G.Z. Insertion of poly(ethylene glycol) derivatized phospholipid into pre-formed liposomes results in prolonged in vivo circulation time. FEBS Lett. 1996, 386, 243–246. [Google Scholar] [CrossRef] [Green Version]
- Moreira, J.N.; Ishida, T.; Gaspar, R.; Allen, T.M. Use of the post-insertion technique to insert peptide ligands into pre-formed stealth liposomes with retention of binding activity and cytotoxicity. Pharm. Res. 2002, 19, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Paxton, J.W.; Wu, Z. Enhanced pH-responsiveness, cellular trafficking, cytotoxicity and long-circulation of PEGylated liposomes with post-insertion technique using gemcitabine as a model drug. Pharm. Res. 2015, 32, 2428–2438. [Google Scholar] [CrossRef] [PubMed]
- Nag, O.K.; Awasthi, V. Surface engineering of liposomes for stealth behavior. Pharmaceutics 2013, 5, 542–569. [Google Scholar] [CrossRef] [Green Version]
- Mack, K.; Rüger, R.; Fellermeier, S.; Seifert, O.; Kontermann, R.E. Dual targeting of tumor cells with bispecific single-chain Fv-immunoliposomes. Antibodies 2012, 1, 199–214. [Google Scholar] [CrossRef] [Green Version]
- Ta, T.; Porter, T.M. Thermosensitive liposomes for localized delivery and triggered release of chemotherapy. J. Control. Release 2013, 169, 112–125. [Google Scholar] [CrossRef] [Green Version]
- Mills, J.K.; Needham, D. Lysolipid incorporation in dipalmitoylphosphatidylcholine bilayer membranes enhances the ion permeability and drug release rates at the membrane phase transition. Biochim. Biophys. Acta-Biomembr. 2005, 1716, 77–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Needham, D.; Anyarambhatla, G.; Kong, G.; Dewhirst, M.W. A new temperature-sensitive liposome for use with mild hyperthermia: Characterization and testing in a human tumor xenograft model. Cancer Res. 2000, 60, 1197–1201. [Google Scholar]
- Evjen, T.J.; Nilssen, E.A.; Fowler, R.A.; Røgnvaldsson, S.; Brandl, M.; Fossheim, S.L. Lipid membrane composition influences drug release from dioleoylphosphatidylethanolamine-based liposomes on exposure to ultrasound. Int. J. Pharm. 2011, 406, 114–116. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-Y.; Thomas, J.L. Factors affecting responsivity of unilamellar liposomes to 20 kHz ultrasound. Langmuir 2004, 20, 6100–6106. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, K.D.; Huang, S.-L.; Kim, H.; McPherson, D.D.; MacDonald, R.C. Encapsulation of NF-κB decoy oligonucleotides within echogenic liposomes and ultrasound-triggered release. J. Control. Release 2010, 141, 193–198. [Google Scholar] [CrossRef] [Green Version]
- Nappini, S.; Bombelli, F.B.; Bonini, M.; Nordèn, B.; Baglioni, P. Magnetoliposomes for controlled drug release in the presence of low-frequency magnetic field. Soft Matter 2010, 6, 154–162. [Google Scholar] [CrossRef]
- Amstad, E.; Kohlbrecher, J.; Müller, E.; Schweizer, T.; Textor, M.; Reimhult, E. Triggered release from liposomes through magnetic actuation of iron oxide nanoparticle containing membranes. Nano Lett. 2011, 11, 1664–1670. [Google Scholar] [CrossRef]
- Podaru, G.; Ogden, S.; Baxter, A.; Shrestha, T.; Ren, S.; Thapa, P.; Dani, R.K.; Wang, H.; Basel, M.T.; Prakash, P.; et al. Pulsed magnetic field induced fast drug release from magneto liposomes via ultrasound generation. J. Phys. Chem. B 2014, 118, 11715–11722. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-Y.; Javadi, M.; Belnap, D.M.; Barrow, J.R.; Pitt, W.G. Ultrasound sensitive eLiposomes containing doxorubicin for drug targeting therapy. Nanomedicine 2014, 10, 67–76. [Google Scholar] [CrossRef]
- Javadi, M.; Pitt, W.G.; Tracy, C.M.; Barrow, J.R.; Willardson, B.M.; Hartley, J.M.; Tsosie, N.H. Ultrasonic gene and drug delivery using eLiposomes. J. Control. Release 2013, 167, 92–100. [Google Scholar] [CrossRef]
- Deng, Z.; Yan, F.; Jin, Q.; Li, F.; Wu, J.; Liu, X.; Zheng, H. Reversal of multidrug resistance phenotype in human breast cancer cells using doxorubicin-liposome–microbubble complexes assisted by ultrasound. J. Control. Release 2014, 174, 109–116. [Google Scholar] [CrossRef]
- Chung, M.-F.; Chen, K.-J.; Liang, H.-F.; Liao, Z.-X.; Chia, W.-T.; Xia, Y.; Sung, H.-W. A liposomal system capable of generating CO2 bubbles to induce transient cavitation, lysosomal rupturing, and cell necrosis. Angew. Chem. Int. Ed. 2012, 51, 10089–10093. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Kang, Z.; Xue, L.; Shang, Y.; Su, Z.; Sun, H.; Ping, Q.; Mo, R.; Zhang, C. A collaborative assembly strategy for tumor-targeted siRNA delivery. J. Am. Chem. Soc. 2015, 137, 6000–6010. [Google Scholar] [CrossRef] [PubMed]
- Yavlovich, A.; Singh, A.; Tarasov, S.; Capala, J.; Blumenthal, R.; Puri, A. Design of liposomes containing photopolymerizable phospholipids for triggered release of contents. J. Therm. Anal. Calorim. 2009, 98, 97. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Zhang, H.; Thor, D.; Rahimian, R.; Guo, X. Novel pH-sensitive cationic lipids with linear ortho ester linkers for gene delivery. Eur. J. Med. Chem. 2012, 52, 159–172. [Google Scholar] [CrossRef] [Green Version]
- Thamphiwatana, S.; Gao, W.; Pornpattananangkul, D.; Zhang, Q.; Fu, V.; Li, J.; Li, J.; Obonyo, M.; Zhang, L. Phospholipase A2-responsive antibiotic delivery via nanoparticle-stabilized liposomes for the treatment of bacterial infection. J. Mater. Chem. B 2014, 2, 8201–8207. [Google Scholar] [CrossRef] [Green Version]
- Castelli, D.D.; Boffa, C.; Giustetto, P.; Terreno, E.; Aime, S. Design and testing of paramagnetic liposome-based CEST agents for MRI visualization of payload release on pH-induced and ultrasound stimulation. J. Biol. Inorg. Chem. 2014, 19, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Torres, E.; Mainini, F.; Napolitano, R.; Fedeli, F.; Cavalli, R.; Aime, S.; Terreno, E. Improved paramagnetic liposomes for MRI visualization of pH triggered release. J. Control. Release 2011, 154, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, M.V.; Monteiro, L.O.F.; Carneiro, G.; Malagutti, A.R.; Vilela, J.M.C.; Andrade, M.S.; Oliveira, M.C.; Carvalho-Junior, A.D.; Leite, E.A. Experimental design of a liposomal lipid system: A potential strategy for paclitaxel-based breast cancer treatment. Colloids Surf. B 2015, 136, 553–561. [Google Scholar] [CrossRef]
- Shimanouchi, T.; Kawasaki, H.; Fuse, M.; Umakoshi, H.; Kuboi, R. Membrane fusion mediated by phospholipase C under endosomal pH conditions. Colloids Surf. B 2013, 103, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, S.M.; Akita, H.; Nakamura, T.; Takayama, S.; Futaki, S.; Yamashita, A.; Katoono, R.; Yui, N.; Harashima, H. KALA-modified multi-layered nanoparticles as gene carriers for MHC class-I mediated antigen presentation for a DNA vaccine. Biomaterials 2011, 32, 6342–6350. [Google Scholar] [CrossRef] [PubMed]
- Hersch, N.; Wolters, B.; Ungvari, Z.; Gautam, T.; Deshpande, D.; Merkel, R.; Csiszar, A.; Hoffmann, B.; Csiszár, A. Biotin-conjugated fusogenic liposomes for high-quality cell purification. J. Biomater. Appl. 2015, 30, 846–856. [Google Scholar] [CrossRef]
- Thamphiwatana, S.; Fu, V.; Zhu, J.; Lu, D.; Gao, W.; Zhang, L. Nanoparticle-stabilized liposomes for pH-responsive gastric drug delivery. Langmuir 2013, 29, 12228–12233. [Google Scholar] [CrossRef]
- Kube, S.; Hersch, N.; Naumovska, E.; Gensch, T.; Hendriks, J.; Franzen, A.; Landvogt, L.; Siebrasse, J.-P.; Kubitscheck, U.; Hoffmann, B.; et al. Fusogenic liposomes as nanocarriers for the delivery of intracellular proteins. Langmuir 2017, 33, 1051–1059. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Pang, H.-B.; Kang, J.; Park, J.-H.; Ruoslahti, E.; Sailor, M.J. Immunogene therapy with fusogenic nanoparticles modulates macrophage response to Staphylococcus aureus. Nat. Commun. 2018, 9, 1969. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Szoka, F.C. Mechanism of DNA release from cationic liposome/DNA complexes used in cell transfection. Biochemistry 1996, 35, 5616–5623. [Google Scholar] [CrossRef]
- Zelphati, O.; Szoka, F.C. Mechanism of oligonucleotide release from cationic liposomes. Proc. Natl. Acad. Sci. USA 1996, 93, 11493. [Google Scholar] [CrossRef] [Green Version]
- Felgner, P.L.; Gadek, T.R.; Holm, M.; Roman, R.; Chan, H.W.; Wenz, M.; Northrop, J.P.; Ringold, G.M.; Danielsen, M. Lipofection: A highly efficient, lipid-mediated DNA-transfection procedure. Proc. Natl. Acad. Sci. USA 1987, 84, 7413–7417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuhorn, I.S.; Bakowsky, U.; Polushkin, E.; Visser, W.H.; Stuart, M.C.A.; Engberts, J.B.F.N.; Hoekstra, D. Nonbilayer phase of lipoplex–membrane mixture determines endosomal escape of genetic cargo and transfection efficiency. Mol. Ther. 2005, 11, 801–810. [Google Scholar] [CrossRef] [Green Version]
- Bartomeu Garcia, C.; Shi, D.; Webster, T.J. Tat-functionalized liposomes for the treatment of meningitis: An in vitro study. Int. J. Nanomedicine 2017, 12, 3009–3021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Yang, S.; Petrenko, V.A.; Torchilin, V.P. Cytoplasmic delivery of liposomes into MCF-7 breast cancer cells mediated by cell-specific phage fusion coat protein. Mol. Pharm. 2010, 7, 1149–1158. [Google Scholar] [CrossRef] [Green Version]
- Csiszár, A.; Hersch, N.; Dieluweit, S.; Biehl, R.; Merkel, R.; Hoffmann, B. Novel fusogenic liposomes for fluorescent cell labeling and membrane modification. Bioconjug. Chem. 2010, 21, 537–543. [Google Scholar] [CrossRef]
- Andreev, O.A.; Engelman, D.M.; Reshetnyak, Y.K. Targeting acidic diseased tissue: New technology based on use of the pH (low) insertion peptide (pHLIP). Chim Oggi 2009, 27, 34–37. [Google Scholar]
- Yao, L.; Daniels, J.; Wijesinghe, D.; Andreev, O.A.; Reshetnyak, Y.K. pHLIP®-mediated delivery of PEGylated liposomes to cancer cells. J. Control. Release 2013, 167, 228–237. [Google Scholar] [CrossRef] [Green Version]
- Sosunov, E.A.; Anyukhovsky, E.P.; Sosunov, A.A.; Moshnikova, A.; Wijesinghe, D.; Engelman, D.M.; Reshetnyak, Y.K.; Andreev, O.A. pH (low) insertion peptide (pHLIP) targets ischemic myocardium. Proc. Natl. Acad. Sci. USA 2013, 110, 82–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Nicol, F.; Szoka, F.C. GALA: A designed synthetic pH-responsive amphipathic peptide with applications in drug and gene delivery. Adv. Drug Deliv. Rev. 2004, 56, 967–985. [Google Scholar] [CrossRef] [PubMed]
- Andreev, O.A.; Dupuy, A.D.; Segala, M.; Sandugu, S.; Serra, D.A.; Chichester, C.O.; Engelman, D.M.; Reshetnyak, Y.K. Mechanism and uses of a membrane peptide that targets tumors and other acidic tissues in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 7893–7898. [Google Scholar] [CrossRef] [Green Version]
- El-Say, K.M.; El-Sawy, H.S. Polymeric nanoparticles: Promising platform for drug delivery. Int. J. Pharm. 2017, 528, 675–691. [Google Scholar] [CrossRef]
- Xu, Y.; Du, Y.; Huang, R.; Gao, L. Preparation and modification of N-(2-hydroxyl) propyl-3-trimethyl ammonium chitosan chloride nanoparticle as a protein carrier. Biomaterials 2003, 24, 5015–5022. [Google Scholar] [CrossRef]
- McNeil, S.E. Nanotechnology for the biologist. J. Leukoc. Biol. 2005, 78, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhai, Y.; Wang, J.; Zhai, G. New progress and prospects: The application of nanogel in drug delivery. Mater. Sci. Eng. C 2016, 60, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Akiyoshi, K. Nanogel engineering for new nanobiomaterials: From chaperoning engineering to biomedical applications. Chem. Rec. 2010, 10, 366–376. [Google Scholar] [CrossRef]
- Steinhilber, D.; Witting, M.; Zhang, X.; Staegemann, M.; Paulus, F.; Friess, W.; Küchler, S.; Haag, R. Surfactant free preparation of biodegradable dendritic polyglycerol nanogels by inverse nanoprecipitation for encapsulation and release of pharmaceutical biomacromolecules. J. Control. Release 2013, 169, 289–295. [Google Scholar] [CrossRef]
- Fujioka-Kobayashi, M.; Ota, M.S.; Shimoda, A.; Nakahama, K.-i.; Akiyoshi, K.; Miyamoto, Y.; Iseki, S. Cholesteryl group- and acryloyl group-bearing pullulan nanogel to deliver BMP2 and FGF18 for bone tissue engineering. Biomaterials 2012, 33, 7613–7620. [Google Scholar] [CrossRef]
- Soppimath, K.S.; Aminabhavi, T.M.; Kulkarni, A.R.; Rudzinski, W.E. Biodegradable polymeric nanoparticles as drug delivery devices. J. Control. Release 2001, 70, 1–20. [Google Scholar] [CrossRef]
- Banik, B.L.; Fattahi, P.; Brown, J.L. Polymeric nanoparticles: The future of nanomedicine. Wiley Interdicsip Rev. Nanomed. Nanobiotechnol. 2016, 8, 271–299. [Google Scholar] [CrossRef]
- Wang, Y.; Li, P.; Truong-Dinh Tran, T.; Zhang, J.; Kong, L. Manufacturing techniques and surface engineering of polymer based nanoparticles for targeted drug delivery to cancer. Nanomaterials 2016, 6, 26. [Google Scholar] [CrossRef] [Green Version]
- Masood, F. Polymeric nanoparticles for targeted drug delivery system for cancer therapy. Mater. Sci. Eng. C 2016, 60, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Ge, Y.; Li, L. Advancement of multifunctional hybrid nanogel systems: Construction and application in drug co-delivery and imaging technique. Mater. Sci. Eng. C 2017, 71, 1281–1292. [Google Scholar] [CrossRef]
- Uhrich, K.E.; Cannizzaro, S.M.; Langer, R.S.; Shakesheff, K.M. Polymeric systems for controlled drug release. Chem. Rev. 1999, 99, 3181–3198. [Google Scholar] [CrossRef] [PubMed]
- Paramonov, S.E.; Bachelder, E.M.; Beaudette, T.T.; Standley, S.M.; Lee, C.C.; Dashe, J.; Fréchet, J.M.J. Fully acid-degradable biocompatible polyacetal microparticles for drug delivery. Bioconjug. Chem. 2008, 19, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Hou, Y.; Tu, Z.; Gao, L.; Haag, R. pH-degradable PVA-based nanogels via photo-crosslinking of thermo-preinduced nanoaggregates for controlled drug delivery. J. Control. Release 2017, 259, 160–167. [Google Scholar] [CrossRef]
- Dong, L.; Xia, S.; Wu, K.; Huang, Z.; Chen, H.; Chen, J.; Zhang, J. A pH/enzyme-responsive tumor-specific delivery system for doxorubicin. Biomaterials 2010, 31, 6309–6316. [Google Scholar] [CrossRef]
- Wong, C.; Stylianopoulos, T.; Cui, J.; Martin, J.; Chauhan, V.P.; Jiang, W.; Popović, Z.; Jain, R.K.; Bawendi, M.G.; Fukumura, D. Multistage nanoparticle delivery system for deep penetration into tumor tissue. Proc. Natl. Acad. Sci. USA 2011, 108, 2426–2431. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Yan, M.; Hu, B.; Joo, K.-I.; Biswas, A.; Huang, Y.; Lu, Y.; Wang, P.; Tang, Y. Protein nanocapsule weaved with enzymatically degradable polymeric network. Nano Lett. 2009, 9, 4533–4538. [Google Scholar] [CrossRef] [PubMed]
- Dorresteijn, R.; Billecke, N.; Schwendy, M.; Pütz, S.; Bonn, M.; Parekh, S.H.; Klapper, M.; Müllen, K. Polylactide-block-polypeptide-block-polylactide copolymer nanoparticles with tunable cleavage and controlled drug release. Adv. Funct. Mater. 2014, 24, 4026–4033. [Google Scholar] [CrossRef]
- Aguirre, G.; Ramos, J.; Forcada, J. Synthesis of new enzymatically degradable thermo-responsive nanogels. Soft Matter 2013, 9, 261–270. [Google Scholar] [CrossRef]
- Thornton, P.D.; Mart, R.J.; Ulijn, R.V. Enzyme-responsive polymer hydrogel particles for controlled release. Adv. Mater. 2007, 19, 1252–1256. [Google Scholar] [CrossRef]
- Kozielski, K.L.; Tzeng, S.Y.; Hurtado De Mendoza, B.A.; Green, J.J. Bioreducible cationic polymer-based nanoparticles for efficient and environmentally triggered cytoplasmic siRNA delivery to primary human brain cancer cells. ACS Nano 2014, 8, 3232–3241. [Google Scholar] [CrossRef]
- Wilson, D.S.; Dalmasso, G.; Wang, L.; Sitaraman, S.V.; Merlin, D.; Murthy, N. Orally delivered thioketal nanoparticles loaded with TNF-α–siRNA target inflammation and inhibit gene expression in the intestines. Nat. Mater. 2010, 9, 923–928. [Google Scholar] [CrossRef]
- Li, Y.-L.; Zhu, L.; Liu, Z.; Cheng, R.; Meng, F.; Cui, J.-H.; Ji, S.-J.; Zhong, Z. Reversibly stabilized multifunctional dextran nanoparticles efficiently deliver doxorubicin into the nuclei of cancer cells. Angew. Chem. Int. Ed. 2009, 48, 9914–9918. [Google Scholar] [CrossRef] [PubMed]
- Pei, M.; Jia, X.; Zhao, X.; Li, J.; Liu, P. Alginate-based cancer-associated, stimuli-driven and turn-on theranostic prodrug nanogel for cancer detection and treatment. Carbohydr. Polym. 2018, 183, 131–139. [Google Scholar] [CrossRef]
- Tian, Y.; Zheng, J.; Tang, X.; Ren, Q.; Wang, Y.; Yang, W. Near-infrared light-responsive nanogels with diselenide-cross-linkers for on-demand degradation and triggered drug release. Part. Pert. Syst. Charact. 2015, 32, 547–551. [Google Scholar] [CrossRef]
- Ohya, Y.; Takahashi, A.; Kuzuya, A. Preparation of biodegradable oligo(lactide)s-grafted dextran nanogels for efficient drug delivery by controlling intracellular traffic. Int. J. Mol. Sci. 2018, 19, 1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Gracia Lux, C.; McFearin, C.L.; Joshi-Barr, S.; Sankaranarayanan, J.; Fomina, N.; Almutairi, A. Single UV or near IR triggering event leads to polymer degradation into small molecules. ACS Macro Lett. 2012, 1, 922–926. [Google Scholar] [CrossRef] [Green Version]
- Fomina, N.; McFearin, C.; Sermsakdi, M.; Edigin, O.; Almutairi, A. UV and near-IR triggered release from polymeric nanoparticles. J. Am. Chem. Soc. 2010, 132, 9540–9542. [Google Scholar] [CrossRef] [Green Version]
- Viger, M.L.; Grossman, M.; Fomina, N.; Almutairi, A. Low power upconverted near-IR light for efficient polymeric nanoparticle degradation and cargo release. Adv. Mater. 2013, 25, 3733–3738. [Google Scholar] [CrossRef]
- Huang, Q.; Bao, C.; Ji, W.; Wang, Q.; Zhu, L. Photocleavable coumarin crosslinkers based polystyrene microgels: Phototriggered swelling and release. J. Mater. Chem. 2012, 22, 18275–18282. [Google Scholar] [CrossRef]
- Lee, Y.; Miyata, K.; Oba, M.; Ishii, T.; Fukushima, S.; Han, M.; Koyama, H.; Nishiyama, N.; Kataoka, K. Charge-conversion ternary polyplex with endosome disruption moiety: A technique for efficient and safe gene delivery. Angew. Chem. Int. Ed. 2008, 47, 5163–5166. [Google Scholar] [CrossRef] [PubMed]
- Morton, S.W.; Poon, Z.; Hammond, P.T. The architecture and biological performance of drug-loaded LbL nanoparticles. Biomaterials 2013, 34, 5328–5335. [Google Scholar] [CrossRef] [Green Version]
- Dreaden, E.C.; Morton, S.W.; Shopsowitz, K.E.; Choi, J.-H.; Deng, Z.J.; Cho, N.-J.; Hammond, P.T. Bimodal tumor-targeting from microenvironment responsive hyaluronan layer-by-layer (LbL) nanoparticles. ACS Nano 2014, 8, 8374–8382. [Google Scholar] [CrossRef] [Green Version]
- Oishi, M.; Sumitani, S.; Nagasaki, Y. On−off regulation of 19F magnetic resonance signals based on pH-sensitive PEGylated nanogels for potential tumor-specific smart 19F MRI probes. Bioconjug. Chem. 2007, 18, 1379–1382. [Google Scholar] [CrossRef] [PubMed]
- Kabanov, A.V.; Vinogradov, S.V. Nanogels as pharmaceutical carriers: Finite networks of infinite capabilities. Angew. Chem., Int. Ed. 2009, 48, 5418–5429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirakura, T.; Nomura, Y.; Aoyama, Y.; Akiyoshi, K. Photoresponsive nanogels formed by the self-assembly of spiropyrane-bearing pullulan that act as artificial molecular chaperones. Biomacromolecules 2004, 5, 1804–1809. [Google Scholar] [CrossRef]
- Liu, Q.; Chen, X.; Jia, J.; Zhang, W.; Yang, T.; Wang, L.; Ma, G. pH-responsive poly(d,l-lactic-co-glycolic acid) nanoparticles with rapid antigen release behavior promote immune response. ACS Nano 2015, 9, 4925–4938. [Google Scholar] [CrossRef]
- Wang, X.-G.; Dong, Z.-Y.; Cheng, H.; Wan, S.-S.; Chen, W.-H.; Zou, M.-Z.; Huo, J.-W.; Deng, H.-X.; Zhang, X.-Z. A multifunctional metal–organic framework based tumor targeting drug delivery system for cancer therapy. Nanoscale 2015, 7, 16061–16070. [Google Scholar] [CrossRef]
- Tan, L.-L.; Li, H.; Qiu, Y.-C.; Chen, D.-X.; Wang, X.; Pan, R.-Y.; Wang, Y.; Zhang, S.X.-A.; Wang, B.; Yang, Y.-W. Stimuli-responsive metal–organic frameworks gated by pillar[5]arene supramolecular switches. Chem. Sci. 2015, 6, 1640–1644. [Google Scholar] [CrossRef] [Green Version]
- Park, C.; Kim, H.; Kim, S.; Kim, C. Enzyme responsive nanocontainers with cyclodextrin gatekeepers and synergistic effects in release of guests. J. Am. Chem. Soc. 2009, 131, 16614–16615. [Google Scholar] [CrossRef]
- Angelos, S.; Choi, E.; Vögtle, F.; De Cola, L.; Zink, J.I. Photo-driven expulsion of molecules from mesostructured silica nanoparticles. J. Phys. Chem. C 2007, 111, 6589–6592. [Google Scholar] [CrossRef]
- Kempen, P.J.; Greasley, S.; Parker, K.A.; Campbell, J.L.; Chang, H.-Y.; Jones, J.R.; Sinclair, R.; Gambhir, S.S.; Jokerst, J.V. Theranostic mesoporous silica nanoparticles biodegrade after pro-survival drug delivery and ultrasound/magnetic resonance imaging of stem cells. Theranostics 2015, 5, 631–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, C.; Liu, T.; Li, L.; Liu, H.; Chen, D.; Tang, F. The absorption, distribution, excretion and toxicity of mesoporous silica nanoparticles in mice following different exposure routes. Biomaterials 2013, 34, 2565–2575. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Li, L.; Teng, X.; Huang, X.; Liu, H.; Chen, D.; Ren, J.; He, J.; Tang, F. Single and repeated dose toxicity of mesoporous hollow silica nanoparticles in intravenously exposed mice. Biomaterials 2011, 32, 1657–1668. [Google Scholar] [CrossRef]
- Shen, D.; Yang, J.; Li, X.; Zhou, L.; Zhang, R.; Li, W.; Chen, L.; Wang, R.; Zhang, F.; Zhao, D. Biphase stratification approach to three-dimensional dendritic biodegradable mesoporous silica nanospheres. Nano Lett. 2014, 14, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Steel, A.; Carr, S.W.; Anderson, M.W. 29Si solid-state NMR study of mesoporous M41S materials. Chem. Mater. 1995, 7, 1829–1832. [Google Scholar] [CrossRef]
- Hao, X.; Hu, X.; Zhang, C.; Chen, S.; Li, Z.; Yang, X.; Liu, H.; Jia, G.; Liu, D.; Ge, K.; et al. Hybrid mesoporous silica-based drug carrier nanostructures with improved degradability by hydroxyapatite. ACS Nano 2015, 9, 9614–9625. [Google Scholar] [CrossRef] [PubMed]
- Fatieiev, Y.; Croissant, J.G.; Julfakyan, K.; Deng, L.; Anjum, D.H.; Gurinov, A.; Khashab, N.M. Enzymatically degradable hybrid organic–inorganic bridged silsesquioxane nanoparticles for in vitro imaging. Nanoscale 2015, 7, 15046–15050. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Shen, C.; Zhao, N.; Xu, F.-J. Redox-responsive and drug-embedded silica nanoparticles with unique self-destruction features for efficient gene/drug codelivery. Adv. Funct. Mater. 2017, 27, 1606229. [Google Scholar] [CrossRef]
- Xu, Z.; Zhang, K.; Liu, X.; Zhang, H. A new strategy to prepare glutathione responsive silica nanoparticles. RSC Adv. 2013, 3, 17700–17702. [Google Scholar] [CrossRef]
- Vivero-Escoto, J.L.; Rieter, W.J.; Lau, H.; Huxford-Phillips, R.C.; Lin, W. Biodegradable polysilsesquioxane nanoparticles as efficient contrast agents for magnetic resonance imaging. Small 2013, 9, 3523–3531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Xu, Z.; Chen, Z.; Liu, X.; Hou, C.; Zhang, X.; Zhang, H. Fabrication of single-hole glutathione-responsive degradable hollow silica nanoparticles for drug delivery. ACS Appl. Mater. Interfaces 2014, 6, 12600–12608. [Google Scholar] [CrossRef] [PubMed]
- Maggini, L.; Cabrera, I.; Ruiz-Carretero, A.; Prasetyanto, E.A.; Robinet, E.; De Cola, L. Breakable mesoporous silica nanoparticles for targeted drug delivery. Nanoscale 2016, 8, 7240–7247. [Google Scholar] [CrossRef]
- Croissant, J.; Cattoën, X.; Man, M.W.C.; Gallud, A.; Raehm, L.; Trens, P.; Maynadier, M.; Durand, J.-O. Biodegradable ethylene-bis(propyl)disulfide-based periodic mesoporous organosilica nanorods and nanospheres for efficient in-vitro drug delivery. Adv. Mater. 2014, 26, 6174–6180. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Williams, G.R.; Niu, S.; Gao, F.; Tang, R.; Zhu, L.-M. A multifunctional biodegradable nanocomposite for cancer theranostics. Adv. Sci. 2019, 6, 1802001. [Google Scholar] [CrossRef]
- Huang, P.; Chen, Y.; Lin, H.; Yu, L.; Zhang, L.; Wang, L.; Zhu, Y.; Shi, J. Molecularly organic/inorganic hybrid hollow mesoporous organosilica nanocapsules with tumor-specific biodegradability and enhanced chemotherapeutic functionality. Biomaterials 2017, 125, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Chu, Z.; Yin, C.; Zhang, C.; Lin, G.; Li, Q. Controllable drug release and simultaneously carrier decomposition of SiO2-drug composite nanoparticles. J. Am. Chem. Soc. 2013, 135, 5709–5716. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.-Y.; Trewyn, B.G.; Jeftinija, D.M.; Jeftinija, K.; Xu, S.; Jeftinija, S.; Lin, V.S.Y. A mesoporous silica nanosphere-based carrier system with chemically removable CdS nanoparticle caps for stimuli-responsive controlled release of neurotransmitters and drug molecules. J. Am. Chem. Soc. 2003, 125, 4451–4459. [Google Scholar] [CrossRef] [PubMed]
- Giri, S.; Trewyn, B.G.; Stellmaker, M.P.; Lin, V.S.-Y. Stimuli-responsive controlled-release delivery system based on mesoporous silica nanorods capped with magnetic nanoparticles. Angew. Chem. Int. Ed. 2005, 44, 5038–5044. [Google Scholar] [CrossRef]
- Gan, Q.; Lu, X.; Yuan, Y.; Qian, J.; Zhou, H.; Lu, X.; Shi, J.; Liu, C. A magnetic, reversible pH-responsive nanogated ensemble based on Fe3O4 nanoparticles-capped mesoporous silica. Biomaterials 2011, 32, 1932–1942. [Google Scholar] [CrossRef]
- Zeng, X.; Liu, G.; Tao, W.; Ma, Y.; Zhang, X.; He, F.; Pan, J.; Mei, L.; Pan, G. A drug-self-gated mesoporous antitumor nanoplatform based on pH-sensitive dynamic covalent bond. Adv. Funct. Mater. 2017, 27, 1605985. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhu, S.; Zhang, D. Grafting of thermo-responsive polymer inside mesoporous silica with large pore size using ATRP and investigation of its use in drug release. J. Mater. Chem. 2007, 17, 2428–2433. [Google Scholar] [CrossRef]
- Bernardos, A.; Mondragón, L.; Aznar, E.; Marcos, M.D.; Martínez-Máñez, R.; Sancenón, F.; Soto, J.; Barat, J.M.; Pérez-Payá, E.; Guillem, C.; et al. Enzyme-responsive intracellular controlled release using nanometric silica mesoporous supports capped with “saccharides”. ACS Nano 2010, 4, 6353–6368. [Google Scholar] [CrossRef]
- Meng, H.; Xue, M.; Xia, T.; Zhao, Y.-L.; Tamanoi, F.; Stoddart, J.F.; Zink, J.I.; Nel, A.E. Autonomous in vitro anticancer drug release from mesoporous silica nanoparticles by pH-sensitive nanovalves. J. Am. Chem. Soc. 2010, 132, 12690–12697. [Google Scholar] [CrossRef] [Green Version]
- Guo, R.; Li, L.-L.; Zhao, W.-H.; Chen, Y.-X.; Wang, X.-Z.; Fang, C.-J.; Feng, W.; Zhang, T.-L.; Ma, X.; Lu, M.; et al. The intracellular controlled release from bioresponsive mesoporous silica with folate as both targeting and capping agent. Nanoscale 2012, 4, 3577–3583. [Google Scholar] [CrossRef]
- Zhang, J.; Yuan, Z.-F.; Wang, Y.; Chen, W.-H.; Luo, G.-F.; Cheng, S.-X.; Zhuo, R.-X.; Zhang, X.-Z. Multifunctional envelope-type mesoporous silica nanoparticles for tumor-triggered targeting drug delivery. J. Am. Chem. Soc. 2013, 135, 5068–5073. [Google Scholar] [CrossRef]
- Patel, K.; Angelos, S.; Dichtel, W.R.; Coskun, A.; Yang, Y.-W.; Zink, J.I.; Stoddart, J.F. Enzyme-responsive snap-top covered silica nanocontainers. J. Am. Chem. Soc. 2008, 130, 2382–2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Zhao, X.; Wu, T.; Feng, P. Tunable redox-responsive hybrid nanogated ensembles. J. Am. Chem. Soc. 2008, 130, 14418–14419. [Google Scholar] [CrossRef] [PubMed]
- He, D.; He, X.; Wang, K.; Cao, J.; Zhao, Y. A light-responsive reversible molecule-gated system using thymine-modified mesoporous silica nanoparticles. Langmuir 2012, 28, 4003–4008. [Google Scholar] [CrossRef]
- Chen, Z.; Li, Z.; Lin, Y.; Yin, M.; Ren, J.; Qu, X. Bioresponsive hyaluronic acid-capped mesoporous silica nanoparticles for targeted drug delivery. Chem. Er. J. 2013, 19, 1778–1783. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.H.; Chen, S.Y.; Liu, D.M.; Hsiao, C.S. Core/single-crystal-shell nanospheres for controlled drug release via a magnetically triggered rupturing mechanism. Adv. Mater. 2008, 20, 2690–2695. [Google Scholar] [CrossRef] [PubMed]
- Paris, J.L.; Cabañas, M.V.; Manzano, M.; Vallet-Regí, M. Polymer-grafted mesoporous silica nanoparticles as ultrasound-responsive drug carriers. ACS Nano 2015, 9, 11023–11033. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Jiang, Q.; Feng, D.; Mao, L.; Zhou, H.-C. Size-controlled synthesis of porphyrinic metal–organic framework and functionalization for targeted photodynamic therapy. J. Am. Chem. Soc. 2016, 138, 3518–3525. [Google Scholar] [CrossRef]
- Wu, M.-X.; Yang, Y.-W. Metal–organic framework (MOF)-based drug/cargo delivery and cancer therapy. Adv. Mater. 2017, 29, 1606134. [Google Scholar] [CrossRef]
- Zhuang, J.; Kuo, C.-H.; Chou, L.-Y.; Liu, D.-Y.; Weerapana, E.; Tsung, C.-K. Optimized metal–organic-framework nanospheres for drug delivery: Evaluation of small-molecule encapsulation. ACS Nano 2014, 8, 2812–2819. [Google Scholar] [CrossRef]
- Lin, W.; Hu, Q.; Jiang, K.; Yang, Y.; Yang, Y.; Cui, Y.; Qian, G. A porphyrin-based metal–organic framework as a pH-responsive drug carrier. J. Solid State Chem. 2016, 237, 307–312. [Google Scholar] [CrossRef]
- Ke, F.; Yuan, Y.-P.; Qiu, L.-G.; Shen, Y.-H.; Xie, A.-J.; Zhu, J.-F.; Tian, X.-Y.; Zhang, L.-D. Facile fabrication of magnetic metal–organic framework nanocomposites for potential targeted drug delivery. J. Mater. Chem. 2011, 21, 3843–3848. [Google Scholar] [CrossRef]
- Tan, L.-L.; Li, H.; Zhou, Y.; Zhang, Y.; Feng, X.; Wang, B.; Yang, Y.-W. Zn2+-triggered drug release from biocompatible zirconium MOFs equipped with supramolecular gates. Small 2015, 11, 3807–3813. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Gui, B.; Yuan, D.; Zeller, M.; Wang, C. Mechanized azobenzene-functionalized zirconium metal-organic framework for on-command cargo release. Sci. Adv. 2016, 2, e1600480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, J.; Yang, Y.; Xiao, W.; Zheng, B.; Lv, Y.-B.; Liu, X.-L.; Ding, J. Extremely low frequency alternating magnetic field–triggered and MRI–traced drug delivery by optimized magnetic zeolitic imidazolate framework-90 nanoparticles. Nanoscale 2016, 8, 3259–3263. [Google Scholar] [CrossRef] [PubMed]
- Bian, R.; Wang, T.; Zhang, L.; Li, L.; Wang, C. A combination of tri-modal cancer imaging and in vivo drug delivery by metal–organic framework based composite nanoparticles. Biomater. Sci. 2015, 3, 1270–1278. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, C.; Chakraborty, A. Smart approach for in situ one-step encapsulation and controlled delivery of a chemotherapeutic drug using metal–organic framework–drug composites in aqueous media. ChemPhysChem 2016, 17, 1070–1077. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Zhang, Y.; Liu, L.; Wan, W.; Guo, P.; Nyström, A.M.; Zou, X. One-pot synthesis of metal–organic frameworks with encapsulated target molecules and their applications for controlled drug delivery. J. Am. Chem. Soc. 2016, 138, 962–968. [Google Scholar] [CrossRef]
- Sun, C.-Y.; Qin, C.; Wang, X.-L.; Yang, G.-S.; Shao, K.-Z.; Lan, Y.-Q.; Su, Z.-M.; Huang, P.; Wang, C.-G.; Wang, E.-B. Zeolitic imidazolate framework-8 as efficient pH-sensitive drug delivery vehicle. Dalton Trans. 2012, 41, 6906–6909. [Google Scholar] [CrossRef] [PubMed]
- Gong, M.; Yang, J.; Li, Y.; Gu, J. Glutathione-responsive nanoscale MOFs for effective intracellular delivery of the anticancer drug 6-mercaptopurine. Chem. Commun. 2020, 56, 6448–6451. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Yang, Y.; Han, X.; Liang, C.; Liu, J.; Song, X.; Ge, Z.; Liu, Z. Redox-sensitive nanoscale coordination polymers for drug delivery and cancer theranostics. ACS Appl. Mater. Interfaces 2017, 9, 23555–23563. [Google Scholar] [CrossRef]
- Lei, B.; Wang, M.; Jiang, Z.; Qi, W.; Su, R.; He, Z. Constructing redox-responsive metal–organic framework nanocarriers for anticancer drug delivery. ACS Appl. Mater. Interfaces 2018, 10, 16698–16706. [Google Scholar] [CrossRef]
- Epley, C.C.; Roth, K.L.; Lin, S.; Ahrenholtz, S.R.; Grove, T.Z.; Morris, A.J. Cargo delivery on demand from photodegradable MOF nano-cages. Dalton Trans. 2017, 46, 4917–4922. [Google Scholar] [CrossRef]
- Paul, M.; Dastidar, P. Coordination polymers derived from non-steroidal anti-inflammatory drugs for cell imaging and drug delivery. Chem. Er. J. 2016, 22, 988–998. [Google Scholar] [CrossRef]
- Liu, J.; Chen, Q.; Zhu, W.; Yi, X.; Yang, Y.; Dong, Z.; Liu, Z. Nanoscale-coordination-polymer-shelled manganese dioxide composite nanoparticles: A multistage redox/pH/H2O2-responsive cancer theranostic nanoplatform. Adv. Funct. Mater. 2017, 27, 1605926. [Google Scholar] [CrossRef]
- Rieter, W.J.; Pott, K.M.; Taylor, K.M.L.; Lin, W. Nanoscale coordination polymers for platinum-based anticancer drug delivery. J. Am. Chem. Soc. 2008, 130, 11584–11585. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, J.; Liang, C.; Feng, L.; Fu, T.; Dong, Z.; Chao, Y.; Li, Y.; Lu, G.; Chen, M.; et al. Nanoscale metal–organic particles with rapid clearance for magnetic resonance imaging-guided photothermal therapy. ACS Nano 2016, 10, 2774–2781. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, L.; Li, Z.; Xie, Z. BODIPY-containing nanoscale metal–organic frameworks for photodynamic therapy. Chem. Commun. 2016, 52, 5402–5405. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhou, J.; Chen, R.; Shi, R.; Xia, G.; Zhou, S.; Liu, Z.; Zhang, N.; Wang, H.; Guo, Z.; et al. Magnetically guided delivery of DHA and Fe ions for enhanced cancer therapy based on pH-responsive degradation of DHA-loaded Fe3O4@C@MIL-100(Fe) nanoparticles. Biomaterials 2016, 107, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Ouyang, J.; Liu, H.; Chen, M.; Zeng, K.; Sheng, J.; Liu, Z.; Han, Y.; Wang, L.; Li, J.; et al. Black phosphorus nanosheet-based drug delivery system for synergistic photodynamic/photothermal/chemotherapy of cancer. Adv. Mater. 2017, 29, 1603864. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, J.; Wang, Y.; Wang, C.; Xiao, J.; Zhang, Q.; Cheng, Y. Multi-responsive photothermal-chemotherapy with drug-loaded melanin-like nanoparticles for synergetic tumor ablation. Biomaterials 2016, 81, 114–124. [Google Scholar] [CrossRef]
- Yu, Y.; Chen, C.-K.; Law, W.-C.; Weinheimer, E.; Sengupta, S.; Prasad, P.N.; Cheng, C. Polylactide-graft-doxorubicin nanoparticles with precisely controlled drug loading for pH-triggered drug delivery. Biomacromolecules 2014, 15, 524–532. [Google Scholar] [CrossRef]
- Zhang, P.; Zhang, H.; He, W.; Zhao, D.; Song, A.; Luan, Y. Disulfide-linked amphiphilic polymer-docetaxel conjugates assembled redox-sensitive micelles for efficient antitumor drug delivery. Biomacromolecules 2016, 17, 1621–1632. [Google Scholar] [CrossRef]
- Lv, S.; Tang, Z.; Zhang, D.; Song, W.; Li, M.; Lin, J.; Liu, H.; Chen, X. Well-defined polymer-drug conjugate engineered with redox and pH-sensitive release mechanism for efficient delivery of paclitaxel. J. Control. Release 2014, 194, 220–227. [Google Scholar] [CrossRef]
- Vasey, P.A.; Kaye, S.B.; Morrison, R.; Twelves, C.; Wilson, P.; Duncan, R.; Thomson, A.H.; Murray, L.S.; Hilditch, T.E.; Murray, T.; et al. Phase I clinical and pharmacokinetic study of PK1 [N-(2-Hydroxypropyl)methacrylamide copolymer doxorubicin]: First member of a new class of chemotherapeutic agents—Drug-polymer conjugates. Clin. 1999, 5, 83. [Google Scholar]
- Zhang, C.; Pan, D.; Luo, K.; She, W.; Guo, C.; Yang, Y.; Gu, Z. Peptide dendrimer–doxorubicin conjugate-based nanoparticles as an enzyme-responsive drug delivery system for cancer therapy. Adv. Helathcare Mater. 2014, 3, 1299–1308. [Google Scholar] [CrossRef] [PubMed]
- Nani, R.R.; Gorka, A.P.; Nagaya, T.; Kobayashi, H.; Schnermann, M.J. Near-IR light-mediated cleavage of antibody–drug conjugates using cyanine photocages. Angew. Chem., Int. Ed. 2015, 54, 13635–13638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Ma, X.; Jin, E.; Tang, J.; Sui, M.; Shen, Y.; Van Kirk, E.A.; Murdoch, W.J.; Radosz, M. Linear-dendritic drug conjugates forming long-circulating nanorods for cancer-drug delivery. Biomaterials 2013, 34, 5722–5735. [Google Scholar] [CrossRef] [PubMed]
- Azagarsamy, M.A.; Anseth, K.S. Wavelength-controlled photocleavage for the orthogonal and sequential release of multiple proteins. Angew. Chem. Int. Ed. 2013, 52, 13803–13807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreaden, E.C.; Alkilany, A.M.; Huang, X.; Murphy, C.J.; El-Sayed, M.A. The golden age: Gold nanoparticles for biomedicine. Chem. Soc. Rev. 2012, 41, 2740–2779. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.K.; Ghosh, P.; Pagliuca, C.; Zhu, Z.-J.; Menichetti, S.; Rotello, V.M. Entrapment of hydrophobic drugs in nanoparticle monolayers with efficient release into cancer cells. J. Am. Chem. Soc. 2009, 131, 1360–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alkilany, A.M.; Frey, R.L.; Ferry, J.L.; Murphy, C.J. Gold nanorods as nanoadmicelles: 1-Naphthol partitioning into a nanorod-bound surfactant bilayer. Langmuir 2008, 24, 10235–10239. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, L.; Wang, J.; Jiang, X.; Li, X.; Hu, Z.; Ji, Y.; Wu, X.; Chen, C. Mesoporous silica-coated gold nanorods as a light-mediated multifunctional theranostic platform for cancer treatment. Adv. Mater. 2012, 24, 1418–1423. [Google Scholar] [CrossRef] [PubMed]
- Kaminskas, L.M.; McLeod, V.M.; Kelly, B.D.; Sberna, G.; Boyd, B.J.; Williamson, M.; Owen, D.J.; Porter, C.J.H. A comparison of changes to doxorubicin pharmacokinetics, antitumor activity, and toxicity mediated by PEGylated dendrimer and PEGylated liposome drug delivery systems. Nanomedicine 2012, 8, 103–111. [Google Scholar] [CrossRef]
- Tao, W.; Zhu, X.; Yu, X.; Zeng, X.; Xiao, Q.; Zhang, X.; Ji, X.; Wang, X.; Shi, J.; Zhang, H.; et al. Black phosphorus nanosheets as a robust delivery platform for cancer theranostics. Adv. Mater. 2017, 29, 1603276. [Google Scholar] [CrossRef]
- Ji, X.; Kong, N.; Wang, J.; Li, W.; Xiao, Y.; Gan, S.T.; Zhang, Y.; Li, Y.; Song, X.; Xiong, Q.; et al. A novel top-down synthesis of ultrathin 2D boron nanosheets for multimodal imaging-guided cancer therapy. Adv. Mater. 2018, 30, 1803031. [Google Scholar] [CrossRef]
- Kannaiyan, D.; Imae, T. pH-dependent encapsulation of pyrene in PPI-core:PAMAM-shell dendrimers. Langmuir 2009, 25, 5282–5285. [Google Scholar] [CrossRef]
- Mendes, L.P.; Jiayi, P.; Torchilin, V.P. Dendrimers as nanocarriers for nucleic acid and drug delivery in cancer therapy. Molecules 2017, 22, 1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; D’Emanuele, A.; Attwood, D. Solubility enhancement of paclitaxel using a linear-dendritic block copolymer. Int. J. Pharm. 2013, 452, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Su, M. Polydopamine nanoparticles for combined chemo-and photothermal cancer therapy. Nanomaterials 2017, 7, 160. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; He, X.; Lei, Z.; Liu, L.; Zhang, J.; You, H.; Zhang, H.; Wang, Z. Facile preparation of doxorubicin-loaded upconversion@polydopamine nanoplatforms for simultaneous in vivo multimodality imaging and chemophotothermal synergistic therapy. Adv. Helathcare Mater. 2015, 4, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Pasut, G.; Scaramuzza, S.; Schiavon, O.; Mendichi, R.; Veronese, F.M. PEG-epirubicin conjugates with high drug loading. J. Bioact. Compat. Polym. 2005, 20, 213–230. [Google Scholar] [CrossRef]
- Soto-Castro, D.; Cruz-Morales, J.A.; Apan, M.T.R.; Guadarrama, P. Solubilization and anticancer-activity enhancement of methotrexate by novel dendrimeric nanodevices synthesized in one-step reaction. Bioorg. Chem. 2012, 41–42, 13–21. [Google Scholar] [CrossRef]
- Zhang, C.; Pan, D.; Luo, K.; Li, N.; Guo, C.; Zheng, X.; Gu, Z. Dendrimer–doxorubicin conjugate as enzyme-sensitive and polymeric nanoscale drug delivery vehicle for ovarian cancer therapy. Polym. Chem. 2014, 5, 5227–5235. [Google Scholar] [CrossRef]
- Cui, J.; Yan, Y.; Such, G.K.; Liang, K.; Ochs, C.J.; Postma, A.; Caruso, F. Immobilization and intracellular delivery of an anticancer drug using mussel-inspired polydopamine capsules. Biomacromolecules 2012, 13, 2225–2228. [Google Scholar] [CrossRef]
- Sun, I.-C.; Lee, S.; Koo, H.; Kwon, I.C.; Choi, K.; Ahn, C.-H.; Kim, K. Caspase sensitive gold nanoparticle for apoptosis imaging in live cells. Bioconjug. Chem. 2010, 21, 1939–1942. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Chen, C.-K.; Law, W.-C.; Sun, H.; Prasad, P.N.; Cheng, C. A degradable brush polymer–drug conjugate for pH-responsive release of doxorubicin. Polym. Chem. 2015, 6, 953–961. [Google Scholar] [CrossRef]
- Gianasi, E.; Wasil, M.; Evagorou, E.G.; Keddle, A.; Wilson, G.; Duncan, R. HPMA copolymer platinates as novel antitumour agents: In vitro properties, pharmacokinetics and antitumour activity in vivo. Eur. J. Cancer 1999, 35, 994–1002. [Google Scholar] [CrossRef]
- Guidry, E.N.; Farand, J.; Soheili, A.; Parish, C.A.; Kevin, N.J.; Pipik, B.; Calati, K.B.; Ikemoto, N.; Waldman, J.H.; Latham, A.H.; et al. Improving the in vivo therapeutic index of siRNA polymer conjugates through increasing pH responsiveness. Bioconjug. Chem. 2014, 25, 296–307. [Google Scholar] [CrossRef]
- Lee, M.H.; Sessler, J.L.; Kim, J.S. Disulfide-based multifunctional conjugates for targeted theranostic drug delivery. Acc. Chem. Res. 2015, 48, 2935–2946. [Google Scholar] [CrossRef]
- Griffin, D.R.; Schlosser, J.L.; Lam, S.F.; Nguyen, T.H.; Maynard, H.D.; Kasko, A.M. Synthesis of photodegradable macromers for conjugation and release of bioactive molecules. Biomacromolecules 2013, 14, 1199–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffin, D.R.; Kasko, A.M. Photoselective delivery of model therapeutics from hydrogels. ACS Macro Lett. 2012, 1, 1330–1334. [Google Scholar] [CrossRef]
- Du, J.-Z.; Du, X.-J.; Mao, C.-Q.; Wang, J. Tailor-made dual pH-sensitive polymer–doxorubicin nanoparticles for efficient anticancer drug delivery. J. Am. Chem. Soc. 2011, 133, 17560–17563. [Google Scholar] [CrossRef] [PubMed]
- Ganivada, M.N.; Rao N, V.; Dinda, H.; Kumar, P.; Das Sarma, J.; Shunmugam, R. Biodegradable magnetic nanocarrier for stimuli responsive drug release. Macromolecules 2014, 47, 2703–2711. [Google Scholar] [CrossRef]
- Aguirre-Chagala, Y.E.; Santos, J.L.; Huang, Y.; Herrera-Alonso, M. Phenylboronic acid-installed polycarbonates for the pH-dependent release of diol-containing molecules. ACS Macro Lett. 2014, 3, 1249–1253. [Google Scholar] [CrossRef]
- Hobbs, S.K.; Monsky, W.L.; Yuan, F.; Roberts, W.G.; Griffith, L.; Torchilin, V.P.; Jain, R.K. Regulation of transport pathways in tumor vessels: Role of tumor type and microenvironment. Proc. Natl. Acad. Sci. USA 1998, 95, 4607–4612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, W.; Kim, B.Y.S.; Rutka, J.T.; Chan, W.C.W. Nanoparticle-mediated cellular response is size-dependent. Nat. Nanotechnol. 2008, 3, 145–150. [Google Scholar] [CrossRef]
- Dos Santos, N.; Allen, C.; Doppen, A.-M.; Anantha, M.; Cox, K.A.K.; Gallagher, R.C.; Karlsson, G.; Edwards, K.; Kenner, G.; Samuels, L.; et al. Influence of poly(ethylene glycol) grafting density and polymer length on liposomes: Relating plasma circulation lifetimes to protein binding. Biochim. Biophys. Acta Biomembr. 2007, 1768, 1367–1377. [Google Scholar] [CrossRef] [Green Version]
- Koide, H.; Asai, T.; Hatanaka, K.; Urakami, T.; Ishii, T.; Kenjo, E.; Nishihara, M.; Yokoyama, M.; Ishida, T.; Kiwada, H.; et al. Particle size-dependent triggering of accelerated blood clearance phenomenon. Int. J. Pharm. 2008, 362, 197–200. [Google Scholar] [CrossRef]
- Zhang, D.; Yang, J.; Guan, J.; Yang, B.; Zhang, S.; Sun, M.; Yang, R.; Zhang, T.; Zhang, R.; Kan, Q.; et al. In vivo tailor-made protein corona of a prodrug-based nanoassembly fabricated by redox dual-sensitive paclitaxel prodrug for the superselective treatment of breast cancer. Biomater. Sci. 2018, 6, 2360–2374. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Luo, C.; Yang, J.; Sun, B.; Zhao, D.; Liu, Y.; Wang, Y.; Yang, W.; Kan, Q.; Sun, J.; et al. Precisely albumin-hitchhiking tumor cell-activated reduction/oxidation-responsive docetaxel prodrugs for the hyperselective treatment of breast cancer. J. Control. Release 2018, 285, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Hui, L.; Qin, S.; Yang, L. Upper critical solution temperature polymer, photothermal agent, and erythrocyte membrane coating: An unexplored recipe for making drug carriers with spatiotemporally controlled cargo release. ACS Biomater. Sci. Eng. 2016, 2, 2127–2132. [Google Scholar] [CrossRef]
- Seynhaeve, A.L.B.; Hoving, S.; Schipper, D.; Vermeulen, C.E.; aan de Wiel-Ambagtsheer, G.; van Tiel, S.T.; Eggermont, A.M.M.; ten Hagen, T.L.M. Tumor necrosis factor α mediates homogeneous distribution of liposomes in murine melanoma that contributes to a better tumor response. Cancer Res. 2007, 67, 9455–9462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eikenes, L.; Tari, M.; Tufto, I.; Bruland, Ø.S.; de Lange Davies, C. Hyaluronidase induces a transcapillary pressure gradient and improves the distribution and uptake of liposomal doxorubicin (Caelyx™) in human osteosarcoma xenografts. Br. J. Cancer 2005, 93, 81–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKee, T.D.; Grandi, P.; Mok, W.; Alexandrakis, G.; Insin, N.; Zimmer, J.P.; Bawendi, M.G.; Boucher, Y.; Breakefield, X.O.; Jain, R.K. Degradation of fibrillar collagen in a human melanoma xenograft improves the efficacy of an oncolytic herpes simplex virus vector. Cancer Res. 2006, 66, 2509–2513. [Google Scholar] [CrossRef] [Green Version]
- Kano, M.R.; Bae, Y.; Iwata, C.; Morishita, Y.; Yashiro, M.; Oka, M.; Fujii, T.; Komuro, A.; Kiyono, K.; Kaminishi, M.; et al. Improvement of cancer-targeting therapy, using nanocarriers for intractable solid tumors by inhibition of TGF-β signaling. Proc. Natl. Acad. Sci. USA 2007, 104, 3460–3465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, R.T.; Boucher, Y.; Kozin, S.V.; Winkler, F.; Hicklin, D.J.; Jain, R.K. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res. 2004, 64, 3731–3736. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Z.; Levy-Nissenbaum, E.; Alexis, F.; Lupták, A.; Teply, B.A.; Chan, J.M.; Shi, J.; Digga, E.; Cheng, J.; Langer, R.; et al. Engineering of targeted nanoparticles for cancer therapy using internalizing aptamers isolated by cell-uptake selection. ACS Nano 2012, 6, 696–704. [Google Scholar] [CrossRef] [Green Version]
- Wan, X.-M.; Chen, Y.-P.; Xu, W.-R.; Yang, W.-J.; Wen, L.-P. Identification of nose-to-brain homing peptide through phage display. Peptides 2009, 30, 343–350. [Google Scholar] [CrossRef]
- Wang, S.; Dormidontova, E.E. Nanoparticle design optimization for enhanced targeting: Monte Carlo simulations. Biomacromolecules 2010, 11, 1785–1795. [Google Scholar] [CrossRef] [Green Version]
- Gu, F.; Zhang, L.; Teply, B.A.; Mann, N.; Wang, A.; Radovic-Moreno, A.F.; Langer, R.; Farokhzad, O.C. Precise engineering of targeted nanoparticles by using self-assembled biointegrated block copolymers. Proc. Natl. Acad. Sci. USA 2008, 105, 2586–2591. [Google Scholar] [CrossRef] [Green Version]
- Hristov, D.R.; Rocks, L.; Kelly, P.M.; Thomas, S.S.; Pitek, A.S.; Verderio, P.; Mahon, E.; Dawson, K.A. Tuning of nanoparticle biological functionality through controlled surface chemistry and characterisation at the bioconjugated nanoparticle surface. Sci. Rep. 2015, 5, 17040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Min, J.; Eghtesadi, S.A.; Kane, R.S.; Chilkoti, A. Quantitative study of the interaction of multivalent ligand-modified nanoparticles with breast cancer cells with tunable receptor density. ACS Nano 2020, 14, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Elias, D.R.; Poloukhtine, A.; Popik, V.; Tsourkas, A. Effect of ligand density, receptor density, and nanoparticle size on cell targeting. Nanomedicine 2013, 9, 194–201. [Google Scholar] [CrossRef] [Green Version]
- Hakem, I.F.; Leech, A.M.; Johnson, J.D.; Donahue, S.J.; Walker, J.P.; Bockstaller, M.R. Understanding ligand distributions in modified particle and particlelike systems. J. Am. Chem. Soc. 2010, 132, 16593–16598. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Tian, S.; Petros, R.A.; Napier, M.E.; DeSimone, J.M. The complex role of multivalency in nanoparticles targeting the transferrin receptor for cancer therapies. J. Am. Chem. Soc. 2010, 132, 11306–11313. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Weller, G.E.R.; Zern, B.; Ayyaswamy, P.S.; Eckmann, D.M.; Muzykantov, V.R.; Radhakrishnan, R. Computational model for nanocarrier binding to endothelium validated using in vivo, in vitro, and atomic force microscopy experiments. Proc. Natl. Acad. Sci. USA 2010, 107, 16530–16535. [Google Scholar] [CrossRef] [Green Version]
- Hlavacek, W.S.; Posner, R.G.; Perelson, A.S. Steric effects on multivalent ligand-receptor binding: Exclusion of ligand sites by bound cell surface receptors. Biophys. J. 1999, 76, 3031–3043. [Google Scholar] [CrossRef] [Green Version]
- Abstiens, K.; Gregoritza, M.; Goepferich, A.M. Ligand density and linker length are critical factors for multivalent nanoparticle–receptor interactions. ACS Appl. Mater. Interfaces 2019, 11, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Bagalkot, V.; Gao, X. siRNA-aptamer chimeras on nanoparticles: Preserving targeting functionality for effective gene silencing. ACS Nano 2011, 5, 8131–8139. [Google Scholar] [CrossRef] [PubMed]
- Poon, Z.; Chen, S.; Engler, A.C.; Lee, H.-I.; Atas, E.; von Maltzahn, G.; Bhatia, S.N.; Hammond, P.T. Ligand-clustered “patchy” nanoparticles for modulated cellular uptake and in vivo tumor targeting. Angew. Chem. Int. Ed. 2010, 49, 7266–7270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefanick, J.F.; Ashley, J.D.; Kiziltepe, T.; Bilgicer, B. A systematic analysis of peptide linker length and liposomal polyethylene glycol coating on cellular uptake of peptide-targeted liposomes. ACS Nano 2013, 7, 2935–2947. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef]
- Haley, B.; Frenkel, E. Nanoparticles for drug delivery in cancer treatment. Urol. Oncol. 2008, 26, 57–64. [Google Scholar] [CrossRef]
- Maeda, H.; Nakamura, H.; Fang, J. The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv. Drug Deliv. Rev. 2013, 65, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Bissery, M.C. Preclinical pharmacology of docetaxel. Eur. J. Cancer 1995, 31, S1–S6. [Google Scholar] [CrossRef]
- Zamboni, W.C.; Strychor, S.; Joseph, E.; Parise, R.A.; Egorin, M.J.; Eiseman, J.L. Tumor, tissue, and plasma pharmacokinetic studies and antitumor response studies of docetaxel in combination with 9-nitrocamptothecin in mice bearing SKOV-3 human ovarian xenografts. Cancer Chemother. Pharmacol. 2008, 62, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.H.; Park, K. Targeted drug delivery to tumors: Myths, reality and possibility. J. Control. Release 2011, 153, 198–205. [Google Scholar] [CrossRef] [Green Version]
- Chrastina, A.; Massey, K.A.; Schnitzer, J.E. Overcoming in vivo barriers to targeted nanodelivery. Wiley Interdicsip Rev. Nanomed. Nanobiotechnol. 2011, 3, 421–437. [Google Scholar] [CrossRef]
- Lammers, T.; Kiessling, F.; Hennink, W.E.; Storm, G. Drug targeting to tumors: Principles, pitfalls and (pre-) clinical progress. J. Control. Release 2012, 161, 175–187. [Google Scholar] [CrossRef]
- Maeda, H. Toward a full understanding of the EPR effect in primary and metastatic tumors as well as issues related to its heterogeneity. Adv. Drug Deliv. Rev. 2015, 91, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Yuan, W.; Li, J.; Yang, L.; Su, Y.; Peng, J.; Chen, R.; Tham, H.P.; Chen, H.; Lim, W.Q.; et al. Independent of EPR effect: A smart delivery nanosystem for tracking and treatment of nonvascularized intra-abdominal metastases. Adv. Funct. Mater. 2018, 28, 1806162. [Google Scholar] [CrossRef]
- Singhana, B.; Slattery, P.; Melancon, M.P. 14—Targeted gold nanoshells. In Applications of Nanoscience in Photomedicine; Hamblin, M.R., Avci, P., Eds.; Chandos Publishing: Oxford, UK, 2015; pp. 267–290. [Google Scholar]
- Maeda, H. Tumor-selective delivery of macromolecular drugs via the EPR effect: Background and future prospects. Bioconjug. Chem. 2010, 21, 797–802. [Google Scholar] [CrossRef]
- Mulder, W.J.M.; Douma, K.; Koning, G.A.; van Zandvoort, M.A.; Lutgens, E.; Daemen, M.J.; Nicolay, K.; Strijkers, G.J. Liposome-enhanced MRI of neointimal lesions in the ApoE-KO mouse. Magn. Reson. Med. 2006, 55, 1170–1174. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.-H.; Rubin, K.; Pietras, K.; Östman, A. High interstitial fluid pressure—An obstacle in cancer therapy. Nat. Rev. Cancer 2004, 4, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K.; Stylianopoulos, T. Delivering nanomedicine to solid tumors. Nat. Rev. Clin. Oncol. 2010, 7, 653–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, M.J.; Jain, R.K.; Langer, R. Engineering and physical sciences in oncology: Challenges and opportunities. Nat. Rev. Cancer 2017, 17, 659–675. [Google Scholar] [CrossRef] [PubMed]
- Libutti, S.K.; Tamarkin, L.; Nilubol, N. Targeting the invincible barrier for drug delivery in solid cancers: Interstitial fluid pressure. Oncotarget 2018, 9, 35723–35725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, J.; Nakamura, H.; Maeda, H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Perentes, J.Y.; McKee, T.D.; Ley, C.D.; Mathiew, H.; Dawson, M.; Padera, T.P.; Munn, L.L.; Jain, R.K.; Boucher, Y. In vivo imaging of extracellular matrix remodeling by tumor-associated fibroblasts. Nat. Methods 2009, 6, 143–145. [Google Scholar] [CrossRef] [Green Version]
- Attia, M.F.; Anton, N.; Wallyn, J.; Omran, Z.; Vandamme, T.F. An overview of active and passive targeting strategies to improve the nanocarriers efficiency to tumour sites. J. Pharm. Pharmacol. 2019, 71, 1185–1198. [Google Scholar] [CrossRef] [Green Version]
- Narum, S.M.; Le, T.; Le, D.P.; Lee, J.C.; Donahue, N.D.; Yang, W.; Wilhelm, S. Chapter 4—Passive targeting in nanomedicine: Fundamental concepts, body interactions, and clinical potential. In Nanoparticles for Biomedical Applications; Chung, E.J., Leon, L., Rinaldi, C., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 37–53. [Google Scholar]
- Ghaffari, M.; Dehghan, G.; Abedi-Gaballu, F.; Kashanian, S.; Baradaran, B.; Ezzati Nazhad Dolatabadi, J.; Losic, D. Surface functionalized dendrimers as controlled-release delivery nanosystems for tumor targeting. Eur. J. Pharm. Sci. 2018, 122, 311–330. [Google Scholar] [CrossRef]
- Sakurai, Y.; Akita, H.; Harashima, H. Targeting tumor endothelial cells with nanoparticles. Int. J. Mol. Sci. 2019, 20, 5819. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xu, J.; Xia, X.; Yang, M.; Vangveravong, S.; Chen, J.; Mach, R.H.; Xia, Y. SV119-gold nanocage conjugates: A new platform for targeting cancer cells via sigma-2 receptors. Nanoscale 2012, 4, 421–424. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Dong, K.; Lin, M.; Dong, G.; Shan, S.; Lawson, T.; Yan, L.; Zhang, W.; Shi, B.; Chou, S.; et al. A transferrin triggered pathway for highly targeted delivery of graphene-based nanodrugs to treat choroidal melanoma. Adv. Helathcare Mater. 2018, 7, 1800377. [Google Scholar] [CrossRef]
- Zhang, H.; Shang, Y.; Li, Y.-H.; Sun, S.-K.; Yin, X.-B. Smart metal–organic framework-based nanoplatforms for imaging-guided precise chemotherapy. ACS Appl. Mater. Interfaces 2019, 11, 1886–1895. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, F.; Nguyen, K.T.; Ma, X.; Wang, X.; Xing, B.; Zhao, Y. Multifunctional mesoporous silica nanoparticles for cancer-targeted and controlled drug delivery. Adv. Funct. Mater. 2012, 22, 5144–5156. [Google Scholar] [CrossRef]
- Palanikumar, L.; Choi, E.S.; Cheon, J.Y.; Joo, S.H.; Ryu, J.-H. Noncovalent polymer-gatekeeper in mesoporous silica nanoparticles as a targeted drug delivery platform. Adv. Funct. Mater. 2015, 25, 957–965. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Y.; Zhang, X.; Wei, X.; Xiong, X.; Zhou, S. Enzyme and redox dual-triggered intracellular release from actively targeted polymeric micelles. ACS Appl. Mater. Interfaces 2017, 9, 3388–3399. [Google Scholar] [CrossRef] [PubMed]
- Ke, W.; Li, J.; Zhao, K.; Zha, Z.; Han, Y.; Wang, Y.; Yin, W.; Zhang, P.; Ge, Z. Modular design and facile synthesis of enzyme-responsive peptide-linked block copolymers for efficient delivery of doxorubicin. Biomacromolecules 2016, 17, 3268–3276. [Google Scholar] [CrossRef]
- Cheng, Y.-J.; Luo, G.-F.; Zhu, J.-Y.; Xu, X.-D.; Zeng, X.; Cheng, D.-B.; Li, Y.-M.; Wu, Y.; Zhang, X.-Z.; Zhuo, R.-X.; et al. Enzyme-induced and tumor-targeted drug delivery system based on multifunctional mesoporous silica nanoparticles. ACS Appl. Mater. Interfaces 2015, 7, 9078–9087. [Google Scholar] [CrossRef]
- Lei, Q.; Qiu, W.-X.; Hu, J.-J.; Cao, P.-X.; Zhu, C.-H.; Cheng, H.; Zhang, X.-Z. Multifunctional mesoporous silica nanoparticles with thermal-responsive gatekeeper for NIR light-triggered chemo/photothermal-therapy. Small 2016, 12, 4286–4298. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, L.; Assanhou, A.G.; Wang, L.; Zhang, Y.; Li, W.; Xue, L.; Mo, R.; Zhang, C. Acid/redox dual-activated liposomes for tumor-targeted drug delivery and enhanced therapeutic efficacy. RSC Adv. 2015, 5, 67803–67808. [Google Scholar] [CrossRef]
- Kim, M.S.; Lee, D.-W.; Park, K.; Park, S.-J.; Choi, E.-J.; Park, E.S.; Kim, H.R. Temperature-triggered tumor-specific delivery of anticancer agents by cRGD-conjugated thermosensitive liposomes. Colloids Surf. B 2014, 116, 17–25. [Google Scholar] [CrossRef]
- Bareford, L.M.; Swaan, P.W. Endocytic mechanisms for targeted drug delivery. Adv. Drug Deliv. Rev. 2007, 59, 748–758. [Google Scholar] [CrossRef] [Green Version]
- Farokhzad, O.C.; Cheng, J.; Teply, B.A.; Sherifi, I.; Jon, S.; Kantoff, P.W.; Richie, J.P.; Langer, R. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 6315–6320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chase, D.M.; Chaplin, D.J.; Monk, B.J. The development and use of vascular targeted therapy in ovarian cancer. Gynecol. Oncol. 2017, 145, 393–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nel, A.E.; Mädler, L.; Velegol, D.; Xia, T.; Hoek, E.M.V.; Somasundaran, P.; Klaessig, F.; Castranova, V.; Thompson, M. Understanding biophysicochemical interactions at the nano–bio interface. Nat. Mater. 2009, 8, 543–557. [Google Scholar] [CrossRef]
- Kaufmann, R.; MÜLler, P.; Hildenbrand, G.; Hausmann, M.; Cremer, C. Analysis of Her2/neu membrane protein clusters in different types of breast cancer cells using localization microscopy. J. Microsc. 2011, 242, 46–54. [Google Scholar] [CrossRef] [Green Version]
- Smart, E.J.; Mineo, C.; Anderson, R.G. Clustered folate receptors deliver 5-methyltetrahydrofolate to cytoplasm of MA104 cells. J. Cell Biol. 1996, 134, 1169–1177. [Google Scholar] [CrossRef]
- Cluzel, C.; Saltel, F.d.r.; Lussi, J.; Paulhe, F.d.r.; Imhof, B.A.; Wehrle-Haller, B. The mechanisms and dynamics of αvβ3 integrin clustering in living cells. J. Cell Biol. 2005, 171, 383–392. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.P.; Aguet, F.; Danuser, G.; Schmid, S.L. Local clustering of transferrin receptors promotes clathrin-coated pit initiation. J. Cell Biol. 2010, 191, 1381–1393. [Google Scholar] [CrossRef] [Green Version]
- Pirollo, K.F.; Chang, E.H. Does a targeting ligand influence nanoparticle tumor localization or uptake? Trends Biotechnol. 2008, 26, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Farokhzad, O.C.; Langer, R. Impact of nanotechnology on drug delivery. ACS Nano 2009, 3, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Kirpotin, D.B.; Drummond, D.C.; Shao, Y.; Shalaby, M.R.; Hong, K.; Nielsen, U.B.; Marks, J.D.; Benz, C.C.; Park, J.W. Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res. 2006, 66, 6732–6740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longmire, M.; Choyke, P.L.; Kobayashi, H. Clearance properties of nano-sized particles and molecules as imaging agents: Considerations and caveats. Nanomedicine 2008, 3, 703–717. [Google Scholar] [CrossRef] [Green Version]
- Moghimi, S.M.; Hunter, A.C.; Murray, J.C. Long-circulating and target-specific nanoparticles: Theory to practice. Pharmacol. Rev. 2001, 53, 283–318. [Google Scholar]
- Schädlich, A.; Caysa, H.; Mueller, T.; Tenambergen, F.; Rose, C.; Göpferich, A.; Kuntsche, J.; Mäder, K. Tumor accumulation of NIR fluorescent PEG–PLA nanoparticles: Impact of particle size and human xenograft tumor model. ACS Nano 2011, 5, 8710–8720. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Stenzel, M.H. Entry of nanoparticles into cells: The importance of nanoparticle properties. Polym. Chem. 2018, 9, 259–272. [Google Scholar] [CrossRef]
- Zhang, S.; Gao, H.; Bao, G. Physical principles of nanoparticle cellular endocytosis. ACS Nano 2015, 9, 8655–8671. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.; Zhang, S. Effects of particle size and ligand density on the kinetics of receptor-mediated endocytosis of nanoparticles. Appl. Phys. Lett. 2010, 96, 033704. [Google Scholar] [CrossRef] [Green Version]
- Chithrani, B.D.; Ghazani, A.A.; Chan, W.C.W. Determining the size and shape dependence of gold nanoparticle uptake into mammalian cells. Nano Lett. 2006, 6, 662–668. [Google Scholar] [CrossRef]
- Lu, F.; Wu, S.-H.; Hung, Y.; Mou, C.-Y. Size effect on cell uptake in well-suspended, uniform mesoporous silica nanoparticles. Small 2009, 5, 1408–1413. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Heller, D.A.; Sharma, R.; Strano, M.S. Size-dependent cellular uptake and expulsion of single-walled carbon nanotubes: Single particle tracking and a generic uptake model for nanoparticles. ACS Nano 2009, 3, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Osaki, F.; Kanamori, T.; Sando, S.; Sera, T.; Aoyama, Y. A quantum dot conjugated sugar ball and its cellular uptake. On the size effects of endocytosis in the subviral region. J. Am. Chem. Soc. 2004, 126, 6520–6521. [Google Scholar] [CrossRef]
- Zhao, J.; Babiuch, K.; Lu, H.; Dag, A.; Gottschaldt, M.; Stenzel, M.H. Fructose-coated nanoparticles: A promising drug nanocarrier for triple-negative breast cancer therapy. Chem. Commun. 2014, 50, 15928–15931. [Google Scholar] [CrossRef]
- Chaudhuri, A.; Battaglia, G.; Golestanian, R. The effect of interactions on the cellular uptake of nanoparticles. Phys. Biol. 2011, 8, 046002. [Google Scholar] [CrossRef]
- Albanese, A.; Chan, W.C.W. Effect of gold nanoparticle aggregation on cell uptake and toxicity. ACS Nano 2011, 5, 5478–5489. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, S.K.; Harth, E. Molecular dendritic transporter nanoparticle vectors provide efficient intracellular delivery of peptides. ACS Nano 2009, 3, 402–410. [Google Scholar] [CrossRef]
- Khine, Y.Y.; Callari, M.; Lu, H.; Stenzel, M.H. Direct correlation between zeta potential and cellular uptake of poly(methacrylic acid) post-modified with guanidinium functionalities. Macromol. Chem. Phys. 2016, 217, 2302–2309. [Google Scholar] [CrossRef]
- Cho, E.C.; Xie, J.; Wurm, P.A.; Xia, Y. Understanding the role of surface charges in cellular adsorption versus internalization by selectively removing gold nanoparticles on the cell surface with a I2/KI etchant. Nano Lett. 2009, 9, 1080–1084. [Google Scholar] [CrossRef]
- Slowing, I.; Trewyn, B.G.; Lin, V.S.Y. Effect of surface functionalization of MCM-41-type mesoporous silica nanoparticles on the endocytosis by human cancer cells. J. Am. Chem. Soc. 2006, 128, 14792–14793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roser, M.; Fischer, D.; Kissel, T. Surface-modified biodegradable albumin nano- and microspheres. II: Effect of surface charges on in vitro phagocytosis and biodistribution in rats. Eur. J. Pharm. Biopharm. 1998, 46, 255–263. [Google Scholar] [CrossRef]
- Cedervall, T.; Lynch, I.; Lindman, S.; Berggård, T.; Thulin, E.; Nilsson, H.; Dawson, K.A.; Linse, S. Understanding the nanoparticle–protein corona using methods to quantify exchange rates and affinities of proteins for nanoparticles. Proc. Natl. Acad. Sci. USA 2007, 104, 2050–2055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Yin, Y.; Wang, L.; Zhang, W.; Chen, X.; Yang, X.; Xu, J.; Ma, G. Surface hydrophobicity of microparticles modulates adjuvanticity. J. Mater. Chem. B 2013, 1, 3888–3896. [Google Scholar] [CrossRef]
- Shahbazi, M.-A.; Fernández, T.D.; Mäkilä, E.M.; Le Guével, X.; Mayorga, C.; Kaasalainen, M.H.; Salonen, J.J.; Hirvonen, J.T.; Santos, H.A. Surface chemistry dependent immunostimulative potential of porous silicon nanoplatforms. Biomaterials 2014, 35, 9224–9235. [Google Scholar] [CrossRef]
- Knop, K.; Hoogenboom, R.; Fischer, D.; Schubert, U.S. Poly(ethylene glycol) in drug delivery: Pros and cons as well as potential alternatives. Angew. Chem., Int. Ed. 2010, 49, 6288–6308. [Google Scholar] [CrossRef] [PubMed]
- Ulbricht, J.; Jordan, R.; Luxenhofer, R. On the biodegradability of polyethylene glycol, polypeptoids and poly(2-oxazoline)s. Biomaterials 2014, 35, 4848–4861. [Google Scholar] [CrossRef] [PubMed]
- Nugraha, B. Application of PEG in drug delivery system. In Gels Handbook; World Scientific: Singapore, 2016; pp. 137–147. [Google Scholar]
- Gulati, N.M.; Stewart, P.L.; Steinmetz, N.F. Bioinspired shielding strategies for nanoparticle drug delivery applications. Mol. Pharm. 2018, 15, 2900–2909. [Google Scholar] [CrossRef]
- Yan, J.; Yu, J.; Wang, C.; Gu, Z. Red blood cells for drug delivery. Small Methods 2017, 1, 1700270. [Google Scholar] [CrossRef]
- Schrade, A.; Mailänder, V.; Ritz, S.; Landfester, K.; Ziener, U. Surface roughness and charge influence the uptake of nanoparticles: Fluorescently labeled pickering-type versus surfactant-stabilized nanoparticles. Macromol. Biosci. 2012, 12, 1459–1471. [Google Scholar] [CrossRef] [PubMed]
- Piloni, A.; Wong, C.K.; Chen, F.; Lord, M.; Walther, A.; Stenzel, M.H. Surface roughness influences the protein corona formation of glycosylated nanoparticles and alter their cellular uptake. Nanoscale 2019, 11, 23259–23267. [Google Scholar] [CrossRef]
- Verma, A.; Uzun, O.; Hu, Y.; Hu, Y.; Han, H.-S.; Watson, N.; Chen, S.; Irvine, D.J.; Stellacci, F. Surface-structure-regulated cell-membrane penetration by monolayer-protected nanoparticles. Nat. Mater. 2008, 7, 588–595. [Google Scholar] [CrossRef]
- Gratton, S.E.A.; Ropp, P.A.; Pohlhaus, P.D.; Luft, J.C.; Madden, V.J.; Napier, M.E.; DeSimone, J.M. The effect of particle design on cellular internalization pathways. Proc. Natl. Acad. Sci. USA 2008, 105, 11613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnida; Malugin, A.; Ghandehari, H. Cellular uptake and toxicity of gold nanoparticles in prostate cancer cells: A comparative study of rods and spheres. J. Appl. Toxicol. 2010, 30, 212–217. [Google Scholar] [CrossRef]
- Arnida; Janát-Amsbury, M.M.; Ray, A.; Peterson, C.M.; Ghandehari, H. Geometry and surface characteristics of gold nanoparticles influence their biodistribution and uptake by macrophages. Eur. J. Pharm Biopharm 2011, 77, 417–423. [Google Scholar] [CrossRef] [Green Version]
- Champion, J.A.; Mitragotri, S. Shape induced inhibition of phagocytosis of polymer particles. Pharm. Res. 2009, 26, 244–249. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Kröger, M.; Liu, W.K. Shape effect in cellular uptake of PEGylated nanoparticles: Comparison between sphere, rod, cube and disk. Nanoscale 2015, 7, 16631–16646. [Google Scholar] [CrossRef]
- Muro, S.; Garnacho, C.; Champion, J.A.; Leferovich, J.; Gajewski, C.; Schuchman, E.H.; Mitragotri, S.; Muzykantov, V.R. Control of endothelial targeting and intracellular delivery of therapeutic enzymes by modulating the size and shape of ICAM-1-targeted carriers. Mol. Ther. 2008, 16, 1450–1458. [Google Scholar] [CrossRef]
- Cho, E.C.; Au, L.; Zhang, Q.; Xia, Y. The effects of size, shape, and surface functional group of gold nanostructures on their adsorption and internalization by cells. Small 2010, 6, 517–522. [Google Scholar] [CrossRef]
- Banerjee, A.; Qi, J.; Gogoi, R.; Wong, J.; Mitragotri, S. Role of nanoparticle size, shape and surface chemistry in oral drug delivery. J. Control. Release 2016, 238, 176–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, J.F.; Kozlovskaya, V.; Chen, J.; Kuncewicz, T.; Kharlampieva, E.; Godin, B. Cubical shape enhances the interaction of layer-by-layer polymeric particles with breast cancer cells. Adv. Helathcare Mater. 2015, 4, 2657–2666. [Google Scholar] [CrossRef] [Green Version]
- Champion, J.A.; Mitragotri, S. Role of target geometry in phagocytosis. Proc. Natl. Acad. Sci. USA 2006, 103, 4930–4934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Champion, J.A.; Katare, Y.K.; Mitragotri, S. Particle shape: A new design parameter for micro- and nanoscale drug delivery carriers. J. Control. Release 2007, 121, 3–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, Y.; Dalhaimer, P.; Cai, S.; Tsai, R.; Tewari, M.; Minko, T.; Discher, D.E. Shape effects of filaments versus spherical particles in flow and drug delivery. Nat. Nanotechnol. 2007, 2, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Shimoni, O.; Yan, Y.; Wang, Y.; Caruso, F. Shape-dependent cellular processing of polyelectrolyte capsules. ACS Nano 2013, 7, 522–530. [Google Scholar] [CrossRef]
- Florez, L.; Herrmann, C.; Cramer, J.M.; Hauser, C.P.; Koynov, K.; Landfester, K.; Crespy, D.; Mailänder, V. How shape influences uptake: Interactions of anisotropic polymer nanoparticles and human mesenchymal stem cells. Small 2012, 8, 2222–2230. [Google Scholar] [CrossRef]
- Black, K.C.L.; Wang, Y.; Luehmann, H.P.; Cai, X.; Xing, W.; Pang, B.; Zhao, Y.; Cutler, C.S.; Wang, L.V.; Liu, Y.; et al. Radioactive 198Au-doped nanostructures with different shapes for in vivo analyses of their biodistribution, tumor uptake, and intratumoral distribution. ACS Nano 2014, 8, 4385–4394. [Google Scholar] [CrossRef]
- Barua, S.; Yoo, J.-W.; Kolhar, P.; Wakankar, A.; Gokarn, Y.R.; Mitragotri, S. Particle shape enhances specificity of antibody-displaying nanoparticles. Proc. Natl. Acad. Sci. USA 2013, 110, 3270–3275. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Rossin, R.; Hagooly, A.; Chen, Z.; Welch, M.J.; Wooley, K.L. Folate-mediated cell uptake of shell-crosslinked spheres and cylinders. J. Polym. Sci. A Polym. Chem. 2008, 46, 7578–7583. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Tang, Z.; Gao, Y.; Sun, H.; Zhou, S. A bio-inspired rod-shaped nanoplatform for strongly infecting tumor cells and enhancing the delivery efficiency of anticancer drugs. Adv. Funct. Mater. 2016, 26, 66–79. [Google Scholar] [CrossRef]
- Chauhan, V.P.; Popović, Z.; Chen, O.; Cui, J.; Fukumura, D.; Bawendi, M.G.; Jain, R.K. Fluorescent nanorods and nanospheres for real-time in vivo probing of nanoparticle shape-dependent tumor penetration. Angew. Chem. Int. Ed. 2011, 50, 11417–11420. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Li, L.; Liu, T.; Hao, N.; Liu, H.; Chen, D.; Tang, F. The shape effect of mesoporous silica nanoparticles on biodistribution, clearance, and biocompatibility in vivo. ACS Nano 2011, 5, 5390–5399. [Google Scholar] [CrossRef]
- Yi, X.; Gao, H. Kinetics of receptor-mediated endocytosis of elastic nanoparticles. Nanoscale 2017, 9, 454–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, X.; Shi, X.; Gao, H. Cellular uptake of elastic nanoparticles. Phys. Rev. Lett. 2011, 107, 098101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Z.; Ye, H.; Li, Y. Understanding receptor-mediated endocytosis of elastic nanoparticles through coarse grained molecular dynamic simulation. Phys. Chem. Chem. Phys. 2018, 20, 16372–16385. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Wong, E.H.H.; Yan, Y.; Cui, J.; Dai, Q.; Guo, J.; Qiao, G.G.; Caruso, F. The role of capsule stiffness on cellular processing. Chem. Sci. 2015, 6, 3505–3514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, R.; Weidenbach, M.; Neubauer, M.; Fery, A.; Parak, W.J. Stiffness-dependent in vitro uptake and lysosomal acidification of colloidal particles. Angew. Chem. Int. Ed. 2015, 54, 1365–1368. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhou, X.; Mao, Z.; Yu, D.; Wang, B.; Gao, C. Uptake of hydrogel particles with different stiffness and its influence on HepG2 cell functions. Soft Matter 2012, 8, 9235–9245. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, L.; Wang, J.; Feng, Q.; Liu, D.; Yin, Q.; Xu, D.; Wei, Y.; Ding, B.; Shi, X.; et al. Tunable rigidity of (polymeric core)–(lipid shell) nanoparticles for regulated cellular uptake. Adv. Mater. 2015, 27, 1402–1407. [Google Scholar] [CrossRef]
- Banquy, X.; Suarez, F.; Argaw, A.; Rabanel, J.-M.; Grutter, P.; Bouchard, J.-F.; Hildgen, P.; Giasson, S. Effect of mechanical properties of hydrogel nanoparticles on macrophage cell uptake. Soft Matter 2009, 5, 3984–3991. [Google Scholar] [CrossRef]
- Sun, H.; Björnmalm, M.; Cui, J.; Wong, E.H.H.; Dai, Y.; Dai, Q.; Qiao, G.G.; Caruso, F. Structure governs the deformability of polymer particles in a microfluidic blood capillary model. ACS Macro Lett. 2015, 4, 1205–1209. [Google Scholar] [CrossRef]
- Alford, A.; Rich, M.; Kozlovskaya, V.; Chen, J.; Sherwood, J.; Bolding, M.; Warram, J.; Bao, Y.; Kharlampieva, E. Ultrasound-triggered delivery of anticancer therapeutics from MRI-visible multilayer microcapsules. Adv. Ther. 2018, 1, 1800051. [Google Scholar] [CrossRef]
- Merkel, T.J.; Jones, S.W.; Herlihy, K.P.; Kersey, F.R.; Shields, A.R.; Napier, M.; Luft, J.C.; Wu, H.; Zamboni, W.C.; Wang, A.Z.; et al. Using mechanobiological mimicry of red blood cells to extend circulation times of hydrogel microparticles. Proc. Natl. Acad. Sci. USA 2011, 108, 586–591. [Google Scholar] [CrossRef] [Green Version]
- Guo, P.; Liu, D.; Subramanyam, K.; Wang, B.; Yang, J.; Huang, J.; Auguste, D.T.; Moses, M.A. Nanoparticle elasticity directs tumor uptake. Nat. Commun. 2018, 9, 130. [Google Scholar] [CrossRef] [Green Version]
- Estrella, V.; Chen, T.; Lloyd, M.; Wojtkowiak, J.; Cornnell, H.H.; Ibrahim-Hashim, A.; Bailey, K.; Balagurunathan, Y.; Rothberg, J.M.; Sloane, B.F.; et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013, 73, 1524–1535. [Google Scholar] [CrossRef] [Green Version]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helmlinger, G.; Yuan, F.; Dellian, M.; Jain, R.K. Interstitial pH and pO2 gradients in solid tumors in vivo: High-resolution measurements reveal a lack of correlation. Nat. Med. 1997, 3, 177–182. [Google Scholar] [CrossRef]
- Gillies, R.J.; Liu, Z.; Bhujwalla, Z. 31P-MRS measurements of extracellular pH of tumors using 3-aminopropylphosphonate. Am. J. Physiol. Cell Phys. 1994, 267, C195–C203. [Google Scholar] [CrossRef]
- Stubbs, M.; McSheehy, P.M.J.; Griffiths, J.R.; Bashford, C.L. Causes and consequences of tumour acidity and implications for treatment. Mol. Med. Today 2000, 6, 15–19. [Google Scholar] [CrossRef]
- Maxfield, F.R.; Yamashiro, D.J. Endosome acidification and the pathways of receptor-mediated endocytosis. In Immunobiology of Proteins and Peptides IV: T-Cell Recognition and Antigen Presentation; Atassi, M.Z., Ed.; Springer: Boston, MA, USA, 1987; pp. 189–198. [Google Scholar]
- Mindell, J.A. Lysosomal acidification mechanisms. Annu. Rev. Physiol. 2012, 74, 69–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgio, M.; Trinei, M.; Migliaccio, E.; Pelicci, P.G. Hydrogen peroxide: A metabolic by-product or a common mediator of ageing signals? Nat. Rev. Mol. Cell Bio. 2007, 8, 722–728. [Google Scholar] [CrossRef]
- Borovanský, J.; Elleder, M. Melanosome degradation: Fact or fiction. Pigment Cell Res. 2003, 16, 280–286. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Kumari, S.; Badana, A.K.; G, M.M.; G, S.; Malla, R. Reactive oxygen species: A key constituent in cancer survival. Biomark. Insights 2018, 13, 1177271918755391. [Google Scholar] [CrossRef] [Green Version]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of glutathione in cancer progression and chemoresistance. Oxid. Med. Cell Longev. 2013, 2013, 972913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Jiang, J.; Lei, Y.; Zhou, S.; Wei, Y.; Huang, C. Targeting metabolic–redox circuits for cancer therapy. Trends Biochem. Sci. 2019, 44, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Manickam, D.S.; Li, J.; Putt, D.A.; Zhou, Q.-H.; Wu, C.; Lash, L.H.; Oupický, D. Effect of innate glutathione levels on activity of redox-responsive gene delivery vectors. J. Control. Release 2010, 141, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Kuppusamy, P.; Li, H.; Ilangovan, G.; Cardounel, A.J.; Zweier, J.L.; Yamada, K.; Krishna, M.C.; Mitchell, J.B. Noninvasive imaging of tumor redox status and its modification by tissue glutathione levels. Cancer Res. 2002, 62, 307. [Google Scholar] [PubMed]
- Touyz, R.M.; Schiffrin, E.L. Reactive oxygen species in vascular biology: Implications in hypertension. Histochem. Cell Biol. 2004, 122, 339–352. [Google Scholar] [CrossRef]
- Lee, S.H.; Boire, T.C.; Lee, J.B.; Gupta, M.K.; Zachman, A.L.; Rath, R.; Sung, H.-J. ROS-cleavable proline oligomer crosslinking of polycaprolactone for pro-angiogenic host response. J. Mater. Chem. B 2014, 2, 7109–7113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugamura, K.; Keaney, J.F. Reactive oxygen species in cardiovascular disease. Free Radical Bio. Med. 2011, 51, 978–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houstis, N.; Rosen, E.D.; Lander, E.S. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 2006, 440, 944–948. [Google Scholar] [CrossRef] [PubMed]
- Rastew, E.; Vicente, J.B.; Singh, U. Oxidative stress resistance genes contribute to the pathogenic potential of the anaerobic protozoan parasite, Entamoeba histolytica. Int. J. Parasitol. 2012, 42, 1007–1015. [Google Scholar] [CrossRef] [Green Version]
- Rahman, I. Oxidative stress, chromatin remodeling and gene transcription in inflammation and chronic lung diseases. J. Biochem Mol. Biol 2003, 36, 95–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008, 4, 278–286. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox. Sign. 2013, 20, 1126–1167. [Google Scholar] [CrossRef] [Green Version]
- Ibañez, I.L.; Notcovich, C.; Catalano, P.N.; Bellino, M.G.; Durán, H. The redox-active nanomaterial toolbox for cancer therapy. Cancer Lett. 2015, 359, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radical Bio. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Itano, N. Simple primary structure, complex turnover regulation and multiple roles of hyaluronan. J. Biochem. 2008, 144, 131–137. [Google Scholar] [CrossRef]
- Graff, J.R.; Konicek, B.W.; Deddens, J.A.; Chedid, M.; Hurst, B.M.; Colligan, B.; Neubauer, B.L.; Carter, H.W.; Carter, J.H. Expression of group IIa secretory phospholipase A2 increases with prostate tumor grade. Clin. Cancer Res. 2001, 7, 3857–3861. [Google Scholar] [PubMed]
- Scott, K.F.; Sajinovic, M.; Hein, J.; Nixdorf, S.; Galettis, P.; Liauw, W.; de Souza, P.; Dong, Q.; Graham, G.G.; Russell, P.J. Emerging roles for phospholipase A2 enzymes in cancer. Biochimie 2010, 92, 601–610. [Google Scholar] [CrossRef]
- Andresen, T.L.; Thompson, D.H.; Kaasgaard, T. Enzyme-triggered nanomedicine: Drug release strategies in cancer therapy (Invited Review). Mol. Membr. Biol. 2010, 27, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Wells, A.; Grandis, J.R. Phospholipase C-γ1 in tumor progression. Clin. Exp. Metastasis 2003, 20, 285. [Google Scholar] [CrossRef]
- Hu, Q.; Katti, P.S.; Gu, Z. Enzyme-responsive nanomaterials for controlled drug delivery. Nanoscale 2014, 6, 12273–12286. [Google Scholar] [CrossRef]
- Sharma, A.; Arambula, J.F.; Koo, S.; Kumar, R.; Singh, H.; Sessler, J.L.; Kim, J.S. Hypoxia-targeted drug delivery. Chem. Soc. Rev. 2019, 48, 771–813. [Google Scholar] [CrossRef]
- Yahara, T.; Koga, T.; Yoshida, S.; Nakagawa, S.; Deguchi, H.; Shirouzu, K. Relationship between microvessel density and thermographic hot areas in breast cancer. Surg. Today 2003, 33, 243–248. [Google Scholar] [CrossRef]
- Stefanadis, C.; Chrysochoou, C.; Markou, D.; Petraki, K.; Panagiotakos, D.B.; Fasoulakis, C.; Kyriakidis, A.; Papadimitriou, C.; Toutouzas, P.K. Increased temperature of malignant urinary bladder tumors in vivo: The application of a new method based on a catheter technique. J. Clin. Oncol. 2001, 19, 676–681. [Google Scholar] [CrossRef]
- Li, H.-J.; Du, J.-Z.; Du, X.-J.; Xu, C.-F.; Sun, C.-Y.; Wang, H.-X.; Cao, Z.-T.; Yang, X.-Z.; Zhu, Y.-H.; Nie, S.; et al. Stimuli-responsive clustered nanoparticles for improved tumor penetration and therapeutic efficacy. Proc. Natl. Acad. Sci. USA 2016, 113, 4164–4169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Gagne, L.; Chen, H.; Szoka, F.C. Novel ortho ester-based, pH-sensitive cationic lipid for gene delivery in vitro and in vivo. J. Liposome Res. 2014, 24, 90–98. [Google Scholar] [CrossRef]
- Petrova, S.; Jäger, E.; Konefał, R.; Jäger, A.; Venturini, C.G.; Spěváček, J.; Pavlova, E.; Štěpánek, P. Novel poly(ethylene oxide monomethyl ether)-b-poly(ε-caprolactone) diblock copolymers containing a pH-acid labile ketal group as a block linkage. Polym. Chem. 2014, 5, 3884–3893. [Google Scholar] [CrossRef]
- Chen, J.; Ding, J.; Zhang, Y.; Xiao, C.; Zhuang, X.; Chen, X. Polyion complex micelles with gradient pH-sensitivity for adjustable intracellular drug delivery. Polym. Chem. 2015, 6, 397–405. [Google Scholar] [CrossRef]
- Mo, R.; Sun, Q.; Li, N.; Zhang, C. Intracellular delivery and antitumor effects of pH-sensitive liposomes based on zwitterionic oligopeptide lipids. Biomaterials 2013, 34, 2773–2786. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Nishida, J.; Ma, W.; Wu, H.; Kobayashi, M.; Otsuka, H.; Takahara, A. Competition between oxidation and coordination in cross-linking of polystyrene copolymer containing catechol groups. ACS Macro Lett. 2012, 1, 457–460. [Google Scholar] [CrossRef]
- Kim, S.; Gim, T.; Kang, S.M. Stability-enhanced polydopamine coatings on solid substrates by iron(III) coordination. Prog. Org. Coat. 2014, 77, 1336–1339. [Google Scholar] [CrossRef]
- Holten-Andersen, N.; Harrington, M.J.; Birkedal, H.; Lee, B.P.; Messersmith, P.B.; Lee, K.Y.C.; Waite, J.H. pH-induced metal-ligand cross-links inspired by mussel yield self-healing polymer networks with near-covalent elastic moduli. Proc. Natl. Acad. Sci. USA 2011, 108, 2651–2655. [Google Scholar] [CrossRef] [Green Version]
- Wei, P.; Gangapurwala, G.; Pretzel, D.; Leiske, M.N.; Wang, L.; Hoeppener, S.; Schubert, S.; Brendel, J.C.; Schubert, U.S. Smart pH-sensitive nanogels for controlled release in an acidic environment. Biomacromolecules 2019, 20, 130–140. [Google Scholar] [CrossRef]
- Li, Y.; Xiao, W.; Xiao, K.; Berti, L.; Luo, J.; Tseng, H.P.; Fung, G.; Lam, K.S. Well-defined, reversible boronate crosslinked nanocarriers for targeted drug delivery in response to acidic pH values and cis-diols. Angew. Chem. Int. Ed. 2012, 51, 2864–2869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Li, Y.; Hu, X.-Y.; Zou, X.; Xiong, S.; Lin, C.; Wang, L. Supramolecular nanoparticles constructed by DOX-based prodrug with water-soluble pillar[6]arene for self-catalyzed rapid drug release. Chem. Mater. 2015, 27, 1110–1119. [Google Scholar] [CrossRef]
- Chen, Q.; Ding, H.; Zhou, J.; Zhao, X.; Zhang, J.; Yang, C.; Li, K.; Qiao, M.; Hu, H.; Ding, P.; et al. Novel glycyrrhetinic acid conjugated pH-sensitive liposomes for the delivery of doxorubicin and its antitumor activities. RSC Adv. 2016, 6, 17782–17791. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, H.; Liu, J.; Deng, L.; Liu, J.; Dong, A.; Zhang, J. Comb-like amphiphilic copolymers bearing acetal-functionalized backbones with the ability of acid-triggered hydrophobic-to-hydrophilic transition as effective nanocarriers for intracellular release of curcumin. Biomacromolecules 2013, 14, 3973–3984. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Bian, Q.; Wang, P.; Zheng, X.; Lv, L.; Dang, Z.; Wang, G. Photo, pH and redox multi-responsive nanogels for drug delivery and fluorescence cell imaging. Polym. Chem. 2017, 8, 6150–6157. [Google Scholar] [CrossRef]
- Chen, S.; Gao, Y.; Cao, Z.; Wu, B.; Wang, L.; Wang, H.; Dang, Z.; Wang, G. Nanocomposites of spiropyran-functionalized polymers and upconversion nanoparticles for controlled release stimulated by near-infrared light and pH. Macromolecules 2016, 49, 7490–7496. [Google Scholar] [CrossRef]
- Chen, Y.; Ai, K.; Liu, J.; Ren, X.; Jiang, C.; Lu, L. Polydopamine-based coordination nanocomplex for T1/T2 dual mode magnetic resonance imaging-guided chemo-photothermal synergistic therapy. Biomaterials 2016, 77, 198–206. [Google Scholar] [CrossRef]
- Yu, X.; Pan, Q.; Zheng, Z.; Chen, Y.; Chen, Y.; Weng, S.; Huang, L. pH-responsive and porous vancomycin-loaded PLGA microspheres: Evidence of controlled and sustained release for localized inflammation inhibition in vitro. RSC Adv. 2018, 8, 37424–37432. [Google Scholar] [CrossRef] [Green Version]
- Nahire, R.; Hossain, R.; Patel, R.; Paul, S.; Meghnani, V.; Ambre, A.H.; Gange, K.N.; Katti, K.S.; Leclerc, E.; Srivastava, D.K.; et al. pH-triggered echogenicity and contents release from liposomes. Mol. Pharm. 2014, 11, 4059–4068. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Li, Z.; Lin, Y.; Yin, M.; Ren, J.; Qu, X. Biomineralization inspired surface engineering of nanocarriers for pH-responsive, targeted drug delivery. Biomaterials 2013, 34, 1364–1371. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zeng, B.; Liang, S.; Long, M.; Xu, H. Synthesis of pH-responsive biodegradable mesoporous silica–calcium phosphate hybrid nanoparticles as a high potential drug carrier. ACS Appl. Mater. Interfaces 2017, 9, 44402–44409. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, F.; Guo, M.; Qi, W.; Sun, F.; Wang, A.; Guo, Y.; Zhu, G. pH-triggered controlled drug release from mesoporous silica nanoparticles via intracelluar dissolution of ZnO nanolids. J. Am. Chem. Soc. 2011, 133, 8778–8781. [Google Scholar] [CrossRef] [PubMed]
- Obata, Y.; Tajima, S.; Takeoka, S. Evaluation of pH-responsive liposomes containing amino acid-based zwitterionic lipids for improving intracellular drug delivery in vitro and in vivo. J. Control. Release 2010, 142, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Hu, Q.; Zhang, Q.; Jiang, K.; Lin, W.; Yang, Y.; Cui, Y.; Qian, G. A large capacity cationic metal–organic framework nanocarrier for physiological pH responsive drug delivery. Mol. Pharm. 2016, 13, 2782–2786. [Google Scholar] [CrossRef] [PubMed]
- Salinas, Y.; Castilla, A.M.; Resmini, M. An l-proline based thermoresponsive and pH-switchable nanogel as a drug delivery vehicle. Polym. Chem. 2018, 9, 2271–2280. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-M.; Chen, H.; Dettmer, C.M.; O’Halloran, T.V.; Nguyen, S.T. Polymer-caged lipsomes: A pH-responsive delivery system with high stability. J. Am. Chem. Soc. 2007, 129, 15096–15097. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Zeng, F.; Wu, S.; Wang, X. Polymer micelle with pH-triggered hydrophobic–hydrophilic transition and de-cross-linking process in the core and its application for targeted anticancer drug delivery. Biomacromolecules 2012, 13, 4126–4137. [Google Scholar] [CrossRef]
- Sun, Y.; Davis, E.W. Facile fabrication of polydopamine nanotubes for combined chemo-photothermal therapy. J. Mater. Chem. B 2019, 7, 6828–6839. [Google Scholar] [CrossRef]
- He, H.; Cattran, A.W.; Nguyen, T.; Nieminen, A.-L.; Xu, P. Triple-responsive expansile nanogel for tumor and mitochondria targeted photosensitizer delivery. Biomaterials 2014, 35, 9546–9553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Du, P.; Liu, P. Layer-by-layer polyelectrolyte complex coated poly(methacrylic acid) nanogels as a drug delivery system for controlled release: Structural effects. RSC Adv. 2014, 4, 56323–56331. [Google Scholar] [CrossRef]
- Deng, Z.; Zhen, Z.; Hu, X.; Wu, S.; Xu, Z.; Chu, P.K. Hollow chitosan–silica nanospheres as pH-sensitive targeted delivery carriers in breast cancer therapy. Biomaterials 2011, 32, 4976–4986. [Google Scholar] [CrossRef]
- Kogure, K.; Akita, H.; Yamada, Y.; Harashima, H. Multifunctional envelope-type nano device (MEND) as a non-viral gene delivery system. Adv. Drug Deliv. Rev. 2008, 60, 559–571. [Google Scholar] [CrossRef]
- Mohammed, M.A.; Syeda, J.T.M.; Wasan, K.M.; Wasan, E.K. An overview of chitosan nanoparticles and its application in non-parenteral drug delivery. Pharmaceutics 2017, 9, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.; Zhao, X.; Ma, P.X.; Guo, B.; Du, Y.; Han, X. pH-responsive injectable hydrogels with mucosal adhesiveness based on chitosan-grafted-dihydrocaffeic acid and oxidized pullulan for localized drug delivery. J. Colloid Interface Sci. 2019, 536, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Lam, J.K. Endosomal escape pathways for non-viral nucleic acid delivery systems. In Molecular Regulation of Endocytosis; Ceresa, B., Ed.; IntechOpen: London, UK, 2012; pp. 429–456. [Google Scholar]
- Le, C.-F.; Fang, C.-M.; Sekaran, S.D. Intracellular targeting mechanisms by antimicrobial peptides. Antimicrob. Agents Chemother. 2017, 61, e02340-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanderson, J.M. Peptide–lipid interactions: Insights and perspectives. Org. Biomol. Chem. 2005, 3, 201–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Yan, J.; Wen, N.; Xiong, H.; Cai, S.; He, Q.; Hu, Y.; Peng, D.; Liu, Z.; Liu, Y. Metal-organic frameworks for stimuli-responsive drug delivery. Biomaterials 2020, 230, 119619. [Google Scholar] [CrossRef] [PubMed]
- Korenchan, D.E.; Flavell, R.R. Spatiotemporal pH heterogeneity as a promoter of cancer progression and therapeutic resistance. Cancers 2019, 11, 1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, M.P.; Langer, R.; Jensen, K.F. Intracellular delivery by membrane disruption: Mechanisms, strategies, and concepts. Chem. Rev. 2018, 118, 7409–7531. [Google Scholar] [CrossRef]
- Stewart, M.P.; Sharei, A.; Ding, X.; Sahay, G.; Langer, R.; Jensen, K.F. In vitro and ex vivo strategies for intracellular delivery. Nature 2016, 538, 183–192. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.A.; Selby, L.I.; Johnston, A.P.; Such, G.K. The endosomal escape of nanoparticles: Toward more efficient cellular delivery. Bioconjug. Chem. 2018, 30, 263–272. [Google Scholar] [CrossRef]
- Libánská, A.; Randárová, E.; Lager, F.; Renault, G.; Scherman, D.; Etrych, T. Polymer nanomedicines with pH-sensitive release of dexamethasone for the localized treatment of inflammation. Pharmaceutics 2020, 12, 700. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Y.; Lin, W.; Gao, J.; Shi, X.; Davaritouchaee, M.; Nielsen, A.E.; Mancini, R.J.; Wang, Z. pH-responsive nanoparticles targeted to lungs for improved therapy of acute lung inflammation/injury. ACS Appl. Mater. Interfaces 2019, 11, 16380–16390. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Li, Y.; Ren, H.; Xu, H.; Li, Z.; Zhang, X. Selenium-containing block copolymers and their oxidation-responsive aggregates. Polym. Chem. 2010, 1, 1609–1614. [Google Scholar] [CrossRef]
- Li, F.; Li, T.; Cao, W.; Wang, L.; Xu, H. Near-infrared light stimuli-responsive synergistic therapy nanoplatforms based on the coordination of tellurium-containing block polymer and cisplatin for cancer treatment. Biomaterials 2017, 133, 208–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Sun, X.; Mao, W.; Sun, W.; Tang, J.; Sui, M.; Shen, Y.; Gu, Z. Tumor redox heterogeneity-responsive prodrug nanocapsules for cancer chemotherapy. Adv. Mater. 2013, 25, 3670–3676. [Google Scholar] [CrossRef]
- Luo, C.; Sun, J.; Liu, D.; Sun, B.; Miao, L.; Musetti, S.; Li, J.; Han, X.; Du, Y.; Li, L.; et al. Self-assembled redox dual-responsive prodrug-nanosystem formed by single thioether-bridged paclitaxel-fatty acid conjugate for cancer chemotherapy. Nano Lett. 2016, 16, 5401–5408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, H.; Mu, S.; Qiu, G.; Liu, X.; Zhang, L.; Yuan, Y.; Astruc, D. Redox-stimuli-responsive drug delivery systems with supramolecular ferrocenyl-containing polymers for controlled release. Coord. Chem. Rev. 2018, 364, 51–85. [Google Scholar] [CrossRef]
- Wang, Z.; Feng, Z.; Gao, C. Stepwise assembly of the same polyelectrolytes using host−guest interaction to obtain microcapsules with multiresponsive properties. Chem. Mater. 2008, 20, 4194–4199. [Google Scholar] [CrossRef]
- Liu, L.; Rui, L.; Gao, Y.; Zhang, W. Self-assembly and disassembly of a redox-responsive ferrocene-containing amphiphilic block copolymer for controlled release. Polym. Chem. 2015, 6, 1817–1829. [Google Scholar] [CrossRef]
- Xu, F.; Li, H.; Luo, Y.-L.; Tang, W. Redox-responsive self-assembly micelles from poly(N-acryloylmorpholine-block-2-acryloyloxyethyl ferrocenecarboxylate) amphiphilic block copolymers as drug release carriers. ACS Appl. Mater. Interfaces 2017, 9, 5181–5192. [Google Scholar] [CrossRef]
- Noyhouzer, T.; L’Homme, C.; Beaulieu, I.; Mazurkiewicz, S.; Kuss, S.; Kraatz, H.-B.; Canesi, S.; Mauzeroll, J. Ferrocene-modified phospholipid: An innovative precursor for redox-triggered drug delivery vesicles selective to cancer cells. Langmuir 2016, 32, 4169–4178. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Jiao, X.; Xu, T.; Wang, W.; Cao, Y.; Wen, Y.; Zhang, X. Free-blockage mesoporous anticancer nanoparticles based on ROS-responsive wetting behavior of nanopores. Small 2017, 13, 1701942. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Luo, C.; Zhang, X.; Guo, M.; Sun, M.; Yu, H.; Chen, Q.; Yang, W.; Wang, M.; Zuo, S.; et al. Probing the impact of sulfur/selenium/carbon linkages on prodrug nanoassemblies for cancer therapy. Nat. Commun. 2019, 10, 3211. [Google Scholar] [CrossRef]
- Ren, H.; Wu, Y.; Ma, N.; Xu, H.; Zhang, X. Side-chain selenium-containing amphiphilic block copolymers: Redox-controlled self-assembly and disassembly. Soft Matter 2012, 8, 1460–1466. [Google Scholar] [CrossRef]
- Kang, Y.; Ju, X.; Ding, L.-S.; Zhang, S.; Li, B.-J. Reactive oxygen species and glutathione dual redox-responsive supramolecular assemblies with controllable release capability. ACS Appl. Mater. Interfaces 2017, 9, 4475–4484. [Google Scholar] [CrossRef] [PubMed]
- Candiani, G.; Pezzoli, D.; Ciani, L.; Chiesa, R.; Ristori, S. Bioreducible liposomes for gene delivery: From the formulation to the mechanism of action. PLoS ONE 2010, 5, e13430. [Google Scholar] [CrossRef] [Green Version]
- Giménez, C.; de la Torre, C.; Gorbe, M.; Aznar, E.; Sancenón, F.; Murguía, J.R.; Martínez-Máñez, R.; Marcos, M.D.; Amorós, P. Gated mesoporous silica nanoparticles for the controlled delivery of drugs in cancer cells. Langmuir 2015, 31, 3753–3762. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Wang, C.; Liu, Y.; Wang, J.; Gao, Y.; Zhang, X.; Jiang, T.; Wang, S. PEGylated mesoporous silica as a redox-responsive drug delivery system for loading thiol-containing drugs. Int. J. Pharm. 2014, 477, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Shen, N.; Lei, B.; Wang, Y.; Xu, S.; Liu, H. Redox/ultrasound dual stimuli-responsive nanogel for precisely controllable drug release. New J. Chem. 2018, 42, 9472–9481. [Google Scholar] [CrossRef]
- Ma, N.; Li, Y.; Xu, H.; Wang, Z.; Zhang, X. Dual redox responsive assemblies formed from diselenide block copolymers. J. Am. Chem. Soc. 2010, 132, 442–443. [Google Scholar] [CrossRef]
- Tian, Y.; Lei, M.; Yan, L.; An, F. Diselenide-crosslinked zwitterionic nanogels with dual redox-labile properties for controlled drug release. Polym. Chem. 2020, 11, 2360–2369. [Google Scholar] [CrossRef]
- Pang, Z.; Zhou, J.; Sun, C. Ditelluride-bridged PEG-PCL copolymer as folic acid-targeted and redox-responsive nanoparticles for enhanced cancer therapy. Front. Chem. 2020, 8. [Google Scholar] [CrossRef] [Green Version]
- Gupta, M.K.; Lee, S.H.; Crowder, S.W.; Wang, X.; Hofmeister, L.H.; Nelson, C.E.; Bellan, L.M.; Duvall, C.L.; Sung, H.-J. Oligoproline-derived nanocarrier for dual stimuli-responsive gene delivery. J. Mater. Chem. B 2015, 3, 7271–7280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Ke, W.; Wang, L.; Huang, M.; Yin, W.; Zhang, P.; Chen, Q.; Ge, Z. Self-sufficing H2O2-responsive nanocarriers through tumor-specific H2O2 production for synergistic oxidation-chemotherapy. J. Control. Release 2016, 225, 64–74. [Google Scholar] [CrossRef]
- Kildahl, N.K. Bond energy data summarized. J.Chem. Educ. 1995, 72, 423. [Google Scholar] [CrossRef]
- Stadtman, E.R.; Levine, R.L. Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids 2003, 25, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Han, Y.; Tang, J.; Piao, Y.; Liu, X.; Zhou, Z.; Gao, J.; Rao, J.; Shen, Y. A tumor-specific cascade amplification drug Release nanoparticle for overcoming multidrug resistance in cancers. Adv. Mater. 2017, 29, 1702342. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Huang, X.; Moore, Z.; Huang, G.; Kilgore, J.A.; Wang, Y.; Hammer, S.; Williams, N.S.; Boothman, D.A.; Gao, J. Esterase-activatable β-lapachone prodrug micelles for NQO1-targeted lung cancer therapy. J. Control. Release 2015, 200, 201–211. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Pang, Y.; Zhu, Z.; Wang, D.; Li, C.; Huang, W.; Zhu, X.; Yan, D. Therapeutic nanocarriers with hydrogen peroxide-triggered drug release for cancer treatment. Biomacromolecules 2013, 14, 1627–1636. [Google Scholar] [CrossRef]
- Ghosh, J. Rapid induction of apoptosis in prostate cancer cells by selenium: Reversal by metabolites of arachidonate 5-lipoxygenase. Biochem. Biophys. Res. Commun. 2004, 315, 624–635. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhao, Y. Controlled release from cleavable polymerized liposomes upon redox and pH stimulation. Bioconjug. Chem. 2011, 22, 523–528. [Google Scholar] [CrossRef] [Green Version]
- Colonna, S.; Gaggero, N.; Carrea, G.; Pasta, P. Enantio and diastereoselectivity of cyclohexanone monooxygenase catalyzed oxidation of 1,3-dithioacetals. Tetrahedron Asymmetry 1996, 7, 565–570. [Google Scholar] [CrossRef]
- Shukla, A.K.; Verma, M.; Singh, K.N. Superoxide induced deprotection of 1, 3-dithiolanes: A convenient method of dedithioacetalization. Indian J. Chem. B 2004, 43B, 1748–1752. [Google Scholar] [CrossRef]
- Ruoslahti, E. Peptides as targeting elements and tissue penetration devices for nanoparticles. Adv. Mater. 2012, 24, 3747–3756. [Google Scholar] [CrossRef]
- Li, X.; Lachmanski, L.; Safi, S.; Sene, S.; Serre, C.; Grenèche, J.M.; Zhang, J.; Gref, R. New insights into the degradation mechanism of metal-organic frameworks drug carriers. Sci. Rep. 2017, 7, 13142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Q.; He, C.; Xiao, C.; Chen, X. Reactive oxygen species (ROS) responsive polymers for biomedical applications. Macromol. Biosci. 2016, 16, 635–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Aspects Med. 2009, 30, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Nagy, P. Kinetics and mechanisms of thiol-disulfide exchange covering direct substitution and thiol oxidation-mediated pathways. Antioxid. Redox. Sign. 2013, 18, 1623–1641. [Google Scholar] [CrossRef] [Green Version]
- Austin, C.D.; Wen, X.; Gazzard, L.; Nelson, C.; Scheller, R.H.; Scales, S.J. Oxidizing potential of endosomes and lysosomes limits intracellular cleavage of disulfide-based antibody–drug conjugates. Proc. Natl. Acad. Sci. USA 2005, 102, 17987–17992. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Y.-J.; Shao, L.-H.; Li, Y. Cathepsin B-cleavable doxorubicin prodrugs for targeted cancer therapy (Review). Int. J. Oncol. 2013, 42, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.-E.; Ho, C.-C.; Yang, S.-F.; Lin, S.-H.; Yeh, K.-T.; Lin, C.-W.; Chen, M.-K. Cathepsin B expression and the correlation with clinical aspects of oral squamous cell carcinoma. PLoS ONE 2016, 11, e0152165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, F.; Peng, X.; Luo, C.; Shen, G.; Zhao, C.; Zou, L.; Li, L.; Sang, Y.; Zhao, Y.; Zhao, X. Cathepsin B as a potential prognostic and therapeutic marker for human lung squamous cell carcinoma. Mol. Cancer 2013, 12, 125. [Google Scholar] [CrossRef] [Green Version]
- Miller, K.; Erez, R.; Segal, E.; Shabat, D.; Satchi-Fainaro, R. Targeting bone metastases with a bispecific anticancer and antiangiogenic polymer–alendronate–taxane conjugate. Angew. Chem. Int. Ed. 2009, 48, 2949–2954. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Zhang, F.; Zhao, C.; Lv, Y.; Ma, G.; Wei, W.; Tian, Z. Beyond a carrier: Graphene quantum dots as a probe for programmatically monitoring anti-cancer drug delivery, release, and response. ACS Appl. Mater. Interfaces 2017, 9, 27396–27401. [Google Scholar] [CrossRef] [PubMed]
- Stern, R.; Jedrzejas, M.J. Hyaluronidases: Their genomics, structures, and mechanisms of action. Chem. Rev. 2006, 106, 818–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendoza, M.F.; Hollabaugh, N.M.; Hettiarachchi, S.U.; McCarley, R.L. Human NAD(P)H:Quinone oxidoreductase type I (hNQO1) activation of quinone propionic acid trigger groups. Biochemistry 2012, 51, 8014–8026. [Google Scholar] [CrossRef] [Green Version]
- Bae, J.; Nael, M.A.; Jiang, L.; Hwang, P.T.; Mahdi, F.; Jun, H.-W.; Elshamy, W.M.; Zhou, Y.-D.; Murthy, S.N.; Doerksen, R.J.; et al. Quinone propionic acid-based redox-triggered polymer nanoparticles for drug delivery: Computational analysis and in vitro evaluation. J. Appl. Polym. Sci. 2014, 131. [Google Scholar] [CrossRef]
- Lee, J.; Oh, E.-T.; Yoon, H.; Woo Kim, C.; Han, Y.; Song, J.; Jang, H.; Joo Park, H.; Kim, C. Mesoporous nanocarriers with a stimulus-responsive cyclodextrin gatekeeper for targeting tumor hypoxia. Nanoscale 2017, 9, 6901–6909. [Google Scholar] [CrossRef]
- Asanuma, D.; Sakabe, M.; Kamiya, M.; Yamamoto, K.; Hiratake, J.; Ogawa, M.; Kosaka, N.; Choyke, P.L.; Nagano, T.; Kobayashi, H.; et al. Sensitive β-galactosidase-targeting fluorescence probe for visualizing small peritoneal metastatic tumours in vivo. Nat. Commun. 2015, 6, 6463. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.-J.; Kumar, R.; Sharma, A.; Yoon, B.; Kim, H.M.; Lee, H.; Hong, K.S.; Kim, J.S. In vivo imaging of β-galactosidase stimulated activity in hepatocellular carcinoma using ligand-targeted fluorescent probe. Biomaterials 2017, 122, 83–90. [Google Scholar] [CrossRef]
- Gu, W.-X.; Li, Q.-L.; Lu, H.; Fang, L.; Chen, Q.; Yang, Y.-W.; Gao, H. Construction of stable polymeric vesicles based on azobenzene and beta-cyclodextrin grafted poly(glycerol methacrylate)s for potential applications in colon-specific drug delivery. Chem. Commun. 2015, 51, 4715–4718. [Google Scholar] [CrossRef]
- Rao, J.; Hottinger, C.; Khan, A. Enzyme-triggered cascade reactions and assembly of abiotic block copolymers into micellar nanostructures. J. Am. Chem. Soc. 2014, 136, 5872–5875. [Google Scholar] [CrossRef]
- Wang, C.; Chen, Q.; Wang, Z.; Zhang, X. An enzyme-responsive polymeric superamphiphile. Angew. Chem. Int. Ed. 2010, 49, 8612–8615. [Google Scholar] [CrossRef]
- Hu, J.; He, J.; Cao, D.; Zhang, M.; Ni, P. Core cross-linked polyphosphoester micelles with folate-targeted and acid-cleavable features for pH-triggered drug delivery. Polym. Chem. 2015, 6, 3205–3216. [Google Scholar] [CrossRef]
- Li, J.; Ge, Z.; Liu, S. PEG-sheddable polyplex micelles as smart gene carriers based on MMP-cleavable peptide-linked block copolymers. Chem. Commun. 2013, 49, 6974–6976. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, B.; Luo, Z.; Ding, X.; Li, J.; Dai, L.; Zhou, J.; Zhao, X.; Ye, J.; Cai, K. Enzyme responsive mesoporous silica nanoparticles for targeted tumor therapy in vitro and in vivo. Nanoscale 2015, 7, 3614–3626. [Google Scholar] [CrossRef]
- Li, J.; Liu, F.; Shao, Q.; Min, Y.; Costa, M.; Yeow, E.K.L.; Xing, B. Enzyme-responsive cell-penetrating peptide conjugated mesoporous silica quantum dot nanocarriers for controlled release of nucleus-targeted drug molecules and real-time intracellular fluorescence imaging of tumor cells. Adv. Helathcare Mater. 2014, 3, 1230–1239. [Google Scholar] [CrossRef] [PubMed]
- Satchi, R.; Connors, T.A.; Duncan, R. PDEPT: Polymer-directed enzyme prodrug therapy. Br. J. Cancer 2001, 85, 1070–1076. [Google Scholar] [CrossRef]
- Duan, Z.; Zhang, Y.; Zhu, H.; Sun, L.; Cai, H.; Li, B.; Gong, Q.; Gu, Z.; Luo, K. Stimuli-sensitive biodegradable and amphiphilic block copolymer-gemcitabine conjugates self-assemble into a nanoscale vehicle for cancer therapy. ACS Appl. Mater. Interfaces 2017, 9, 3474–3486. [Google Scholar] [CrossRef] [PubMed]
- Shim, M.K.; Park, J.; Yoon, H.Y.; Lee, S.; Um, W.; Kim, J.-H.; Kang, S.-W.; Seo, J.-W.; Hyun, S.-W.; Park, J.H.; et al. Carrier-free nanoparticles of cathepsin B-cleavable peptide-conjugated doxorubicin prodrug for cancer targeting therapy. J. Control. Release 2019, 294, 376–389. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.Y.; Min, K.H.; Yoon, H.Y.; Kim, K.; Park, J.H.; Kwon, I.C.; Choi, K.; Jeong, S.Y. PEGylation of hyaluronic acid nanoparticles improves tumor targetability in vivo. Biomaterials 2011, 32, 1880–1889. [Google Scholar] [CrossRef]
- Choi, K.Y.; Yoon, H.Y.; Kim, J.-H.; Bae, S.M.; Park, R.-W.; Kang, Y.M.; Kim, I.-S.; Kwon, I.C.; Choi, K.; Jeong, S.Y.; et al. Smart nanocarrier based on PEGylated hyaluronic acid for cancer therapy. ACS Nano 2011, 5, 8591–8599. [Google Scholar] [CrossRef]
- Kim, K.; Lee, S.; Jin, E.; Palanikumar, L.; Lee, J.H.; Kim, J.C.; Nam, J.S.; Jana, B.; Kwon, T.-H.; Kwak, S.K.; et al. MOF × biopolymer: Collaborative combination of metal–organic framework and biopolymer for advanced anticancer therapy. ACS Appl. Mater. Interfaces 2019, 11, 27512–27520. [Google Scholar] [CrossRef]
- Ong, W.; Yang, Y.; Cruciano, A.C.; McCarley, R.L. Redox-triggered contents release from liposomes. J. Am. Chem. Soc. 2008, 130, 14739–14744. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Li, T.; Han, F.; Wang, Y.; Gan, Y.; Shi, J.; Wang, T.; Akhtar, M.L.; Li, Y. A cascade-reaction enabled synergistic cancer starvation/ROS-mediated/chemo-therapy with an enzyme modified Fe-based MOF. Biomater. Sci. 2019, 7, 3683–3692. [Google Scholar] [CrossRef]
- Gu, Z.; Aimetti, A.A.; Wang, Q.; Dang, T.T.; Zhang, Y.; Veiseh, O.; Cheng, H.; Langer, R.S.; Anderson, D.G. Injectable nano-network for glucose-mediated insulin delivery. ACS Nano 2013, 7, 4194–4201. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Xu, D.; Pan, X.; Ma, X. Self-propelled enzymatic nanomotors for enhancing synergetic photodynamic and starvation therapy by self-accelerated cascade reactions. Appl. Mater. Today 2019, 16, 508–517. [Google Scholar] [CrossRef]
- Fu, L.-H.; Qi, C.; Lin, J.; Huang, P. Catalytic chemistry of glucose oxidase in cancer diagnosis and treatment. Chem. Soc. Rev. 2018, 47, 6454–6472. [Google Scholar] [CrossRef]
- Samarajeewa, S.; Shrestha, R.; Li, Y.; Wooley, K.L. Degradability of poly(lactic acid)-containing nanoparticles: Enzymatic access through a cross-linked shell barrier. J. Am. Chem. Soc. 2012, 134, 1235–1242. [Google Scholar] [CrossRef] [Green Version]
- Samarajeewa, S.; Zentay, R.P.; Jhurry, N.D.; Li, A.; Seetho, K.; Zou, J.; Wooley, K.L. Programmed hydrolysis of nanoassemblies by electrostatic interaction-mediated enzymatic-degradation. Chem. Commun. 2014, 50, 968–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harnoy, A.J.; Rosenbaum, I.; Tirosh, E.; Ebenstein, Y.; Shaharabani, R.; Beck, R.; Amir, R.J. Enzyme-responsive amphiphilic PEG-dendron hybrids and their assembly into smart micellar nanocarriers. J. Am. Chem. Soc. 2014, 136, 7531–7534. [Google Scholar] [CrossRef]
- Vadas, P.; Browning, J.; Edelson, J.; Pruzanski, W. Extracellular phospholipase A2 expression and inflammation: The relationship with associated disease states. J. Lipid Mediat. 1993, 8, 1–30. [Google Scholar] [PubMed]
- Pak, C.C.; Ali, S.; Janoff, A.S.; Meers, P. Triggerable liposomal fusion by enzyme cleavage of a novel peptide–lipid conjugate. Biochim. Biophys. Acta Biomembr. 1998, 1372, 13–27. [Google Scholar] [CrossRef] [Green Version]
- Toole, B.P. Hyaluronan: From extracellular glue to pericellular cue. Nat. Rev. Cancer 2004, 4, 528–539. [Google Scholar] [CrossRef]
- Birkedal-Hansen, H.; Moore, W.G.I.; Bodden, M.K.; Windsor, L.J.; Birkedal-Hansen, B.; DeCarlo, A.; Engler, J.A. Matrix metalloproteinases: A Review. Crit. Rev. Oral Biol. Med. 1993, 4, 197–250. [Google Scholar] [CrossRef] [Green Version]
- Duarte, S.; Baber, J.; Fujii, T.; Coito, A.J. Matrix metalloproteinases in liver injury, repair and fibrosis. Matrix Biol. 2015, 44–46, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Paik, W.K.; Kim, S. Comparison of the acetylation of proteins and nucleic acids. Biochem. J. 1970, 116, 611–615. [Google Scholar] [CrossRef] [Green Version]
- Manchanda, M.; Das, P.; Gahlot, G.P.S.; Singh, R.; Roeb, E.; Roderfeld, M.; Datta Gupta, S.; Saraya, A.; Pandey, R.M.; Chauhan, S.S. Cathepsin L and B as potential markers for liver fibrosis: Insights from patients and experimental models. Clin. Transl. Gastroenterol. 2017, 8, e99. [Google Scholar] [CrossRef] [PubMed]
- Wust, P.; Hildebrandt, B.; Sreenivasa, G.; Rau, B.; Gellermann, J.; Riess, H.; Felix, R.; Schlag, P.M. Hyperthermia in combined treatment of cancer. Lancet Oncol. 2002, 3, 487–497. [Google Scholar] [CrossRef]
- Zhao, Y.; Fan, X.; Liu, D.; Wang, Z. PEGylated thermo-sensitive poly(amidoamine) dendritic drug delivery systems. Int. J. Pharm. 2011, 409, 229–236. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, H.; Wang, J.; Ge, L.; Zhu, J. Development of a thermally responsive nanogel based on chitosan–poly(N-isopropylacrylamide-co-acrylamide) for paclitaxel delivery. J. Pharm. Sci. 2014, 103, 2012–2021. [Google Scholar] [CrossRef] [PubMed]
- You, Y.-Z.; Kalebaila, K.K.; Brock, S.L.; Oupický, D. Temperature-controlled uptake and release in PNIPAM-modified porous silica nanoparticles. Chem. Mater. 2008, 20, 3354–3359. [Google Scholar] [CrossRef]
- Nagata, S.; Kokado, K.; Sada, K. Metal–organic framework tethering PNIPAM for ON–OFF controlled release in solution. Chem. Commun. 2015, 51, 8614–8617. [Google Scholar] [CrossRef] [PubMed]
- Brunella, V.; Jadhav, S.A.; Miletto, I.; Berlier, G.; Ugazio, E.; Sapino, S.; Scalarone, D. Hybrid drug carriers with temperature-controlled on–off release: A simple and reliable synthesis of PNIPAM-functionalized mesoporous silica nanoparticles. React. Funct. Polym. 2016, 98, 31–37. [Google Scholar] [CrossRef]
- Aoki, I.; Yoneyama, M.; Hirose, J.; Minemoto, Y.; Koyama, T.; Kokuryo, D.; Bakalova, R.; Murayama, S.; Saga, T.; Aoshima, S.; et al. Thermoactivatable polymer-grafted liposomes for low-invasive image-guided chemotherapy. Transl. Res. 2015, 166, 660–673.e661. [Google Scholar] [CrossRef] [PubMed]
- Kono, K.; Ozawa, T.; Yoshida, T.; Ozaki, F.; Ishizaka, Y.; Maruyama, K.; Kojima, C.; Harada, A.; Aoshima, S. Highly temperature-sensitive liposomes based on a thermosensitive block copolymer for tumor-specific chemotherapy. Biomaterials 2010, 31, 7096–7105. [Google Scholar] [CrossRef]
- Liu, J.; Detrembleur, C.; De Pauw-Gillet, M.-C.; Mornet, S.; Duguet, E.; Jérôme, C. Gold nanorods coated with a thermo-responsive poly(ethylene glycol)-b-poly(N-vinylcaprolactam) corona as drug delivery systems for remotely near infrared-triggered release. Polym. Chem. 2014, 5, 799–813. [Google Scholar] [CrossRef] [Green Version]
- Huang, G.; Li, H.; Feng, S.-T.; Li, X.; Tong, G.; Liu, J.; Quan, C.; Jiang, Q.; Zhang, C.; Li, Z. Self-assembled UCST-type micelles as potential drug carriers for cancer therapeutics. Macromol. Chem. Phys. 2015, 216, 1014–1023. [Google Scholar] [CrossRef]
- Li, W.; Huang, L.; Ying, X.; Jian, Y.; Hong, Y.; Hu, F.; Du, Y. Antitumor drug delivery modulated by a polymeric micelle with an upper critical solution temperature. Angew. Chem., Int. Ed. 2015, 54, 3126–3131. [Google Scholar] [CrossRef] [PubMed]
- Hei, M.; Wang, J.; Wang, K.; Zhu, W.; Ma, P.X. Dually responsive mesoporous silica nanoparticles regulated by upper critical solution temperature polymers for intracellular drug delivery. J. Mater. Chem. B 2017, 5, 9497–9501. [Google Scholar] [CrossRef]
- Wu, L.; Zong, L.; Ni, H.; Liu, X.; Wen, W.; Feng, L.; Cao, J.; Qi, X.; Ge, Y.; Shen, S. Magnetic thermosensitive micelles with upper critical solution temperature for NIR triggered drug release. Biomater. Sci. 2019, 7, 2134–2143. [Google Scholar] [CrossRef]
- Fujihara, A.; Shimada, N.; Maruyama, A.; Ishihara, K.; Nakai, K.; Yusa, S.-i. Preparation of upper critical solution temperature (UCST) responsive diblock copolymers bearing pendant ureido groups and their micelle formation behavior in water. Soft Matter 2015, 11, 5204–5213. [Google Scholar] [CrossRef]
- Kojima, C.; Tsumura, S.; Harada, A.; Kono, K. A collagen-mimic dendrimer capable of controlled release. J. Am. Chem. Soc. 2009, 131, 6052–6053. [Google Scholar] [CrossRef]
- Park, S.M.; Kim, M.S.; Park, S.-J.; Park, E.S.; Choi, K.-S.; Kim, Y.-S.; Kim, H.R. Novel temperature-triggered liposome with high stability: Formulation, in vitro evaluation, and in vivo study combined with high-intensity focused ultrasound (HIFU). J. Control. Release 2013, 170, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, S.V.; Singh, S.K.; Reja, R.M.; Gopi, H.N. γ-Amino acid mutated α-coiled coils as mild thermal triggers for liposome delivery. Chem. Commun. 2013, 49, 11065–11067. [Google Scholar] [CrossRef]
- Bothun, G.D. Hydrophobic silver nanoparticles trapped in lipid bilayers: Size distribution, bilayer phase behavior, and optical properties. J. Nanobiotechnol. 2008, 6, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, J.; Zhang, P.; Hu, F.; Du, Y.; Yuan, H.; Zhu, J.; Wang, Z.; Zhou, J.; Li, C. Near-infrared light-sensitive liposomes for the enhanced photothermal tumor treatment by the combination with chemotherapy. Pharm. Res. 2014, 31, 554–565. [Google Scholar] [CrossRef]
- Béalle, G.; Di Corato, R.; Kolosnjaj-Tabi, J.; Dupuis, V.; Clément, O.; Gazeau, F.; Wilhelm, C.; Ménager, C. Ultra magnetic liposomes for MR imaging, targeting, and hyperthermia. Langmuir 2012, 28, 11834–11842. [Google Scholar] [CrossRef]
- Qiu, D.; An, X.; Chen, Z.; Ma, X. Microstructure study of liposomes decorated by hydrophobic magnetic nanoparticles. Chem. Phys. Lipids 2012, 165, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Needham, D.; Park, J.-Y.; Wright, A.M.; Tong, J. Materials characterization of the low temperature sensitive liposome (LTSL): Effects of the lipid composition (lysolipid and DSPE–PEG2000) on the thermal transition and release of doxorubicin. Faraday Discuss. 2013, 161, 515–534. [Google Scholar] [CrossRef] [PubMed]
- Banno, B.; Ickenstein, L.M.; Chiu, G.N.C.; Bally, M.B.; Thewalt, J.; Brief, E.; Wasan, E.K. The functional roles of poly(ethylene glycol)-lipid and lysolipid in the drug retention and release from lysolipid-containing thermosensitive liposomes in vitro and in vivo. J. Pharm. Sci. 2010, 99, 2295–2308. [Google Scholar] [CrossRef] [PubMed]
- Wood, B.J.; Poon, R.T.; Locklin, J.K.; Dreher, M.R.; Ng, K.K.; Eugeni, M.; Seidel, G.; Dromi, S.; Neeman, Z.; Kolf, M.; et al. Phase I study of heat-deployed liposomal doxorubicin during radiofrequency ablation for hepatic malignancies. J. Vasc. Interv. Radiol. 2012, 23, 248–255.e247. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.-J.; Liang, H.-F.; Chen, H.-L.; Wang, Y.; Cheng, P.-Y.; Liu, H.-L.; Xia, Y.; Sung, H.-W. A thermoresponsive bubble-generating liposomal system for triggering localized extracellular drug delivery. ACS Nano 2013, 7, 438–446. [Google Scholar] [CrossRef]
- Chen, K.-J.; Chaung, E.-Y.; Wey, S.-P.; Lin, K.-J.; Cheng, F.; Lin, C.-C.; Liu, H.-L.; Tseng, H.-W.; Liu, C.-P.; Wei, M.-C.; et al. Hyperthermia-mediated local drug delivery by a bubble-generating liposomal system for tumor-specific chemotherapy. ACS Nano 2014, 8, 5105–5115. [Google Scholar] [CrossRef] [PubMed]
- Schlossbauer, A.; Warncke, S.; Gramlich, P.M.E.; Kecht, J.; Manetto, A.; Carell, T.; Bein, T. A programmable DNA-based molecular valve for colloidal mesoporous silica. Angew. Chem. Int. Ed. 2010, 49, 4734–4737. [Google Scholar] [CrossRef]
- Chen, C.; Geng, J.; Pu, F.; Yang, X.; Ren, J.; Qu, X. Polyvalent nucleic acid/mesoporous silica nanoparticle conjugates: Dual stimuli-responsive vehicles for intracellular drug delivery. Angew. Chem. Int. Ed. 2011, 50, 882–886. [Google Scholar] [CrossRef]
- Chang, Y.-T.; Liao, P.-Y.; Sheu, H.-S.; Tseng, Y.-J.; Cheng, F.-Y.; Yeh, C.-S. Near-infrared light-responsive intracellular drug and siRNA release using Au nanoensembles with oligonucleotide-capped silica shell. Adv. Mater. 2012, 24, 3309–3314. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-H.; Yang, C.-X.; Qiu, W.-X.; Luo, G.-F.; Jia, H.-Z.; Lei, Q.; Wang, X.-Y.; Liu, G.; Zhuo, R.-X.; Zhang, X.-Z. Multifunctional theranostic nanoplatform for cancer combined therapy based on gold nanorods. Adv. Helathcare Mater. 2015, 4, 2247–2259. [Google Scholar] [CrossRef]
- Riedinger, A.; Guardia, P.; Curcio, A.; Garcia, M.A.; Cingolani, R.; Manna, L.; Pellegrino, T. Subnanometer local temperature probing and remotely controlled drug release based on azo-functionalized iron oxide nanoparticles. Nano Lett. 2013, 13, 2399–2406. [Google Scholar] [CrossRef]
- Jiang, K.; Zhang, L.; Hu, Q.; Zhang, Q.; Lin, W.; Cui, Y.; Yang, Y.; Qian, G. Thermal stimuli-triggered drug release from a biocompatible porous metal–organic framework. Chem. Er. J. 2017, 23, 10215–10221. [Google Scholar] [CrossRef] [PubMed]
- Lanzalaco, S.; Armelin, E. Poly(N-isopropylacrylamide) and copolymers: A review on recent progresses in biomedical applications. Gels 2017, 3, 36. [Google Scholar] [CrossRef]
- Cortez-Lemus, N.A.; Licea-Claverie, A. Poly(N-vinylcaprolactam), a comprehensive review on a thermoresponsive polymer becoming popular. Prog. Polym. Sci. 2016, 53, 1–51. [Google Scholar] [CrossRef]
- Badi, N. Non-linear PEG-based thermoresponsive polymer systems. Prog. Polym. Sci. 2017, 66, 54–79. [Google Scholar] [CrossRef]
- Bodratti, A.M.; Alexandridis, P. Formulation of poloxamers for drug delivery. J. Funt. Biomater. 2018, 9, 11. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.; Hoffman, A.S.; Stayton, P.S. Poly(N-isopropylacrylamide-co-propylacrylic acid) copolymers that respond sharply to temperature and pH. Biomacromolecules 2006, 7, 1381–1385. [Google Scholar] [CrossRef]
- Bordat, A.; Boissenot, T.; Nicolas, J.; Tsapis, N. Thermoresponsive polymer nanocarriers for biomedical applications. Adv. Drug Deliv. Rev. 2019, 138, 167–192. [Google Scholar] [CrossRef] [PubMed]
- Sponchioni, M.; Capasso Palmiero, U.; Moscatelli, D. Thermo-responsive polymers: Applications of smart materials in drug delivery and tissue engineering. Mater. Sci. Eng. C 2019, 102, 589–605. [Google Scholar] [CrossRef]
- Andrew Mackay, J.; Chilkoti, A. Temperature sensitive peptides: Engineering hyperthermia-directed therapeutics. Int. J. Hyperthermia 2008, 24, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Na, K.; Lee, S.A.; Jung, S.H.; Hyun, J.; Shin, B.C. Elastin-like polypeptide modified liposomes for enhancing cellular uptake into tumor cells. Colloids Surf. B 2012, 91, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Hossann, M.; Syunyaeva, Z.; Schmidt, R.; Zengerle, A.; Eibl, H.; Issels, R.D.; Lindner, L.H. Proteins and cholesterol lipid vesicles are mediators of drug release from thermosensitive liposomes. J. Control. Release 2012, 162, 400–406. [Google Scholar] [CrossRef]
- De Smet, M.; Langereis, S.; den Bosch, S.v.; Grüll, H. Temperature-sensitive liposomes for doxorubicin delivery under MRI guidance. J. Control. Release 2010, 143, 120–127. [Google Scholar] [CrossRef]
- Ales, P.; Jeffrey M, D. Chapter thirty-five-Gene delivery into cells and tissues. In Principles of Tissue Engineering, 3rd ed.; Lanza, R., Langer, R., Vacanti, J., Eds.; Academic Press: Burlington, ON, Canada, 2007; pp. 493–515. [Google Scholar]
- Mailänder, V.; Landfester, K. Interaction of nanoparticles with cells. Biomacromolecules 2009, 10, 2379–2400. [Google Scholar] [CrossRef]
- Souris, J.S.; Lee, C.-H.; Cheng, S.-H.; Chen, C.-T.; Yang, C.-S.; Ho, J.-a.A.; Mou, C.-Y.; Lo, L.-W. Surface charge-mediated rapid hepatobiliary excretion of mesoporous silica nanoparticles. Biomaterials 2010, 31, 5564–5574. [Google Scholar] [CrossRef] [Green Version]
- Brigham, K. Estimations of permeability properties of pulmonary capillaries (continuous endothelium). Physiologist 1980, 23, 44–46. [Google Scholar]
- Wang, H.; Yang, X.; Shao, W.; Chen, S.; Xie, J.; Zhang, X.; Wang, J.; Xie, Y. Ultrathin black phosphorus nanosheets for efficient singlet oxygen generation. J. Am. Chem. Soc. 2015, 137, 11376–11382. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Chen, G.Y.; Owens, G.; Chu, D.; Xu, H. Photothermal materials: A key platform enabling highly efficient water evaporation driven by solar energy. Mater. Today Energy 2019, 12, 277–296. [Google Scholar] [CrossRef]
- Polenz, I.; Laue, A.; Uhrin, T.; Rüffer, T.; Lang, H.; Schmidt, F.G.; Spange, S. Thermally cleavable imine base/isocyanate adducts and oligomers suitable as initiators for radical homo- and copolymerization. Polym. Chem. 2014, 5, 6678–6686. [Google Scholar] [CrossRef] [Green Version]
- Poon, L.; Zandberg, W.; Hsiao, D.; Erno, Z.; Sen, D.; Gates, B.D.; Branda, N.R. Photothermal release of single-stranded DNA from the surface of gold nanoparticles through controlled denaturating and Au−S bond breaking. ACS Nano 2010, 4, 6395–6403. [Google Scholar] [CrossRef]
- Michal, T.; Asaf, S.; Oshrit, H.; Hila, C.; Michael, S.; Itzhak, K.; Yona, K.; Israel, G. Thermographic investigation of tumor size, and its correlation to tumor relative temperature, in mice with transplantable solid breast carcinoma. J. Biomed. Opt. 2013, 18, 1–11. [Google Scholar] [CrossRef]
- Mohammed, L.; Gomaa, H.G.; Ragab, D.; Zhu, J. Magnetic nanoparticles for environmental and biomedical applications: A review. Particuology 2017, 30, 1–14. [Google Scholar] [CrossRef]
- Yang, D.; Yang, G.; Gai, S.; He, F.; An, G.; Dai, Y.; Lv, R.; Yang, P. Au25 cluster functionalized metal–organic nanostructures for magnetically targeted photodynamic/photothermal therapy triggered by single wavelength 808 nm near-infrared light. Nanoscale 2015, 7, 19568–19578. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, P.; Giri, J.; Rieken, F.; Koch, C.; Mykhaylyk, O.; Döblinger, M.; Banerjee, R.; Bahadur, D.; Plank, C. Targeted temperature sensitive magnetic liposomes for thermo-chemotherapy. J. Control. Release 2010, 142, 108–121. [Google Scholar] [CrossRef]
- Dobson, J. Magnetic nanoparticles for drug delivery. Drug Dev. Res. 2006, 67, 55–60. [Google Scholar] [CrossRef]
- Hu, S.-H.; Liao, B.-J.; Chiang, C.-S.; Chen, P.-J.; Chen, I.W.; Chen, S.-Y. Core-shell nanocapsules stabilized by single-component polymer and nanoparticles for magneto-chemotherapy/hyperthermia with multiple drugs. Adv. Mater. 2012, 24, 3627–3632. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-Y.; Hu, S.-H.; Chian, C.-S.; Chen, S.-Y.; Lai, H.-Y.; Chen, Y.-Y. Self-assembling PVA-F127 thermosensitive nanocarriers with highly sensitive magnetically-triggered drug release for epilepsy therapy in vivo. J. Mater. Chem. 2012, 22, 8566–8573. [Google Scholar] [CrossRef]
- Qiu, D.; An, X. Controllable release from magnetoliposomes by magnetic stimulation and thermal stimulation. Colloids Surf. B 2013, 104, 326–329. [Google Scholar] [CrossRef]
- Katagiri, K.; Imai, Y.; Koumoto, K.; Kaiden, T.; Kono, K.; Aoshima, S. Magnetoresponsive on-demand release of hybrid liposomes formed from Fe3O4 nanoparticles and thermosensitive block copolymers. Small 2011, 7, 1683–1689. [Google Scholar] [CrossRef]
- Hu, S.-H.; Chen, S.-Y.; Gao, X. Multifunctional nanocapsules for simultaneous encapsulation of hydrophilic and hydrophobic compounds and on-demand release. ACS Nano 2012, 6, 2558–2565. [Google Scholar] [CrossRef]
- Cazares-Cortes, E.; Espinosa, A.; Guigner, J.-M.; Michel, A.; Griffete, N.; Wilhelm, C.; Ménager, C. Doxorubicin intracellular remote release from biocompatible oligo(ethylene glycol) methyl ether methacrylate-based magnetic nanogels triggered by magnetic hyperthermia. ACS Appl. Mater. Interfaces 2017, 9, 25775–25788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cazares-Cortes, E.; Nerantzaki, M.; Fresnais, J.; Wilhelm, C.; Griffete, N.; Ménager, C. Magnetic nanoparticles create hot spots in polymer matrix for controlled drug release. Nanomaterials 2018, 8, 850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Hernández, E.; Baeza, A.; Vallet-Regí, M. Smart drug delivery through DNA/magnetic nanoparticle gates. ACS Nano 2011, 5, 1259–1266. [Google Scholar] [CrossRef]
- Hu, S.-H.; Liu, D.-M.; Tung, W.-L.; Liao, C.-F.; Chen, S.-Y. Surfactant-free, self-assembled PVA-iron oxide/silica core–shell nanocarriers for highly sensitive, magnetically controlled drug release and ultrahigh cancer cell uptake efficiency. Adv. Funct. Mater. 2008, 18, 2946–2955. [Google Scholar] [CrossRef]
- Thomas, C.R.; Ferris, D.P.; Lee, J.-H.; Choi, E.; Cho, M.H.; Kim, E.S.; Stoddart, J.F.; Shin, J.-S.; Cheon, J.; Zink, J.I. Noninvasive remote-controlled release of drug molecules in vitro using magnetic actuation of mechanized nanoparticles. J. Am. Chem. Soc. 2010, 132, 10623–10625. [Google Scholar] [CrossRef]
- Derfus, A.M.; von Maltzahn, G.; Harris, T.J.; Duza, T.; Vecchio, K.S.; Ruoslahti, E.; Bhatia, S.N. Remotely triggered release from magnetic nanoparticles. Adv. Mater. 2007, 19, 3932–3936. [Google Scholar] [CrossRef]
- Bringas, E.; Köysüren, Ö.; Quach, D.V.; Mahmoudi, M.; Aznar, E.; Roehling, J.D.; Marcos, M.D.; Martínez-Máñez, R.; Stroeve, P. Triggered release in lipid bilayer-capped mesoporous silica nanoparticles containing SPION using an alternating magnetic field. Chem. Commun. 2012, 48, 5647–5649. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-H.; Vitol, E.A.; Liu, J.; Balasubramanian, S.; Gosztola, D.J.; Cohen, E.E.; Novosad, V.; Rozhkova, E.A. Stimuli-responsive magnetic nanomicelles as multifunctional heat and cargo delivery vehicles. Langmuir 2013, 29, 7425–7432. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-C.; Kim, E.; Jeong, S.W.; Ha, T.-L.; Park, S.-I.; Lee, S.G.; Lee, S.J.; Lee, S.W. Magnetic nanoparticle-conjugated polymeric micelles for combined hyperthermia and chemotherapy. Nanoscale 2015, 7, 16470–16480. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, K.; Nakamura, M.; Miki, H.; Ozaki, S.; Abe, M.; Matsumoto, T.; Sakamoto, W.; Yogo, T.; Ishimura, K. Magnetically responsive smart nanoparticles for cancer treatment with a combination of magnetic hyperthermia and remote-control drug release. Theranostics 2014, 4, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Glover, A.L.; Nikles, S.M.; Nikles, J.A.; Brazel, C.S.; Nikles, D.E. Polymer micelles with crystalline cores for thermally triggered release. Langmuir 2012, 28, 10653–10660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glover, A.L.; Bennett, J.B.; Pritchett, J.S.; Nikles, S.M.; Nikles, D.E.; Nikles, J.A.; Brazel, C.S. Magnetic heating of iron oxide nanoparticles and magnetic micelles for cancer therapy. IEEE Trans. Magn. 2013, 49, 231–235. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wang, T.; Yang, L.; Liu, C.; Wang, C.; Liu, H.; Wang, Y.A.; Su, Z. General route to multifunctional uniform yolk/mesoporous silica shell nanocapsules: A platform for simultaneous cancer-targeted imaging and magnetically guided drug delivery. Chem. Er. J. 2012, 18, 12512–12521. [Google Scholar] [CrossRef]
- Hua, M.-Y.; Liu, H.-L.; Yang, H.-W.; Chen, P.-Y.; Tsai, R.-Y.; Huang, C.-Y.; Tseng, I.C.; Lyu, L.-A.; Ma, C.-C.; Tang, H.-J.; et al. The effectiveness of a magnetic nanoparticle-based delivery system for BCNU in the treatment of gliomas. Biomaterials 2011, 32, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Braun, G.B.; Pallaoro, A.; Zhang, Y.; Shi, Y.; Cui, D.; Moskovits, M.; Zhao, D.; Stucky, G.D. Mesoporous multifunctional upconversion luminescent and magnetic “nanorattle” materials for targeted chemotherapy. Nano Lett. 2012, 12, 61–67. [Google Scholar] [CrossRef]
- Prijic, S.; Prosen, L.; Cemazar, M.; Scancar, J.; Romih, R.; Lavrencak, J.; Bregar, V.B.; Coer, A.; Krzan, M.; Znidarsic, A.; et al. Surface modified magnetic nanoparticles for immuno-gene therapy of murine mammary adenocarcinoma. Biomaterials 2012, 33, 4379–4391. [Google Scholar] [CrossRef] [Green Version]
- Al-Deen, F.N.; Ho, J.; Selomulya, C.; Ma, C.; Coppel, R. Superparamagnetic nanoparticles for effective delivery of malaria DNA vaccine. Langmuir 2011, 27, 3703–3712. [Google Scholar] [CrossRef]
- Park, J.W.; Bae, K.H.; Kim, C.; Park, T.G. Clustered magnetite nanocrystals cross-linked with PEI for efficient siRNA delivery. Biomacromolecules 2011, 12, 457–465. [Google Scholar] [CrossRef]
- Estelrich, J.; Escribano, E.; Queralt, J.; Busquets, A.M. Iron oxide nanoparticles for magnetically-guided and magnetically-responsive drug delivery. Int. J. Mol. Sci. 2015, 16, 8070–8101. [Google Scholar] [CrossRef] [Green Version]
- Arias, L.S.; Pessan, J.P.; Vieira, A.P.M.; Lima, T.M.T.d.; Delbem, A.C.B.; Monteiro, D.R. Iron oxide nanoparticles for biomedical applications: A perspective on synthesis, drugs, antimicrobial activity, and toxicity. Antibiotics 2018, 7, 46. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Liu, Q.; Li, G.; Shi, J.; Liu, J.; Wang, T.; Jiang, G. A mussel-inspired polydopamine coating as a versatile platform for the in situ synthesis of graphene-based nanocomposites. Nanoscale 2012, 4, 5864–5867. [Google Scholar] [CrossRef]
- Xia, B.; Li, J.; Shi, J.; Zhang, Y.; Zhang, Q.; Chen, Z.; Wang, B. Biodegradable and magnetic-fluorescent porous silicon@iron oxide nanocomposites for fluorescence/magnetic resonance bimodal imaging of tumor in vivo. ACS Biomater. Sci. Eng. 2017, 3, 2579–2587. [Google Scholar] [CrossRef] [PubMed]
- Satarkar, N.S.; Zach Hilt, J. Hydrogel nanocomposites as remote-controlled biomaterials. Acta Biomaterialia 2008, 4, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Purushotham, S.; Ramanujan, R.V. Thermoresponsive magnetic composite nanomaterials for multimodal cancer therapy. Acta Biomaterialia 2010, 6, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Griffete, N.; Fresnais, J.; Espinosa, A.; Wilhelm, C.; Bée, A.; Ménager, C. Design of magnetic molecularly imprinted polymer nanoparticles for controlled release of doxorubicin under an alternative magnetic field in athermal conditions. Nanoscale 2015, 7, 18891–18896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larumbe, S.; Gómez-Polo, C.; Pérez-Landazábal, J.I.; Pastor, J.M. Effect of a SiO2 coating on the magnetic properties of Fe3O4 nanoparticles. J. Phys. Condens. Mat. 2012, 24, 266007. [Google Scholar] [CrossRef]
- Kafrouni, L.; Savadogo, O. Recent progress on magnetic nanoparticles for magnetic hyperthermia. Prog. Biomater. 2016, 5, 147–160. [Google Scholar] [CrossRef] [Green Version]
- Formica, D.; Silvestri, S. Biological effects of exposure to magnetic resonance imaging: An overview. Biomed. Eng. Online 2004, 3, 11. [Google Scholar] [CrossRef] [Green Version]
- Mura, S.; Nicolas, J.; Couvreur, P. Stimuli-responsive nanocarriers for drug delivery. Nat. Mater. 2013, 12, 991–1003. [Google Scholar] [CrossRef]
- Han, D.; Tong, X.; Zhao, Y. Block copolymer micelles with a dual-stimuli-responsive core for fast or slow degradation. Langmuir 2012, 28, 2327–2331. [Google Scholar] [CrossRef] [PubMed]
- Linsley, C.S.; Wu, B.M. Recent advances in light-responsive on-demand drug-delivery systems. Ther. Deliv. 2017, 8, 89–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayer, A.M.; Alam, S.; Mattern-Schain, S.I.; Best, M.D. Triggered liposomal release through a synthetic phosphatidylcholine analogue bearing a photocleavable moiety embedded within the sn-2 acyl chain. Chem. Er. J. 2014, 20, 3350–3357. [Google Scholar] [CrossRef]
- Chandra, B.; Mallik, S.; Srivastava, D.K. Design of photocleavable lipids and their application in liposomal “uncorking”. Chem. Commun. 2005. [Google Scholar] [CrossRef]
- Zhao, L.; Peng, J.; Huang, Q.; Li, C.; Chen, M.; Sun, Y.; Lin, Q.; Zhu, L.; Li, F. Near-infrared photoregulated drug release in living tumor tissue via yolk-shell upconversion nanocages. Adv. Funct. Mater. 2014, 24, 363–371. [Google Scholar] [CrossRef]
- Gorka, A.P.; Nani, R.R.; Zhu, J.; Mackem, S.; Schnermann, M.J. A near-IR uncaging strategy based on cyanine photochemistry. J. Am. Chem. Soc. 2014, 136, 14153–14159. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Krippes, K.; Ritz, S.; Chen, Z.; Best, A.; Butt, H.-J.; Mailänder, V.; Wu, S. Ultralow-intensity near-infrared light induces drug delivery by upconverting nanoparticles. Chem. Commun. 2015, 51, 431–434. [Google Scholar] [CrossRef] [Green Version]
- Tong, R.; Chiang, H.H.; Kohane, D.S. Photoswitchable nanoparticles for in vivo cancer chemotherapy. Proc. Natl. Acad. Sci. USA 2013, 110, 19048–19053. [Google Scholar] [CrossRef] [Green Version]
- Tong, R.; Hemmati, H.D.; Langer, R.; Kohane, D.S. Photoswitchable nanoparticles for triggered tissue penetration and drug delivery. J. Am. Chem. Soc. 2012, 134, 8848–8855. [Google Scholar] [CrossRef]
- Liu, X.-M.; Yang, B.; Wang, Y.-L.; Wang, J.-Y. Photoisomerisable cholesterol derivatives as photo-trigger of liposomes: Effect of lipid polarity, temperature, incorporation ratio, and cholesterol. Biochim. Biophys. Acta Biomembr. 2005, 1720, 28–34. [Google Scholar] [CrossRef] [Green Version]
- Kauscher, U.; Samanta, A.; Ravoo, B.J. Photoresponsive vesicle permeability based on intramolecular host–guest inclusion. Org. Biomol. Chem. 2014, 12, 600–606. [Google Scholar] [CrossRef]
- Peng, K.; Tomatsu, I.; Kros, A. Light controlled protein release from a supramolecular hydrogel. Chem. Commun. 2010, 46, 4094–4096. [Google Scholar] [CrossRef]
- Liu, J.; Bu, W.; Pan, L.; Shi, J. NIR-triggered anticancer drug delivery by upconverting nanoparticles with integrated azobenzene-modified mesoporous silica. Angew. Chem. Int. Ed. 2013, 52, 4375–4379. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Choi, E.; Tamanoi, F.; Zink, J.I. Light-activated nanoimpeller-controlled drug release in cancer cells. Small 2008, 4, 421–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saravanakumar, G.; Lee, J.; Kim, J.; Kim, W.J. Visible light-induced singlet oxygen-mediated intracellular disassembly of polymeric micelles co-loaded with a photosensitizer and an anticancer drug for enhanced photodynamic therapy. Chem. Commun. 2015, 51, 9995–9998. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Zhang, C.-J.; Liu, B. A photoactivatable AIE polymer for light-controlled gene delivery: Concurrent endo/lysosomal escape and DNA unpacking. Angew. Chem. Int. Ed. 2015, 54, 11419–11423. [Google Scholar] [CrossRef]
- Xu, L.; Zhao, M.; Yang, Y.; Liang, Y.; Sun, C.; Gao, W.; Li, S.; He, B.; Pu, Y. A reactive oxygen species (ROS)-responsive low molecular weight gel co-loaded with doxorubicin and Zn(ii) phthalocyanine tetrasulfonic acid for combined chemo-photodynamic therapy. J. Mater. Chem. B 2017, 5, 9157–9164. [Google Scholar] [CrossRef]
- Wang, H.; Han, R.-l.; Yang, L.-m.; Shi, J.-h.; Liu, Z.-j.; Hu, Y.; Wang, Y.; Liu, S.-j.; Gan, Y. Design and synthesis of core–shell–shell upconversion nanoparticles for NIR-induced drug release, photodynamic therapy, and cell imaging. ACS Appl. Mater. Interfaces 2016, 8, 4416–4423. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.Y.; Dolphin, D. Site-specific prodrug release using visible light. J. Am. Chem. Soc. 2008, 130, 4236–4237. [Google Scholar] [CrossRef] [PubMed]
- Murthy, R.S.; Bio, M.; You, Y. Low energy light-triggered oxidative cleavage of olefins. Tetrahedron Lett. 2009, 50, 1041–1044. [Google Scholar] [CrossRef]
- Bio, M.; Nkepang, G.; You, Y. Click and photo-unclick chemistry of aminoacrylate for visible light-triggered drug release. Chem. Commun. 2012, 48, 6517–6519. [Google Scholar] [CrossRef]
- Hossion, A.M.L.; Bio, M.; Nkepang, G.; Awuah, S.G.; You, Y. Visible light controlled release of anticancer drug through double activation of prodrug. ACS Med. Chem. Lett. 2013, 4, 124–127. [Google Scholar] [CrossRef] [Green Version]
- Rwei, A.Y.; Lee, J.-J.; Zhan, C.; Liu, Q.; Ok, M.T.; Shankarappa, S.A.; Langer, R.; Kohane, D.S. Repeatable and adjustable on-demand sciatic nerve block with phototriggerable liposomes. Proc. Natl. Acad. Sci. USA 2015, 112, 15719–15724. [Google Scholar] [CrossRef] [Green Version]
- Luo, D.; Li, N.; Carter, K.A.; Lin, C.; Geng, J.; Shao, S.; Huang, W.-C.; Qin, Y.; Atilla-Gokcumen, G.E.; Lovell, J.F. Rapid light-triggered drug release in liposomes containing small amounts of unsaturated and porphyrin–phospholipids. Small 2016, 12, 3039–3047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, L.; Yu, Y.; Luo, Z.; Li, M.; Chen, W.; Shen, X.; Chen, F.; Sun, Q.; Zhang, Q.; Gu, H.; et al. Photosensitizer enhanced disassembly of amphiphilic micelle for ROS-response targeted tumor therapy in vivo. Biomaterials 2016, 104, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Meng, X.; Deng, J.; Lu, D.; Zhang, X.; Chen, Y.; Zhu, J.; Fan, A.; Ding, D.; Kong, D.; et al. Multifunctional micelles dually responsive to hypoxia and singlet oxygen: Enhanced photodynamic therapy via interactively triggered photosensitizer delivery. ACS Appl. Mater. Interfaces 2018, 10, 17117–17128. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Zhan, F.; Zhu, Y. Smart photothermal-triggered bilayer phase transition in AuNPs–liposomes to release drug. Langmuir 2013, 29, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Viger, M.L.; Sheng, W.; Doré, K.; Alhasan, A.H.; Carling, C.-J.; Lux, J.; de Gracia Lux, C.; Grossman, M.; Malinow, R.; Almutairi, A. Near-infrared-induced heating of confined water in polymeric particles for efficient payload release. ACS Nano 2014, 8, 4815–4826. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Yan, L.; Yu, J.; Tian, G.; Zhou, L.; Zheng, X.; Zhang, X.; Yong, Y.; Li, J.; Gu, Z.; et al. High-throughput synthesis of single-layer MoS2 nanosheets as a near-infrared photothermal-triggered drug delivery for effective cancer therapy. ACS Nano 2014, 8, 6922–6933. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, J.; Nie, X.; Wen, T.; Ji, Y.; Wu, X.; Zhao, Y.; Chen, C. Near infrared laser-induced targeted cancer therapy using thermoresponsive polymer encapsulated gold nanorods. J. Am. Chem. Soc. 2014, 136, 7317–7326. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Tan, L.-L.; Jia, P.; Li, Q.-L.; Sun, Y.-L.; Zhang, J.; Ning, Y.-Q.; Yu, J.; Yang, Y.-W. Near-infrared light-responsive supramolecular nanovalve based on mesoporous silica-coated gold nanorods. Chem. Sci. 2014, 5, 2804–2808. [Google Scholar] [CrossRef]
- Zhang, M.; Chen, X.; Zhang, L.; Li, L.; Su, Z.-M.; Wang, C. Spadix-bract structured nanobowls for bimodal imaging-guided multidrug chemo-photothermal synergistic therapy. Chem. Mater. 2018, 30, 3722–3733. [Google Scholar] [CrossRef]
- Wu, G.; Mikhailovsky, A.; Khant, H.A.; Fu, C.; Chiu, W.; Zasadzinski, J.A. Remotely triggered liposome release by near-infrared light absorption via hollow gold nanoshells. J. Am. Chem. Soc. 2008, 130, 8175–8177. [Google Scholar] [CrossRef] [Green Version]
- Forbes, N.; Pallaoro, A.; Reich, N.O.; Zasadzinski, J.A. Rapid, reversible release from thermosensitive liposomes triggered by near-infra-red light. Part. Pert. Syst. Charact. 2014, 31, 1158–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelliccioli, A.P.; Wirz, J. Photoremovable protecting groups: Reaction mechanisms and applications. Photochem. Photobiol. Sci. 2002, 1, 441–458. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Sterner, E.S.; Coughlin, E.B.; Theato, P. o-Nitrobenzyl alcohol derivatives: Opportunities in polymer and materials science. Macromolecules 2012, 45, 1723–1736. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, W.; Gao, C. Shape transformation of light-responsive pyrene-containing micelles and their influence on cytoviability. Biomacromolecules 2015, 16, 2276–2281. [Google Scholar] [CrossRef] [PubMed]
- Bléger, D.; Hecht, S. Visible-light-activated molecular switches. Angew. Chem. Int. Ed. 2015, 54, 11338–11349. [Google Scholar] [CrossRef] [PubMed]
- Moratz, J.; Samanta, A.; Voskuhl, J.; Mohan Nalluri, S.K.; Ravoo, B.J. Light-triggered capture and release of DNA and proteins by host–guest binding and electrostatic interaction. Chem. Er. J. 2015, 21, 3271–3277. [Google Scholar] [CrossRef]
- Zhu, T.C.; Finlay, J.C. The role of photodynamic therapy (PDT) physics. Med. Phys. 2008, 35, 3127–3136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Jiang, C.; Figueiró Longo, J.P.; Azevedo, R.B.; Zhang, H.; Muehlmann, L.A. An updated overview on the development of new photosensitizers for anticancer photodynamic therapy. Acta Pharm. Sin. B 2018, 8, 137–146. [Google Scholar] [CrossRef]
- Hu, F.; Xu, S.; Liu, B. Photosensitizers with aggregation-induced emission: Materials and biomedical applications. Adv. Mater. 2018, 30, 1801350. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Lu, J.; Gao, X.; Li, P.; Zhang, W.; Ma, Y.; Wang, H.; Tang, B. Enhanced photodynamic therapy by reduced levels of intracellular glutathione obtained by employing a nano-MOF with CuII as the active center. Angew. Chem. Int. Ed. 2018, 57, 4891–4896. [Google Scholar] [CrossRef]
- Jia, Q.; Ge, J.; Liu, W.; Zheng, X.; Chen, S.; Wen, Y.; Zhang, H.; Wang, P. A magnetofluorescent carbon dot assembly as an acidic H2O2-driven oxygenerator to regulate tumor hypoxia for simultaneous bimodal imaging and enhanced photodynamic therapy. Adv. Mater. 2018, 30, 1706090. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-H.; Ding, Y.-X.; Li, Y.-J.; Wu, P.; Guo, J.; Wang, C.-C. Modulation of glutathione levels by redox-active nanogel carriers for the synergistic enhancement of photodynamic therapy. Adv. Ther. 2019, 2, 1800113. [Google Scholar] [CrossRef]
- Jiang, F.; Lilge, L.; Belcuig, M.; Singh, G.; Grenier, J.; Li, Y.; Chopp, M. Photodynamic therapy using Photofrin in combination with buthionine sulfoximine (BSO) to treat 9L gliosarcoma in rat brain. Lasers Surg. Med. 1998, 23, 161–166. [Google Scholar] [CrossRef]
- Tang, W.; Zhen, Z.; Wang, M.; Wang, H.; Chuang, Y.-J.; Zhang, W.; Wang, G.D.; Todd, T.; Cowger, T.; Chen, H.; et al. Red blood cell-facilitated photodynamic therapy for cancer treatment. Adv. Funct. Mater. 2016, 26, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Cheng, H.; Jiang, C.; Qiu, X.; Wang, K.; Huan, W.; Yuan, A.; Wu, J.; Hu, Y. Perfluorocarbon nanoparticles enhance reactive oxygen levels and tumour growth inhibition in photodynamic therapy. Nat. Commun. 2015, 6, 8785. [Google Scholar] [CrossRef]
- Liu, C.-P.; Wu, T.-H.; Liu, C.-Y.; Chen, K.-C.; Chen, Y.-X.; Chen, G.-S.; Lin, S.-Y. Self-supplying O2 through the catalase-like activity of gold nanoclusters for photodynamic therapy against hypoxic cancer cells. Small 2017, 13, 1700278. [Google Scholar] [CrossRef]
- Mathiyazhakan, M.; Yang, Y.; Liu, Y.; Zhu, C.; Liu, Q.; Ohl, C.-D.; Tam, K.C.; Gao, Y.; Xu, C. Non-invasive controlled release from gold nanoparticle integrated photo-responsive liposomes through pulse laser induced microbubble cavitation. Colloids Surf. B 2015, 126, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.T.; Choi, S.K. Mechanisms of drug release in nanotherapeutic delivery systems. Chem. Rev. 2015, 115, 3388–3432. [Google Scholar] [CrossRef] [PubMed]
- Bisby, R.H.; Mead, C.; Morgan, C.G. Active uptake of drugs into photosensitive liposomes and rapid release on UV photolysis. Photochem. Photobiol. 2000, 72, 57–61. [Google Scholar] [CrossRef]
- Fomina, N.; McFearin, C.L.; Sermsakdi, M.; Morachis, J.M.; Almutairi, A. Low power, biologically benign NIR light triggers polymer disassembly. Macromolecules 2011, 44, 8590–8597. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Deng, X.; Xie, Z.; Shi, Y.; Pang, M.; Lin, J. Controllable synthesis of highly monodispersed nanoscale Fe-soc-MOF and the construction of Fe-soc-MOF@polypyrrole core-shell nanohybrids for cancer therapy. Chem. Eng. J. 2019, 358, 369–378. [Google Scholar] [CrossRef]
- Hoang, L.T.M.; Ngo, L.H.; Nguyen, H.L.; Nguyen, H.T.H.; Nguyen, C.K.; Nguyen, B.T.; Ton, Q.T.; Nguyen, H.K.D.; Cordova, K.E.; Truong, T. An azobenzene-containing metal–organic framework as an efficient heterogeneous catalyst for direct amidation of benzoic acids: Synthesis of bioactive compounds. Chem. Commun. 2015, 51, 17132–17135. [Google Scholar] [CrossRef]
- Whitmore, S.E.; Potten, C.S.; Chadwick, C.A.; Strickland, P.T.; Morison, W.L. Effect of photoreactivating light on UV radiation-induced alterations in human skin. Photodermatol. Photoimmunol. Photomed. 2001, 17, 213–217. [Google Scholar] [CrossRef]
- Merve, M.; Ronald, K.; Angelika, A.; Ulrike, H.; Hagen, T. Wavelength-dependent penetration depths of ultraviolet radiation in human skin. J. Biomed. Opt. 2008, 13, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Mao, D.; Hu, F.; Xu, S.; Chen, C.; Zhang, C.-J.; Cheng, X.; Yuan, Y.; Ding, D.; Kong, D.; et al. A highly efficient and photostable photosensitizer with near-infrared aggregation-induced emission for image-guided photodynamic anticancer therapy. Adv. Mater. 2017, 29, 1700548. [Google Scholar] [CrossRef]
- Soldatova, A.V.; Kim, J.; Rizzoli, C.; Kenney, M.E.; Rodgers, M.A.J.; Rosa, A.; Ricciardi, G. Near-infrared-emitting phthalocyanines. A combined experimental and density functional theory study of the structural, optical, and photophysical properties of Pd(II) and Pt(II) α-butoxyphthalocyanines. Inorg. Chem. 2011, 50, 1135–1149. [Google Scholar] [CrossRef]
- Dong, M.; Babalhavaeji, A.; Samanta, S.; Beharry, A.A.; Woolley, G.A. Red-shifting azobenzene photoswitches for in vivo use. Acc. Chem. Res. 2015, 48, 2662–2670. [Google Scholar] [CrossRef]
- Cao, J.; Huang, S.; Chen, Y.; Li, S.; Li, X.; Deng, D.; Qian, Z.; Tang, L.; Gu, Y. Near-infrared light-triggered micelles for fast controlled drug release in deep tissue. Biomaterials 2013, 34, 6272–6283. [Google Scholar] [CrossRef]
- Gorka, A.P.; Schnermann, M.J. Harnessing cyanine photooxidation: From slowing photobleaching to near-IR uncaging. Curr. Opin. Chem. Biol. 2016, 33, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Barhoumi, A.; Liu, Q.; Kohane, D.S. Ultraviolet light-mediated drug delivery: Principles, applications, and challenges. J. Control. Release 2015, 219, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Jo, H.; Jeon, M.; Choi, M.-G.; Hahn, S.K.; Yun, S.-H. Luciferase–rose bengal conjugates for singlet oxygen generation by bioluminescence resonance energy transfer. Chem. Commun. 2017, 53, 4569–4572. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.; DeFrancesco, L. Five more years of Nature Biotechnology research. Nat. Biotechnol. 2011, 29, 221–227. [Google Scholar] [CrossRef]
- Pan, Y.; Neuss, S.; Leifert, A.; Fischler, M.; Wen, F.; Simon, U.; Schmid, G.; Brandau, W.; Jahnen-Dechent, W. Size-dependent cytotoxicity of gold nanoparticles. Small 2007, 3, 1941–1949. [Google Scholar] [CrossRef] [PubMed]
- Scholle, K.; Lamrini, S.; Koopmann, P.; Fuhrberg, P. 2 µm laser sources and their possible applications. In Frontiers in Guided Wave Optics and Optoelectronics; Pal, B., Ed.; IntechOpen: London, UK, 2010; pp. 471–500. [Google Scholar]
- Kennedy, J.E. High-intensity focused ultrasound in the treatment of solid tumours. Nat. Rev. Cancer 2005, 5, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Pitt, W.G.; Husseini, G.A.; Kherbeck, L.N. Chapter 6 Ultrasound-triggered release from micelles. In Smart Materials for Drug Delivery: Volume 1; The Royal Society of Chemistry: London, UK, 2013; Volume 1, pp. 148–178. [Google Scholar]
- Halliwell, M. A tutorial on ultrasonic physics and imaging techniques. Proc. Inst. Mech. Eng. H 2009, 224, 127–142. [Google Scholar] [CrossRef]
- Pruitt, J.D.; Pitt, W.G. Sequestration and ultrasound-induced release of doxorubicin from stabilized Pluronic P105 micelles. Drug Deliv. 2002, 9, 253–258. [Google Scholar] [CrossRef]
- Hasanzadeh, H.; Mokhtari-Dizaji, M.; Zahra Bathaie, S.; Hassan, Z.M. Effect of local dual frequency sonication on drug distribution from polymeric nanomicelles. Ultrason. Sonochem. 2011, 18, 1165–1171. [Google Scholar] [CrossRef]
- Husseini, G.A.; Velluto, D.; Kherbeck, L.; Pitt, W.G.; Hubbell, J.A.; Christensen, D.A. Investigating the acoustic release of doxorubicin from targeted micelles. Colloids Surf. B 2013, 101, 153–155. [Google Scholar] [CrossRef]
- Evjen, T.J.; Nilssen, E.A.; Rögnvaldsson, S.; Brandl, M.; Fossheim, S.L. Distearoylphosphatidylethanolamine-based liposomes for ultrasound-mediated drug delivery. Eur. J. Pharm. Biopharm. 2010, 75, 327–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mujoo, H.; Reynolds, J.N.J.; Tucker, I.G. The influence of bile salts on the response of liposomes to ultrasound. J. Liposome Res. 2016, 26, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xie, C.; Xia, H.; Wang, Z. pH and ultrasound dual-responsive polydopamine-coated mesoporous silica nanoparticles for controlled drug delivery. Langmuir 2018, 34, 9974–9981. [Google Scholar] [CrossRef]
- Mohan, P.; Rapoport, N. Doxorubicin as a molecular nanotheranostic agent: Effect of doxorubicin encapsulation in micelles or nanoemulsions on the ultrasound-mediated intracellular delivery and nuclear trafficking. Mol. Pharm. 2010, 7, 1959–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.-L.; McPherson, D.D.; MacDonald, R.C. A method to co-encapsulate gas and drugs in liposomes for ultrasound-controlled drug delivery. Ultrasound Med. Biol. 2008, 34, 1272–1280. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y.; Rim, Y.; McPherson, D.D.; Huang, S.-L.; Kim, H. A novel liposomal nanomedicine for nitric oxide delivery and breast cancer treatment. Bio-Med. Mater. Eng. 2014, 24, 61–67. [Google Scholar] [CrossRef]
- Novell, A.; Al Sabbagh, C.; Escoffre, J.-M.; Gaillard, C.; Tsapis, N.; Fattal, E.; Bouakaz, A. Focused ultrasound influence on calcein-loaded thermosensitive stealth liposomes. Int. J. Hyperthermia 2015, 31, 349–358. [Google Scholar] [CrossRef]
- Liang, X.; Gao, J.; Jiang, L.; Luo, J.; Jing, L.; Li, X.; Jin, Y.; Dai, Z. Nanohybrid liposomal cerasomes with good physiological stability and rapid temperature responsiveness for high intensity focused ultrasound triggered local chemotherapy of cancer. ACS Nano 2015, 9, 1280–1293. [Google Scholar] [CrossRef]
- Demirel, G.B.; von Klitzing, R. A new multiresponsive drug delivery system using smart nanogels. ChemPhysChem 2013, 14, 2833–2840. [Google Scholar] [CrossRef] [PubMed]
- Oerlemans, C.; Deckers, R.; Storm, G.; Hennink, W.E.; Nijsen, J.F.W. Evidence for a new mechanism behind HIFU-triggered release from liposomes. J. Control. Release 2013, 168, 327–333. [Google Scholar] [CrossRef]
- Li, F.; Xie, C.; Cheng, Z.; Xia, H. Ultrasound responsive block copolymer micelle of poly(ethylene glycol)–poly(propylene glycol) obtained through click reaction. Ultrason. Sonochem. 2016, 30, 9–17. [Google Scholar] [CrossRef]
- Liang, B.; Tong, R.; Wang, Z.; Guo, S.; Xia, H. High intensity focused ultrasound responsive metallo-supramolecular block copolymer micelles. Langmuir 2014, 30, 9524–9532. [Google Scholar] [CrossRef]
- Şen, T.; Tüfekçioğlu, O.; Koza, Y. Mechanical index. Anatol J. Cardiol 2015, 15, 334–336. [Google Scholar] [CrossRef]
- Coussios, C.C.; Roy, R.A. Applications of acoustics and cavitation to noninvasive therapy and drug delivery. Annu. Rev. Fluid Mech. 2008, 40, 395–420. [Google Scholar] [CrossRef]
- Marin, A.; Sun, H.; Husseini, G.A.; Pitt, W.G.; Christensen, D.A.; Rapoport, N.Y. Drug delivery in pluronic micelles: Effect of high-frequency ultrasound on drug release from micelles and intracellular uptake. J. Control. Release 2002, 84, 39–47. [Google Scholar] [CrossRef]
- Villeneuve, L.; Alberti, L.; Steghens, J.P.; Lancelin, J.M.; Mestas, J.L. Assay of hydroxyl radicals generated by focused ultrasound. Ultrason. Sonochem. 2009, 16, 339–344. [Google Scholar] [CrossRef]
- Krasovitski, B.; Frenkel, V.; Shoham, S.; Kimmel, E. Intramembrane cavitation as a unifying mechanism for ultrasound-induced bioeffects. Proc. Natl. Acad. Sci. USA 2011, 108, 3258–3263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirsi, S.R.; Borden, M.A. State-of-the-art materials for ultrasound-triggered drug delivery. Adv. Drug Deliv. Rev. 2014, 72, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Potisek, S.L.; Davis, D.A.; Sottos, N.R.; White, S.R.; Moore, J.S. Mechanophore-linked addition polymers. J. Am. Chem. Soc. 2007, 129, 13808–13809. [Google Scholar] [CrossRef]
- Caruso, M.M.; Davis, D.A.; Shen, Q.; Odom, S.A.; Sottos, N.R.; White, S.R.; Moore, J.S. Mechanically-induced chemical changes in polymeric materials. Chem. Rev. 2009, 109, 5755–5798. [Google Scholar] [CrossRef]
- Berkowski, K.L.; Potisek, S.L.; Hickenboth, C.R.; Moore, J.S. Ultrasound-induced site-specific cleavage of azo-functionalized poly(ethylene glycol). Macromolecules 2005, 38, 8975–8978. [Google Scholar] [CrossRef]
- Hickenboth, C.R.; Moore, J.S.; White, S.R.; Sottos, N.R.; Baudry, J.; Wilson, S.R. Biasing reaction pathways with mechanical force. Nature 2007, 446, 423–427. [Google Scholar] [CrossRef]
- Shi, X.; Martin, R.W.; Vaezy, S.; Crum, L.A. Quantitative investigation of acoustic streaming in blood. J. Acoust. Soc. Am. 2002, 111, 1110–1121. [Google Scholar] [CrossRef]
- Doinikov, A.A.; Bouakaz, A. Acoustic microstreaming around an encapsulated particle. J. Acoust. Soc. Am. 2010, 127, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Diederich, C.J.; Hynynen, K. Ultrasound technology for hyperthermia. Ultrasound Med. Biol. 1999, 25, 871–887. [Google Scholar] [CrossRef]
- Partanen, A.; Tillander, M.; Yarmolenko, P.S.; Wood, B.J.; Dreher, M.R.; Köhler, M.O. Reduction of peak acoustic pressure and shaping of heated region by use of multifoci sonications in MR-guided high-intensity focused ultrasound mediated mild hyperthermia. Med. Phys. 2013, 40, 013301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Smet, M.; Heijman, E.; Langereis, S.; Hijnen, N.M.; Grüll, H. Magnetic resonance imaging of high intensity focused ultrasound mediated drug delivery from temperature-sensitive liposomes: An in vivo proof-of-concept study. J. Control. Release 2011, 150, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Sirsi, S.R.; Borden, M.A. Advances in ultrasound mediated gene therapy using microbubble contrast agents. Theranostics 2012, 2, 1208–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, A.; Liu, M.; Ojha, T.; Storm, G.; Kiessling, F.; Lammers, T. Ultrasound-mediated drug delivery to the brain: Principles, progress and prospects. Drug Discov. Today Technol. 2016, 20, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Bussy, C.; Alexiou, C.; Petros, R.A.; Nyström, A.M.; Methven, L.; Kostarelos, K. Chapter 16—Therapeutic applications. In Adverse Effects of Engineered Nanomaterials; Fadeel, B., Pietroiusti, A., Shvedova, A.A., Eds.; Academic Press: Boston, MA, USA, 2012; pp. 285–313. [Google Scholar]
- El-Sherif, D.M.; Lathia, J.D.; Le, N.T.; Wheatley, M.A. Ultrasound degradation of novel polymer contrast agents. J. Biomed. Mater. Res. A 2004, 68A, 71–78. [Google Scholar] [CrossRef]
- Schroeder, A.; Kost, J.; Barenholz, Y. Ultrasound, liposomes, and drug delivery: Principles for using ultrasound to control the release of drugs from liposomes. Chem. Phys. Lipids 2009, 162, 1–16. [Google Scholar] [CrossRef]
- Sirsi, S.; Borden, M. Microbubble compositions, properties and biomedical applications. Bubble Sci. Eng. Technol. 2009, 1, 3–17. [Google Scholar] [CrossRef]
- Gao, Z.; Kennedy, A.M.; Christensen, D.A.; Rapoport, N.Y. Drug-loaded nano/microbubbles for combining ultrasonography and targeted chemotherapy. Ultrasonics 2008, 48, 260–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheeran, P.S.; Luois, S.; Dayton, P.A.; Matsunaga, T.O. Formulation and acoustic studies of a new phase-shift agent for diagnostic and therapeutic ultrasound. Langmuir 2011, 27, 10412–10420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheeran, P.S.; Dayton, P.A. Phase-change contrast agents for imaging and therapy. Curr. Pharm. Des. 2012, 18, 2152–2165. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, N. Phase-shift, stimuli-responsive perfluorocarbon nanodroplets for drug delivery to cancer. Wiley Interdicsip, Rev. Nanomed. Nanobiotechnol. 2012, 4, 492–510. [Google Scholar] [CrossRef] [Green Version]
- Grapentin, C.; Mayenfels, F.; Barnert, S.; Süss, R.; Schubert, R.; Temme, S.; Jacoby, C.; Schrader, J.; Flögel, U. Optimization of perfluorocarbon nanoemulsions for molecular imaging by 19F MRI. In Nanomedicine; Seifalian, A., Mel, A.D., Kalaskar, D.M., Eds.; One Central Press: Cheshire, UK, 2014; pp. 268–286. [Google Scholar]
- Ahrens, E.T.; Helfer, B.M.; O’Hanlon, C.F.; Schirda, C. Clinical cell therapy imaging using a perfluorocarbon tracer and fluorine-19 MRI. Magn. Reson. Med. 2014, 72, 1696–1701. [Google Scholar] [CrossRef] [PubMed]
- Kwan, J.J.; Myers, R.; Coviello, C.M.; Graham, S.M.; Shah, A.R.; Stride, E.; Carlisle, R.C.; Coussios, C.C. Ultrasound-propelled nanocups for drug delivery. Small 2015, 11, 5305–5314. [Google Scholar] [CrossRef] [Green Version]
- Dalecki, D. Mechanical bioeffects of ultrasound. Annu. Rev. Biomed. Eng. 2004, 6, 229–248. [Google Scholar] [CrossRef]
- Liu, D.; Auguste, D.T. Cancer targeted therapeutics: From molecules to drug delivery vehicles. J. Control. Release 2015, 219, 632–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrault, S.D.; Walkey, C.; Jennings, T.; Fischer, H.C.; Chan, W.C.W. Mediating tumor targeting efficiency of nanoparticles through design. Nano Lett. 2009, 9, 1909–1915. [Google Scholar] [CrossRef]
- Wang, J.; Mao, W.; Lock, L.L.; Tang, J.; Sui, M.; Sun, W.; Cui, H.; Xu, D.; Shen, Y. The role of micelle size in tumor accumulation, penetration, and treatment. ACS Nano 2015, 9, 7195–7206. [Google Scholar] [CrossRef] [PubMed]
- Popović, Z.; Liu, W.; Chauhan, V.P.; Lee, J.; Wong, C.; Greytak, A.B.; Insin, N.; Nocera, D.G.; Fukumura, D.; Jain, R.K.; et al. A nanoparticle size series for in vivo fluorescence imaging. Angew. Chem., Int. Ed. 2010, 49, 8649–8652. [Google Scholar] [CrossRef] [Green Version]
- Dreher, M.R.; Liu, W.; Michelich, C.R.; Dewhirst, M.W.; Yuan, F.; Chilkoti, A. Tumor vascular permeability, accumulation, and penetration of macromolecular drug carriers. J. Natl. Cancer Inst. 2006, 98, 335–344. [Google Scholar] [CrossRef] [Green Version]
- Choksakulnimitr, S.; Masuda, S.; Tokuda, H.; Takakura, Y.; Hashida, M. In vitro cytotoxicity of macromolecules in different cell culture systems. J. Control. Release 1995, 34, 233–241. [Google Scholar] [CrossRef]
- Wang, S.; Guo, H.; Li, Y.; Li, X. Penetration of nanoparticles across a lipid bilayer: Effects of particle stiffness and surface hydrophobicity. Nanoscale 2019, 11, 4025–4034. [Google Scholar] [CrossRef]
- Du, J.-Z.; Sun, T.-M.; Song, W.-J.; Wu, J.; Wang, J. A tumor-acidity-activated charge-conversional nanogel as an intelligent vehicle for promoted tumoral-cell uptake and drug delivery. Angew. Chem. Int. Ed. 2010, 49, 3621–3626. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.S.; Shin, H.J.; Na, K.; Bae, Y.H. Poly(l-histidine)–PEG block copolymer micelles and pH-induced destabilization. J. Control. Release 2003, 90, 363–374. [Google Scholar] [CrossRef]
- Min, K.H.; Kim, J.-H.; Bae, S.M.; Shin, H.; Kim, M.S.; Park, S.; Lee, H.; Park, R.-W.; Kim, I.-S.; Kim, K.; et al. Tumoral acidic pH-responsive MPEG-poly(β-amino ester) polymeric micelles for cancer targeting therapy. J. Control. Release 2010, 144, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Sethuraman, V.A.; Bae, Y.H. TAT peptide-based micelle system for potential active targeting of anti-cancer agents to acidic solid tumors. J. Control. Release 2007, 118, 216–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, J.; Sanoj Rejinold, N.; Lee, D.; Jon, S.; Kim, Y.-C. Protease-activatable cell-penetrating peptide possessing ROS-triggered phase transition for enhanced cancer therapy. J. Control. Release 2017, 264, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Jin, E.; Zhang, B.; Sun, X.; Zhou, Z.; Ma, X.; Sun, Q.; Tang, J.; Shen, Y.; Van Kirk, E.; Murdoch, W.J.; et al. Acid-active cell-penetrating peptides for in vivo tumor-targeted drug delivery. J. Am. Chem. Soc. 2013, 135, 933–940. [Google Scholar] [CrossRef]
- Liu, Z.; Xiong, M.; Gong, J.; Zhang, Y.; Bai, N.; Luo, Y.; Li, L.; Wei, Y.; Liu, Y.; Tan, X.; et al. Legumain protease-activated TAT-liposome cargo for targeting tumours and their microenvironment. Nat. Commun. 2014, 5, 4280. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Olson, E.S.; Nguyen, Q.T.; Roy, M.; Jennings, P.A.; Tsien, R.Y. Tumor imaging by means of proteolytic activation of cell-penetrating peptides. Proc. Natl. Acad. Sci. USA 2004, 101, 17867–17872. [Google Scholar] [CrossRef] [Green Version]
- Bode, S.A.; Hansen, M.B.; Oerlemans, R.A.J.F.; van Hest, J.C.M.; Löwik, D.W.P.M. Enzyme-activatable cell-penetrating peptides through a minimal side chain modification. Bioconjug. Chem. 2015, 26, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Wang, Y.; He, Q.; Gao, H. Multistage drug delivery system based on microenvironment-responsive dendrimer–gelatin nanoparticles for deep tumor penetration. RSC Adv. 2015, 5, 85933–85937. [Google Scholar] [CrossRef]
- Cun, X.; Chen, J.; Ruan, S.; Zhang, L.; Wan, J.; He, Q.; Gao, H. A novel strategy through combining iRGD peptide with tumor-microenvironment-responsive and multistage nanoparticles for deep tumor penetration. ACS Appl. Mater. Interfaces 2015, 7, 27458–27466. [Google Scholar] [CrossRef]
- Cun, X.; Ruan, S.; Chen, J.; Zhang, L.; Li, J.; He, Q.; Gao, H. A dual strategy to improve the penetration and treatment of breast cancer by combining shrinking nanoparticles with collagen depletion by losartan. Acta Biomaterialia 2016, 31, 186–196. [Google Scholar] [CrossRef]
- Hu, C.; Cun, X.; Ruan, S.; Liu, R.; Xiao, W.; Yang, X.; Yang, Y.; Yang, C.; Gao, H. Enzyme-triggered size shrink and laser-enhanced NO release nanoparticles for deep tumor penetration and combination therapy. Biomaterials 2018, 168, 64–75. [Google Scholar] [CrossRef]
- Feng, T.; Ai, X.; An, G.; Yang, P.; Zhao, Y. Charge-convertible carbon dots for imaging-guided drug delivery with enhanced in vivo cancer therapeutic efficiency. ACS Nano 2016, 10, 4410–4420. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Huang-Fu, M.-Y.; Guo, W.-W.; Guo, N.-N.; Chen, J.; Liu, H.-N.; Xie, Z.-Q.; Lin, M.-T.; Wei, Q.-C.; Gao, J.-Q. MMP-2-sensitive HA end-conjugated poly(amidoamine) dendrimers via click reaction to enhance drug penetration into solid tumor. ACS Appl. Mater. Interfaces 2017, 9, 42459–42470. [Google Scholar] [CrossRef]
- Yao, Q.; Dai, Z.; Hoon Choi, J.; Kim, D.; Zhu, L. Building stable MMP2-responsive multifunctional polymeric micelles by an all-in-one polymer–lipid conjugate for tumor-targeted intracellular drug delivery. ACS Appl. Mater. Interfaces 2017, 9, 32520–32533. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.-L.; Fang, J.-H.; Liao, C.-Y.; Lin, C.-T.; Li, Y.-T.; Hu, S.-H. Targeted mesoporous iron oxide nanoparticles-encapsulated perfluorohexane and a hydrophobic drug for deep tumor penetration and therapy. Theranostics 2015, 5, 1233–1248. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Xu, Y.; Gao, C.; Zhou, Y.; Zhang, N.; Dai, Z. Ultrasound contrast agent microbubbles with ultrahigh loading capacity of camptothecin and floxuridine for enhancing tumor accumulation and combined chemotherapeutic efficacy. NPG Asia Mater. 2018, 10, 761–774. [Google Scholar] [CrossRef] [Green Version]
- Hatakeyama, H.; Akita, H.; Ito, E.; Hayashi, Y.; Oishi, M.; Nagasaki, Y.; Danev, R.; Nagayama, K.; Kaji, N.; Kikuchi, H.; et al. Systemic delivery of siRNA to tumors using a lipid nanoparticle containing a tumor-specific cleavable PEG-lipid. Biomaterials 2011, 32, 4306–4316. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.-Y.; Liu, Y.; Du, J.-Z.; Cao, Z.-T.; Xu, C.-F.; Wang, J. Facile generation of tumor-pH-labile linkage-bridged block copolymers for chemotherapeutic delivery. Angew. Chem. Int. Ed. 2016, 55, 1010–1014. [Google Scholar] [CrossRef] [PubMed]
- Angelos, S.; Yang, Y.-W.; Khashab, N.M.; Stoddart, J.F.; Zink, J.I. Dual-controlled nanoparticles exhibiting AND logic. J. Am. Chem. Soc. 2009, 131, 11344–11346. [Google Scholar] [CrossRef]
- Ashley, C.E.; Carnes, E.C.; Epler, K.E.; Padilla, D.P.; Phillips, G.K.; Castillo, R.E.; Wilkinson, D.C.; Wilkinson, B.S.; Burgard, C.A.; Kalinich, R.M.; et al. Delivery of small interfering RNA by peptide-targeted mesoporous silica nanoparticle-supported lipid bilayers. ACS Nano 2012, 6, 2174–2188. [Google Scholar] [CrossRef]
- Li, J.; Sun, L.; Liu, Y.; Yao, H.; Jiang, S.; Yunzhu, P.; Li, Y.; Zhang, Y. To reduce premature drug release while ensuring burst intracellular drug release of solid lipid nanoparticle-based drug delivery system with clathrin modification. Nanomedicine 2019, 15, 108–118. [Google Scholar] [CrossRef]
- Jiang, T.; Mo, R.; Bellotti, A.; Zhou, J.; Gu, Z. Gel–liposome-mediated co-delivery of anticancer membrane-associated proteins and small-molecule drugs for enhanced therapeutic efficacy. Adv. Funct. Mater. 2014, 24, 2295–2304. [Google Scholar] [CrossRef]
- Naumovska, E.; Ludwanowski, S.; Hersch, N.; Braun, T.; Merkel, R.; Hoffmann, B.; Csiszár, A. Plasma membrane functionalization using highly fusogenic immune activator liposomes. Acta Biomaterialia 2014, 10, 1403–1411. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy - Assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef]
- Song, Q.; Yin, Y.; Shang, L.; Wu, T.; Zhang, D.; Kong, M.; Zhao, Y.; He, Y.; Tan, S.; Guo, Y.; et al. Tumor microenvironment responsive nanogel for the combinatorial antitumor effect of chemotherapy and immunotherapy. Nano Lett. 2017, 17, 6366–6375. [Google Scholar] [CrossRef] [PubMed]
- Benien, P.; Swami, A. 3D tumor models: History, advances and future perspectives. Future Oncol. 2014, 10, 1311–1327. [Google Scholar] [CrossRef] [PubMed]
- Bhise, N.S.; Ribas, J.; Manoharan, V.; Zhang, Y.S.; Polini, A.; Massa, S.; Dokmeci, M.R.; Khademhosseini, A. Organ-on-a-chip platforms for studying drug delivery systems. J. Control. Release 2014, 190, 82–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Lenaghan, S.C.; Zhang, M. A data-driven predictive approach for drug delivery using machine learning techniques. PLoS ONE 2012, 7, e31724. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Y.; Davis, E. Nanoplatforms for Targeted Stimuli-Responsive Drug Delivery: A Review of Platform Materials and Stimuli-Responsive Release and Targeting Mechanisms. Nanomaterials 2021, 11, 746. https://doi.org/10.3390/nano11030746
Sun Y, Davis E. Nanoplatforms for Targeted Stimuli-Responsive Drug Delivery: A Review of Platform Materials and Stimuli-Responsive Release and Targeting Mechanisms. Nanomaterials. 2021; 11(3):746. https://doi.org/10.3390/nano11030746
Chicago/Turabian StyleSun, Yuzhe, and Edward Davis. 2021. "Nanoplatforms for Targeted Stimuli-Responsive Drug Delivery: A Review of Platform Materials and Stimuli-Responsive Release and Targeting Mechanisms" Nanomaterials 11, no. 3: 746. https://doi.org/10.3390/nano11030746