
Tunable Electronic Properties of Lateral Monolayer Transition Metal Dichalcogenide Superlattice Nanoribbons


Abstract
:1. Introduction
2. Method
3. Results and Discussion
3.1. MoS2/WS2 System
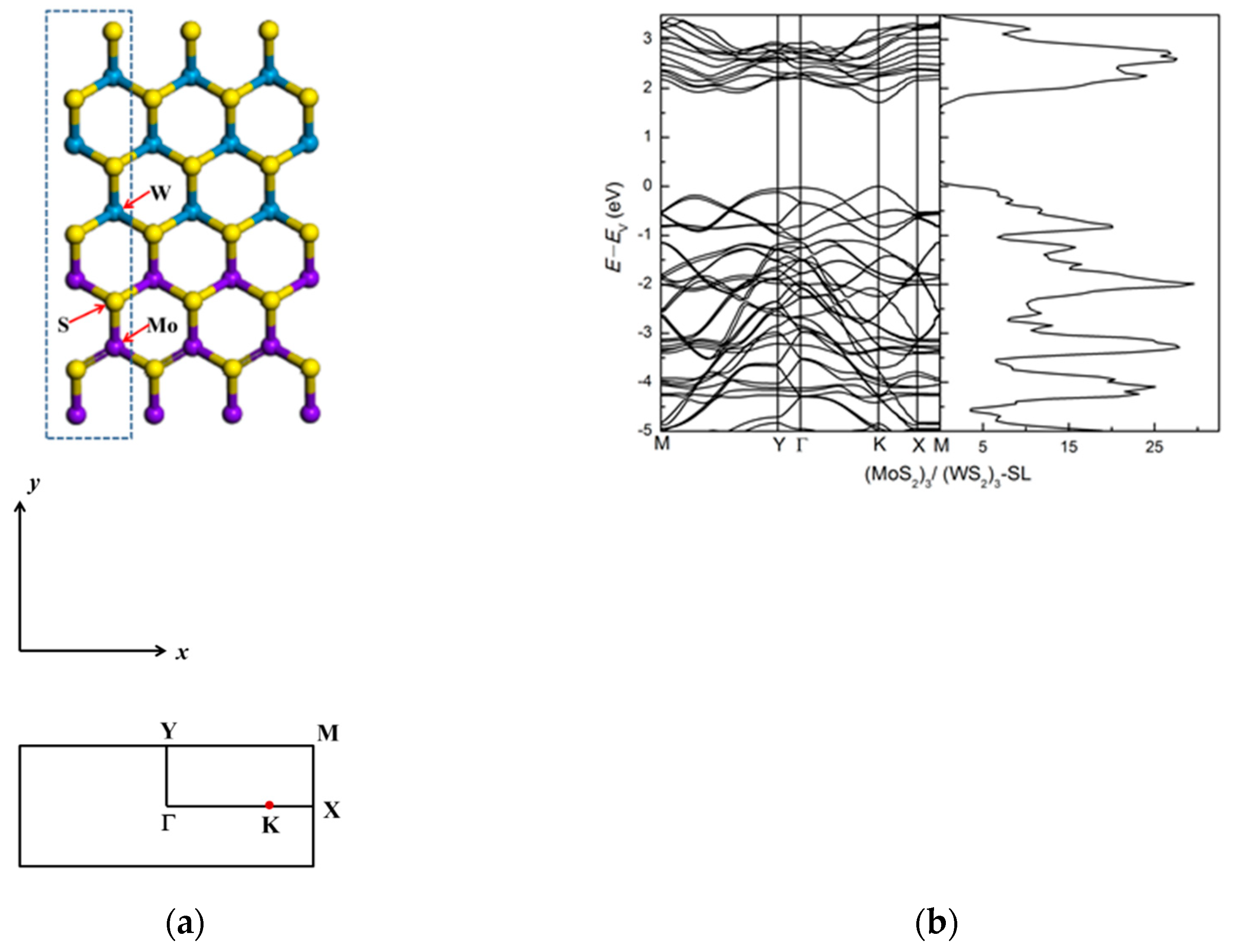
3.1.1. (MoS2)3/(WS2)3 Lateral Superlattice
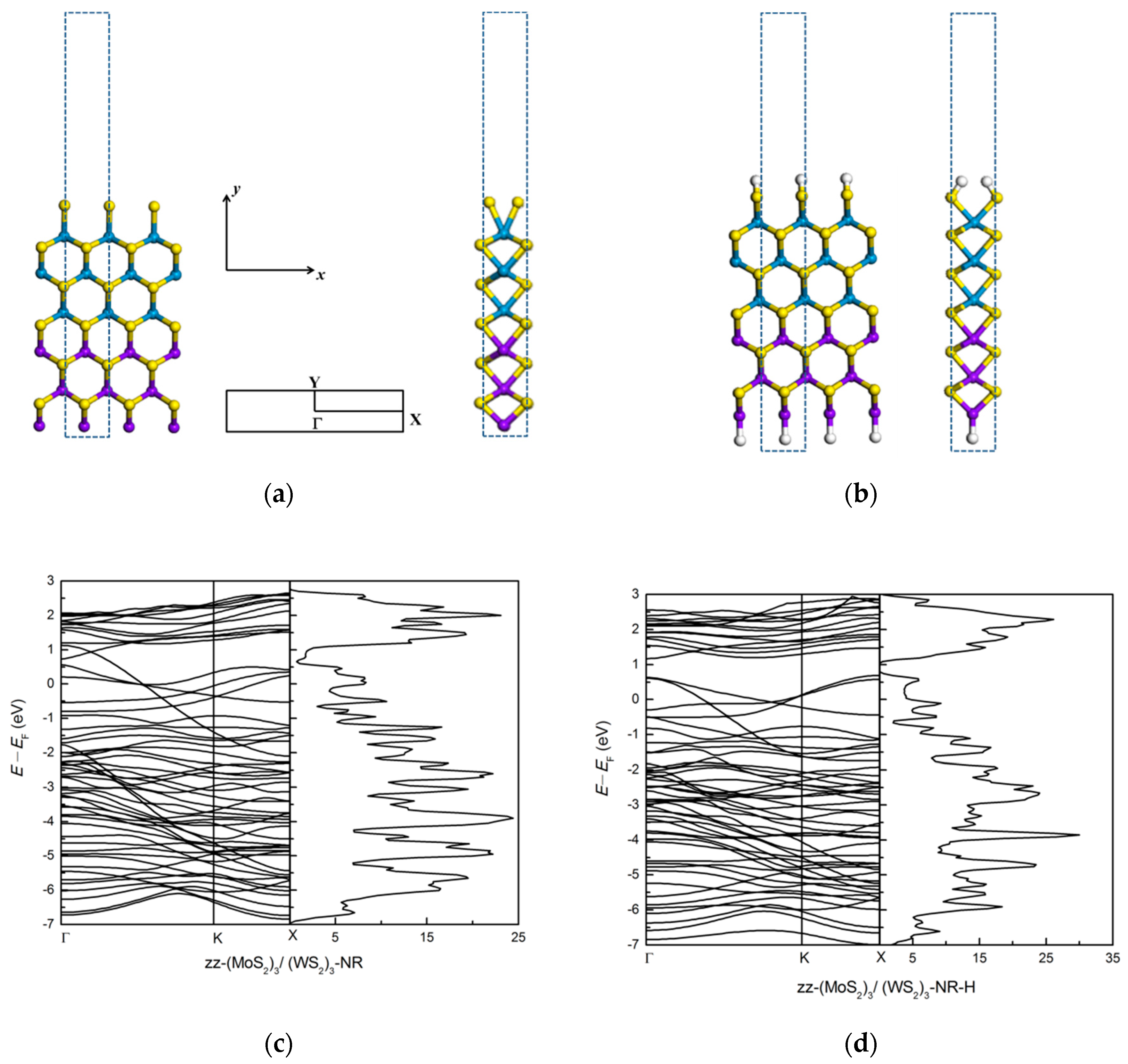
3.1.2. Zigzag (MoS2)3/(WS2)3 Lateral Nanoribbons
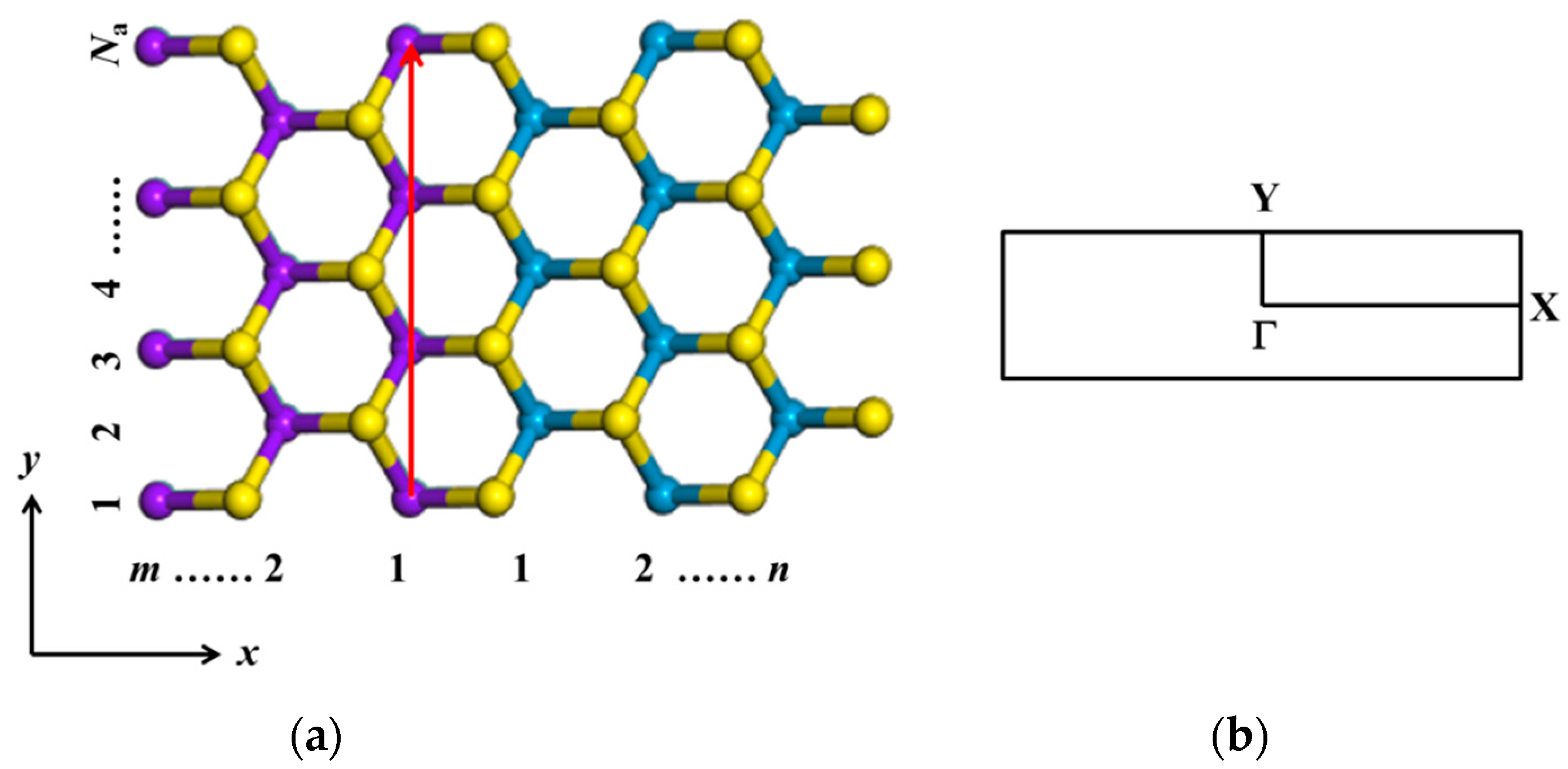
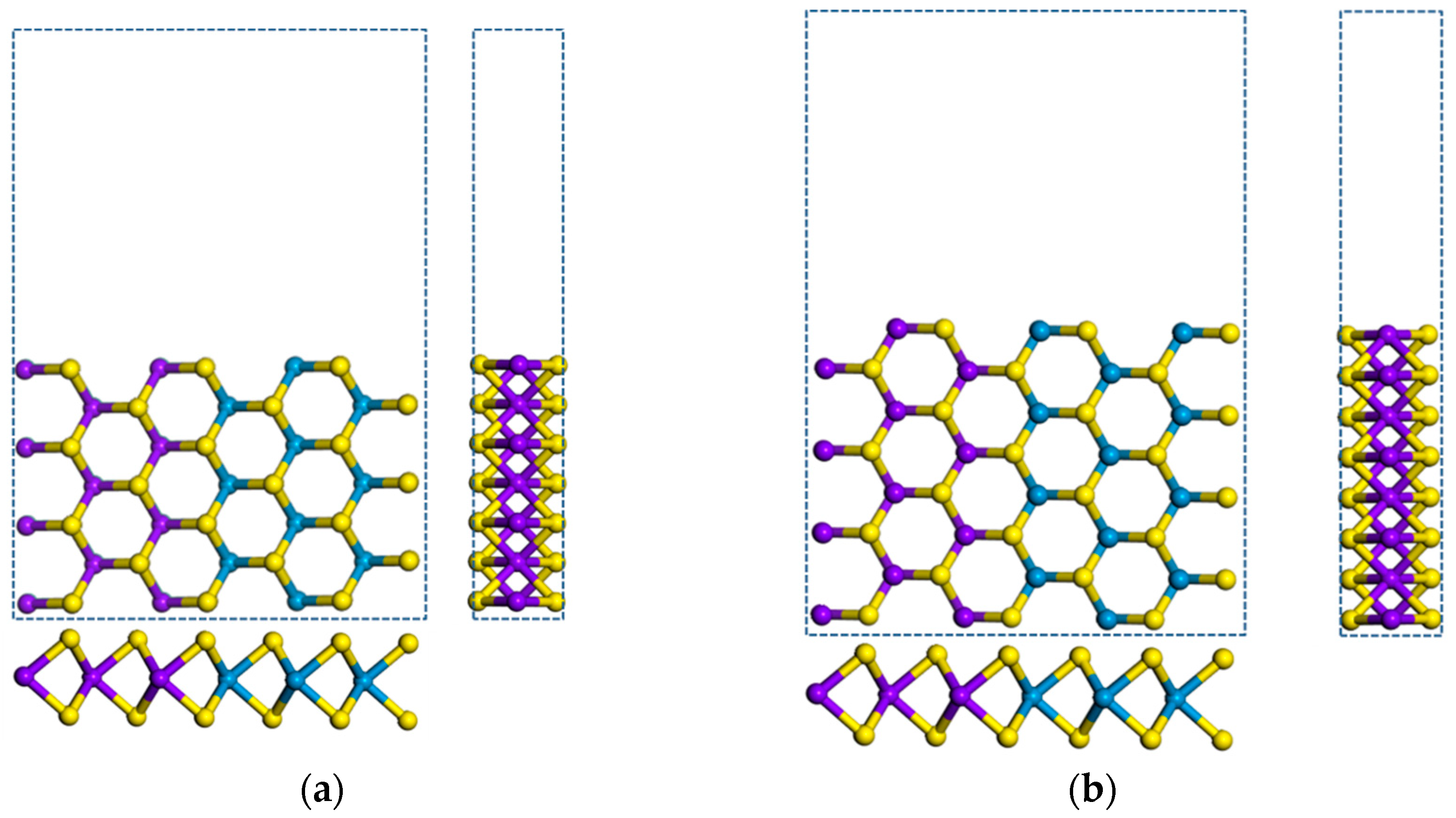
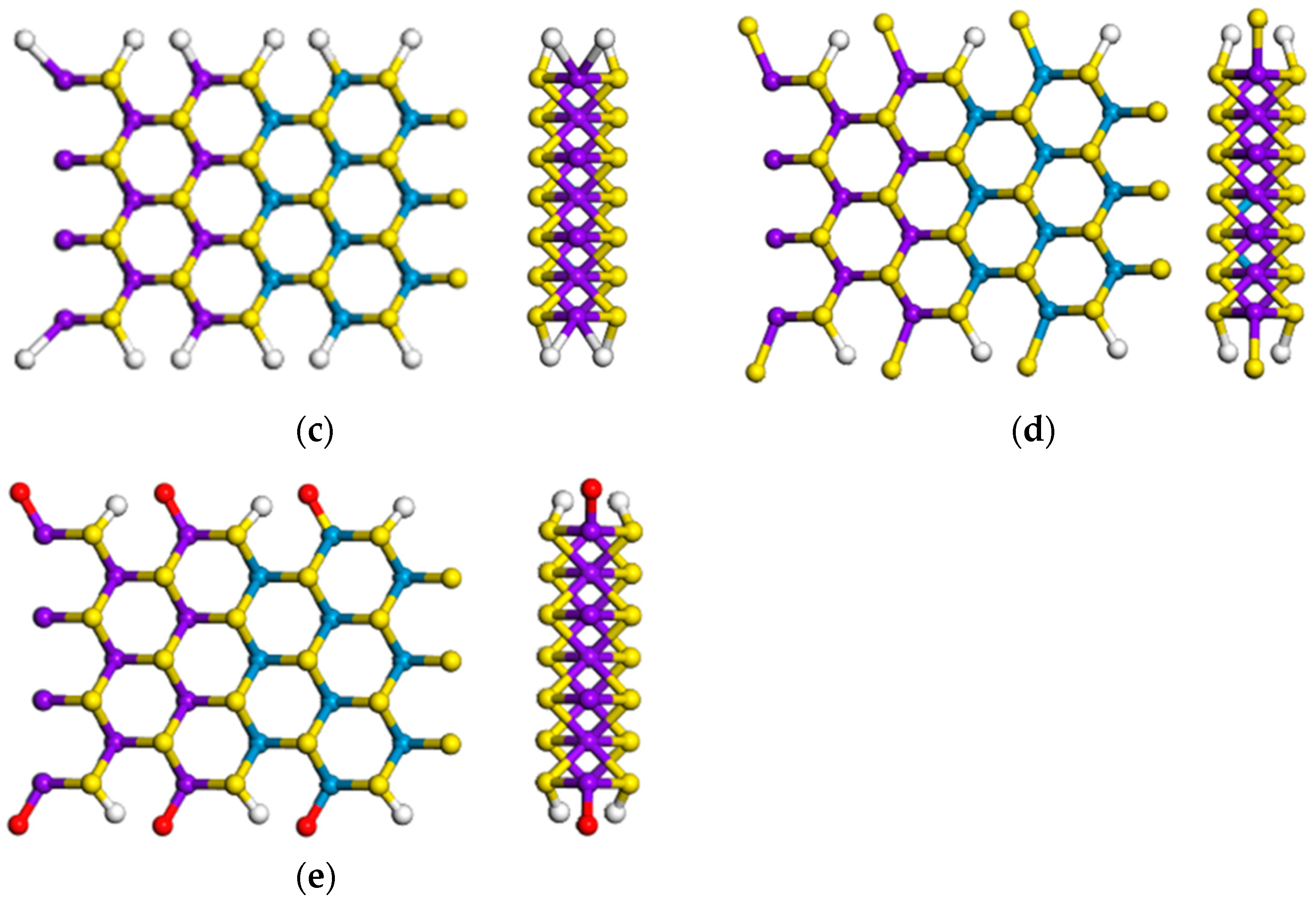
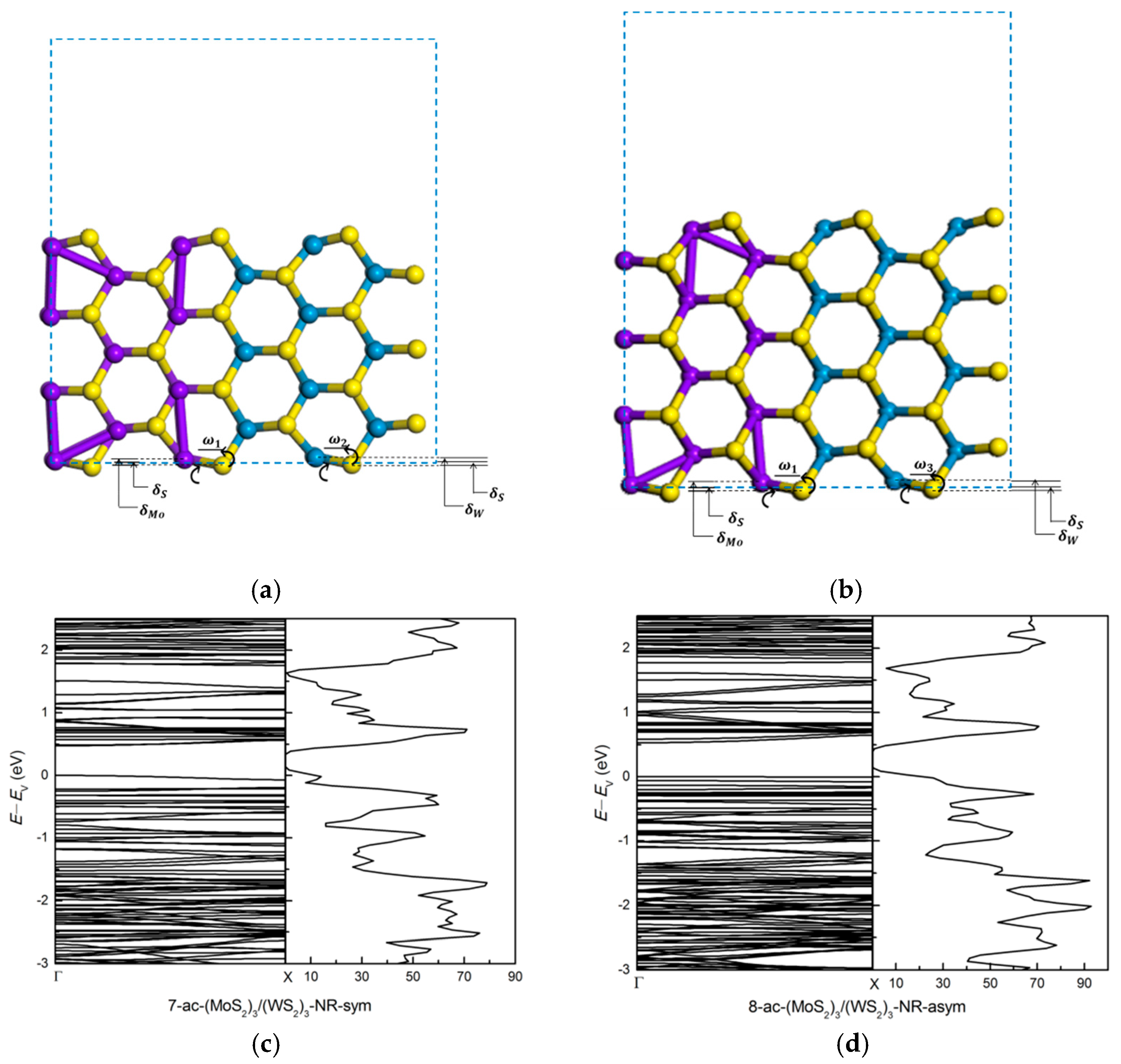
3.1.3. Armchair (MoS2)m/(WS2)n Lateral Nanoribbons
3.2. MoS2/MoTe2 System
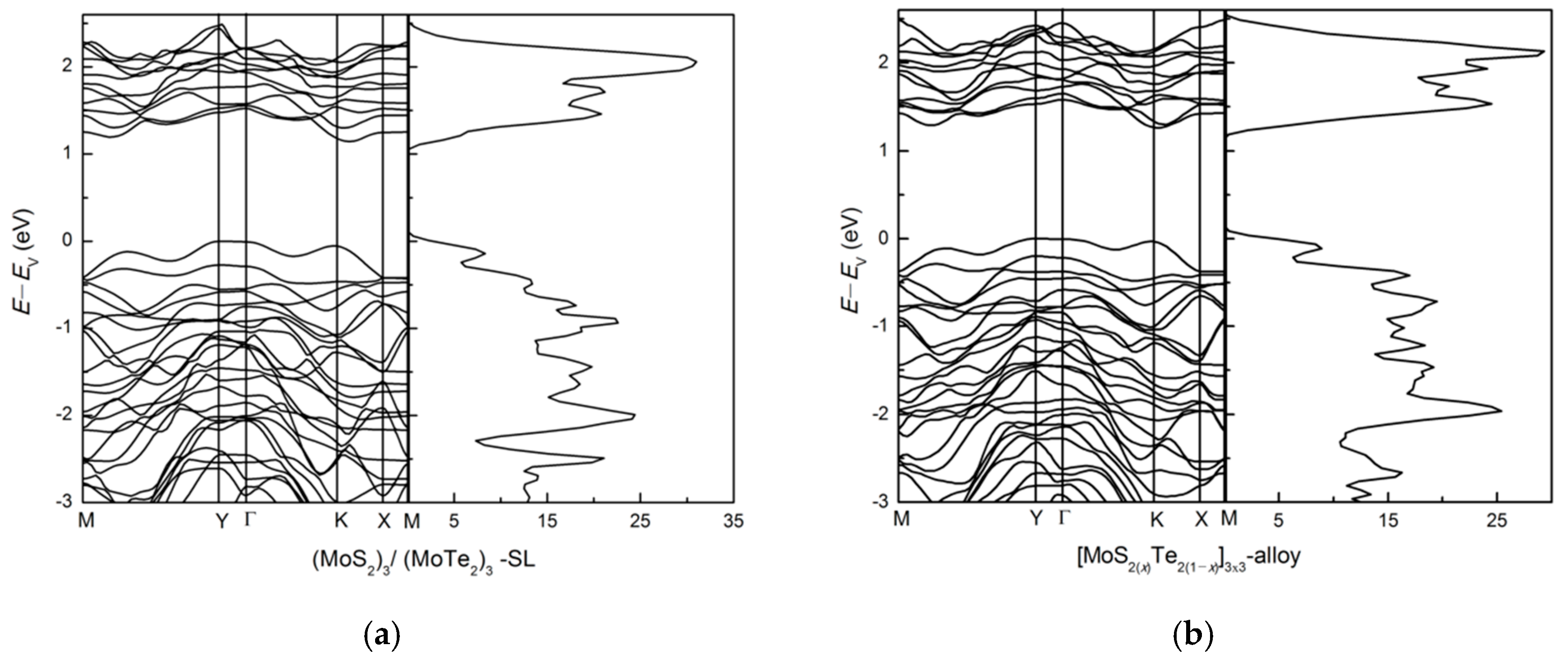
3.2.1. (MoS2)3/(MoTe2)3 Lateral Superlattice and Alloy
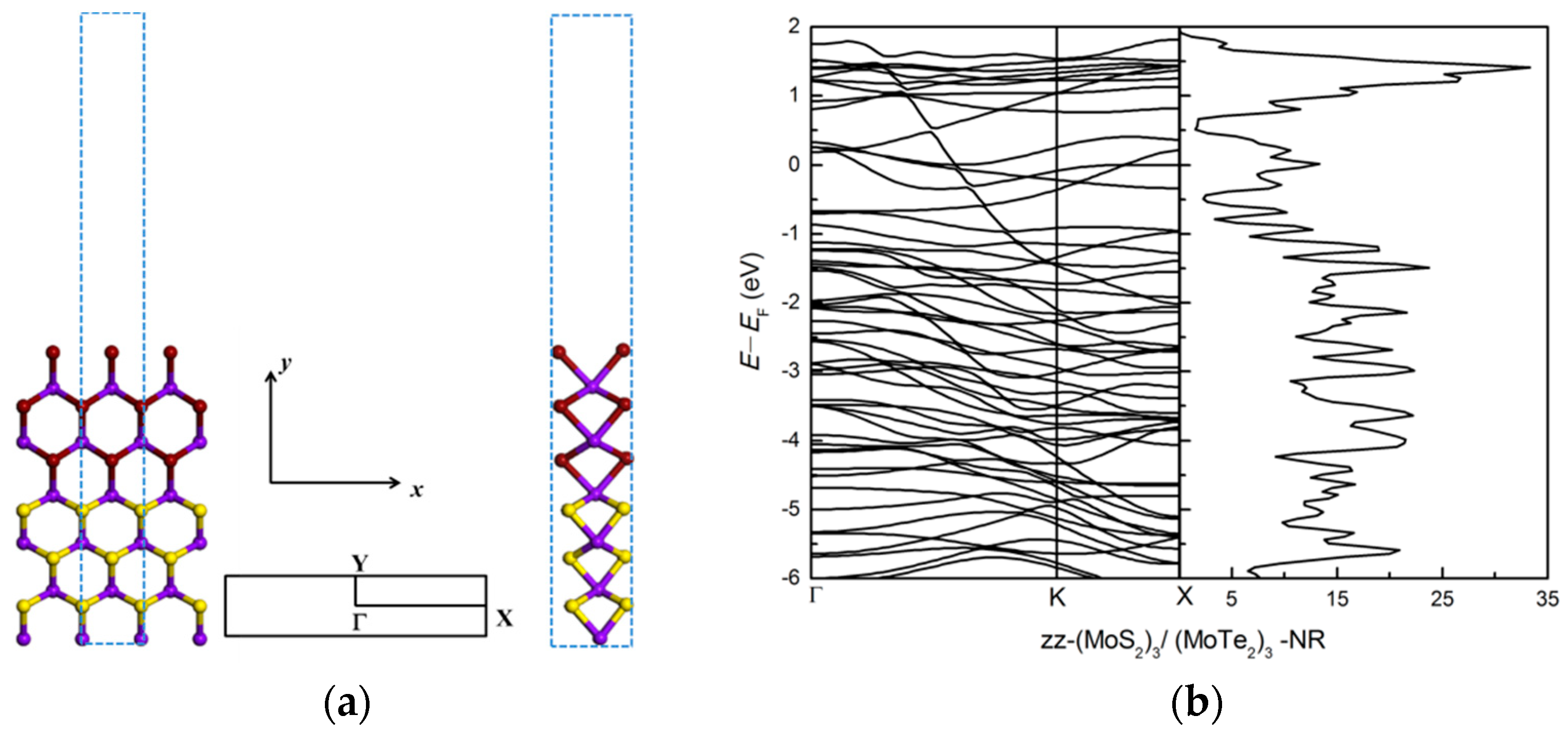
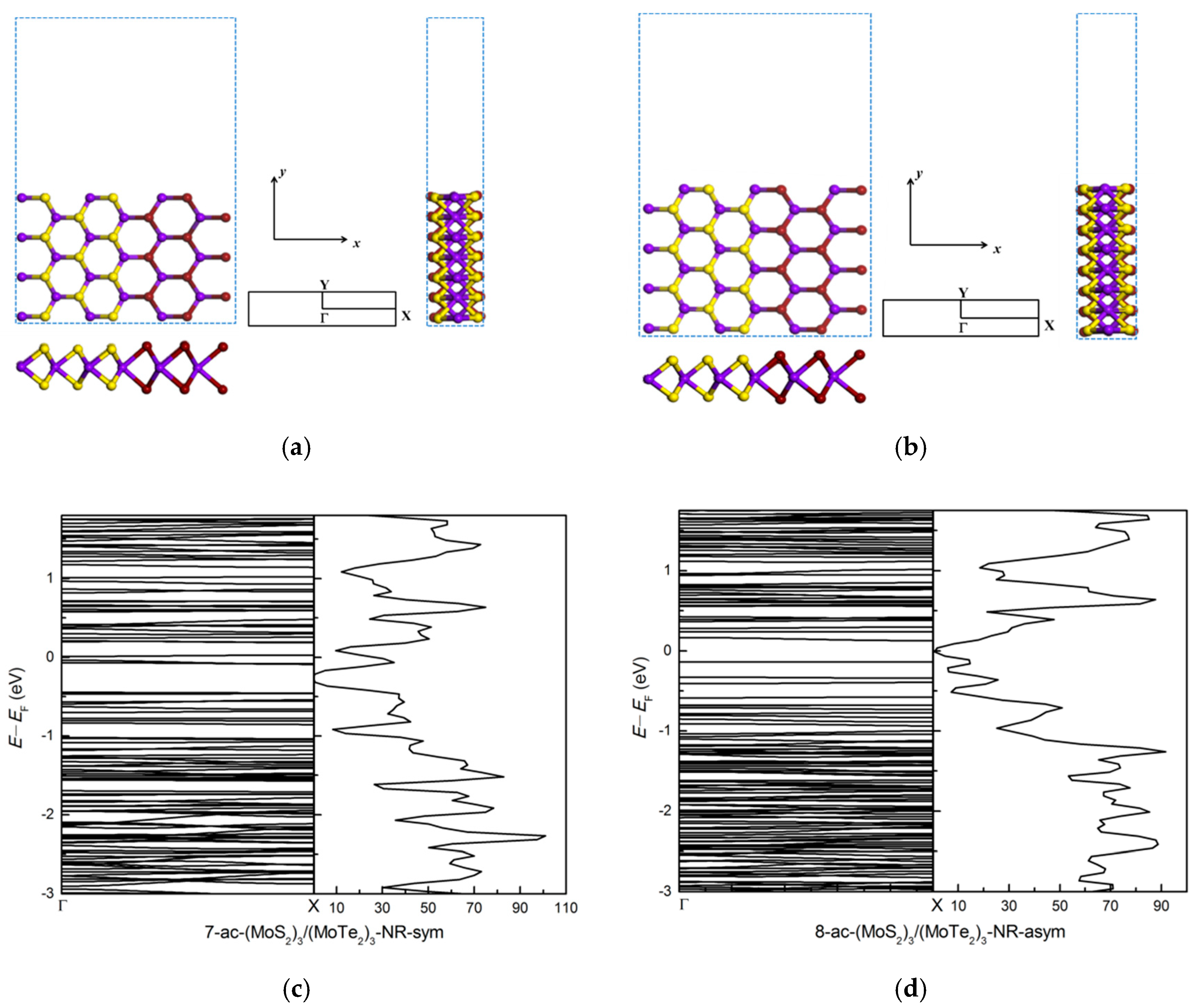
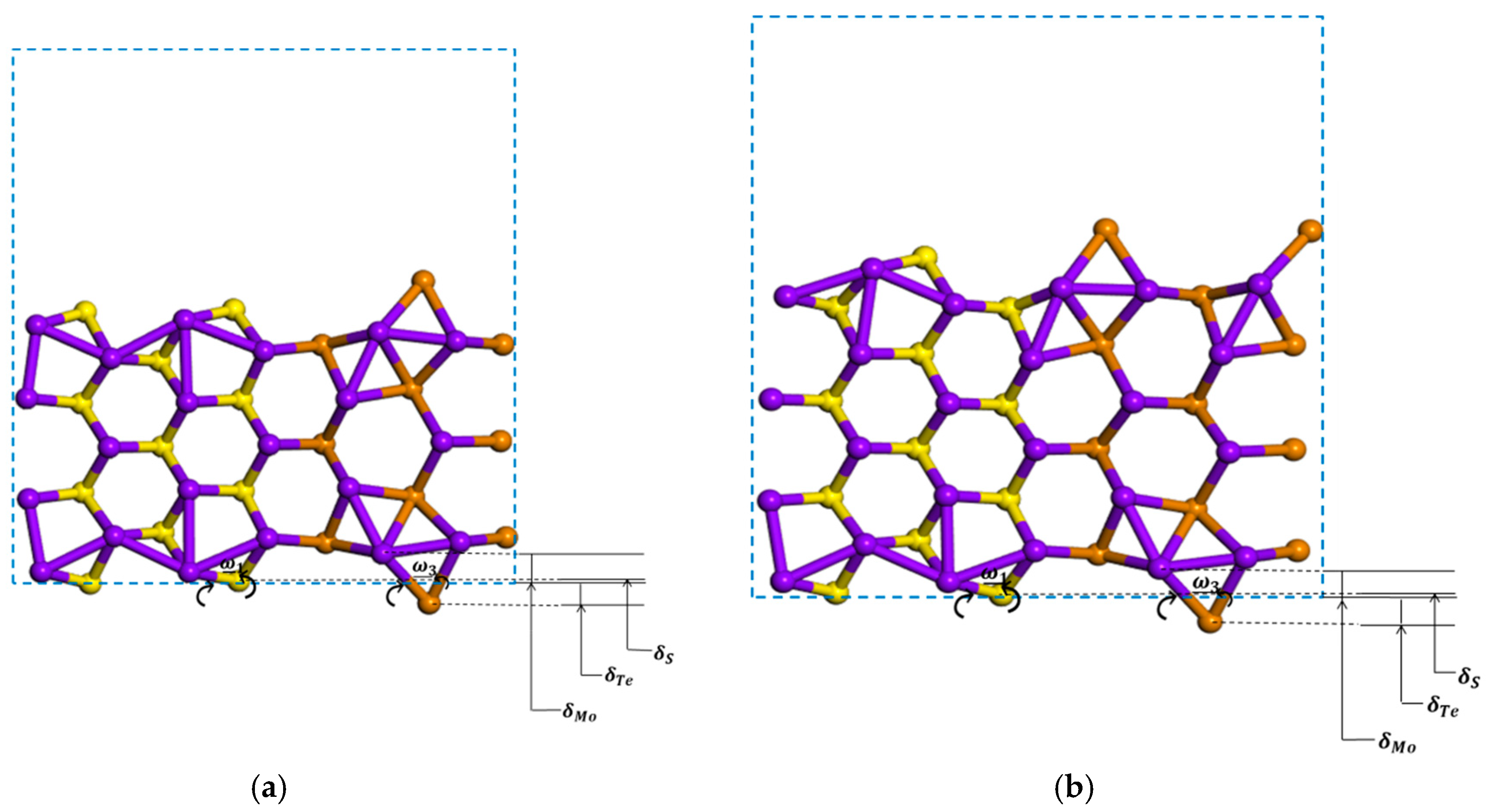
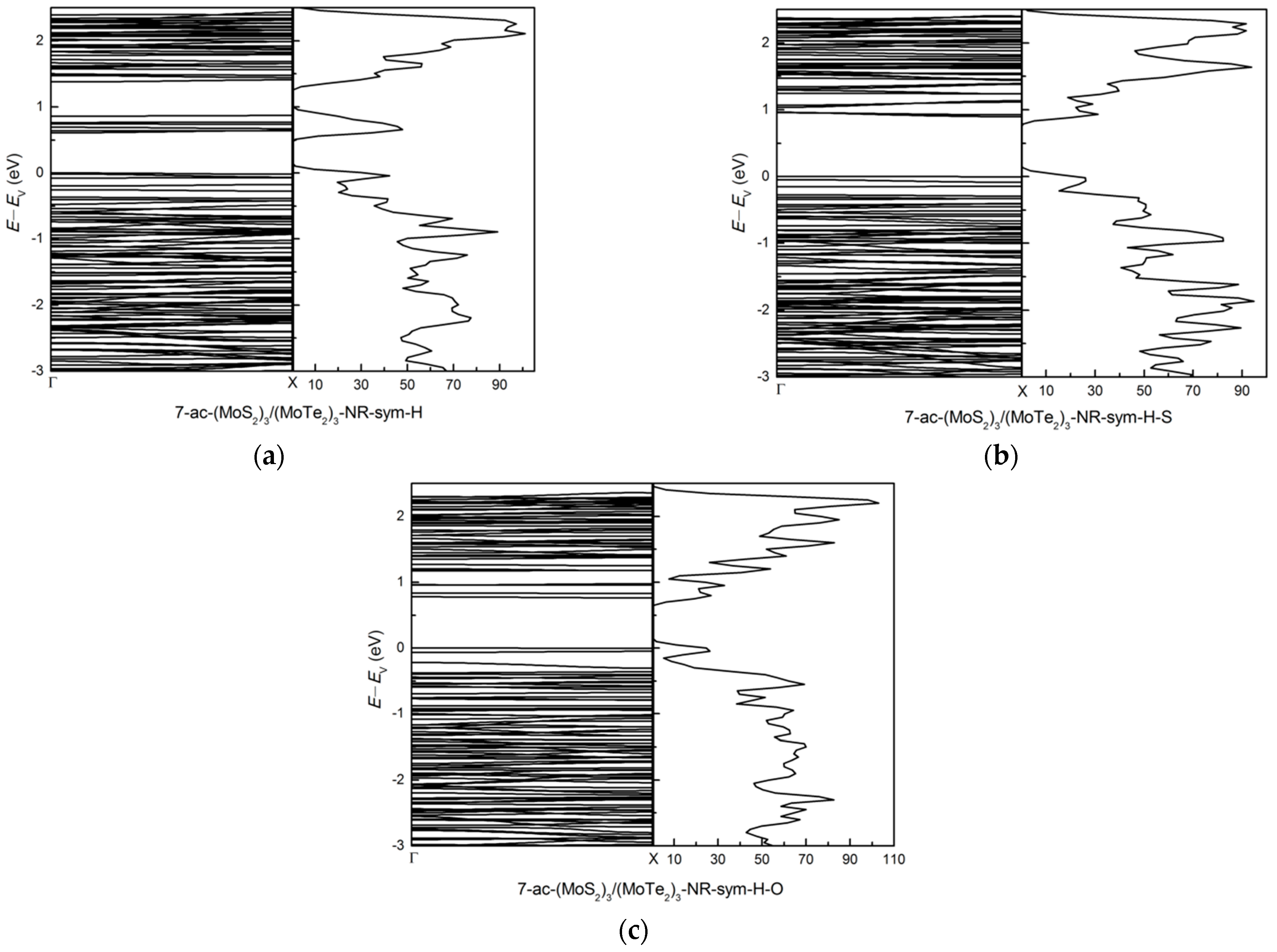
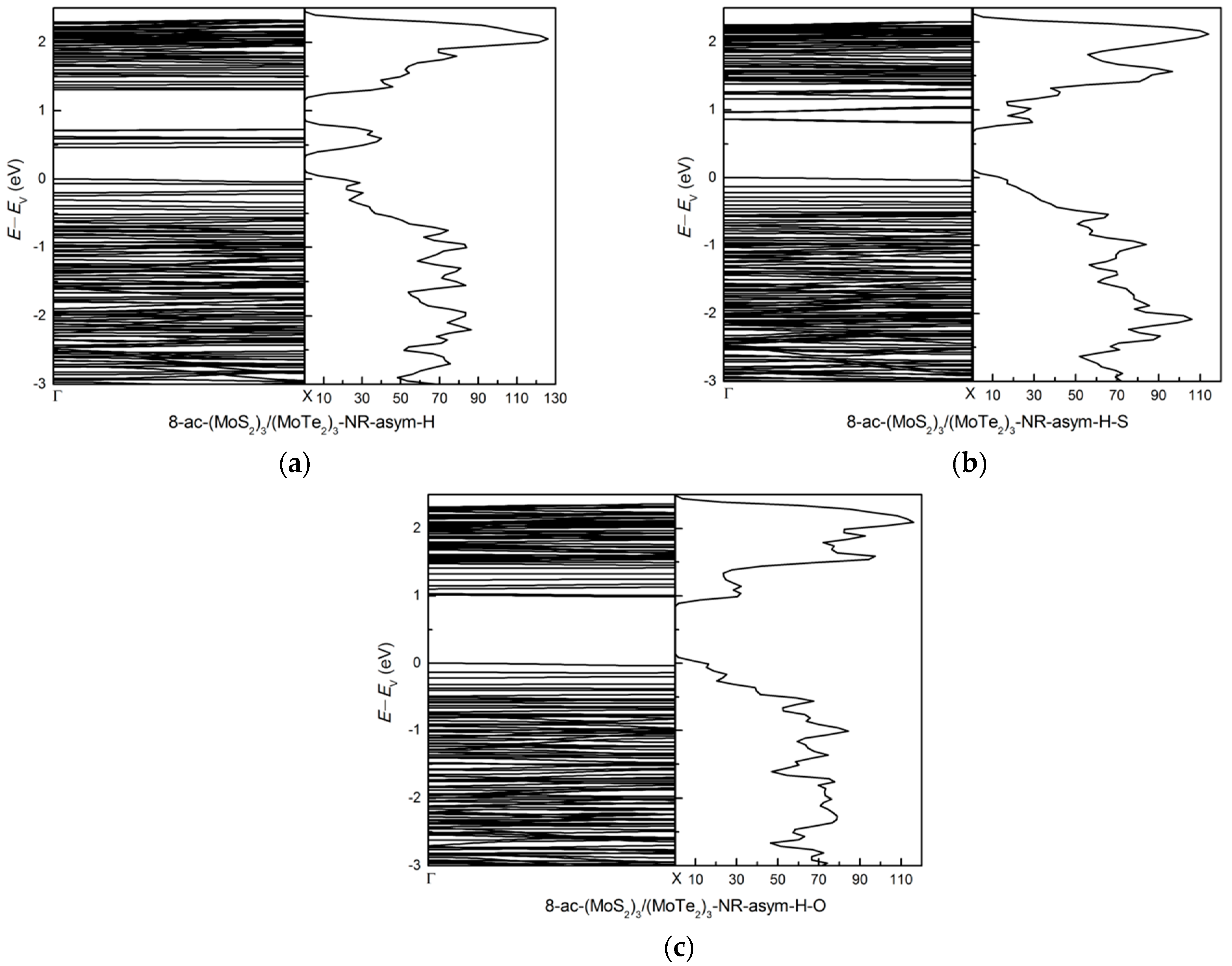
3.2.2. Zigzag and Armchair (MoS2)3/(MoTe2)3 Lateral Nanoribbons
3.2.3. Symmetric and Asymmetric MoS2(x)Te2(1−x) Alloy (x = 0.5) Armchair Nanoribbons
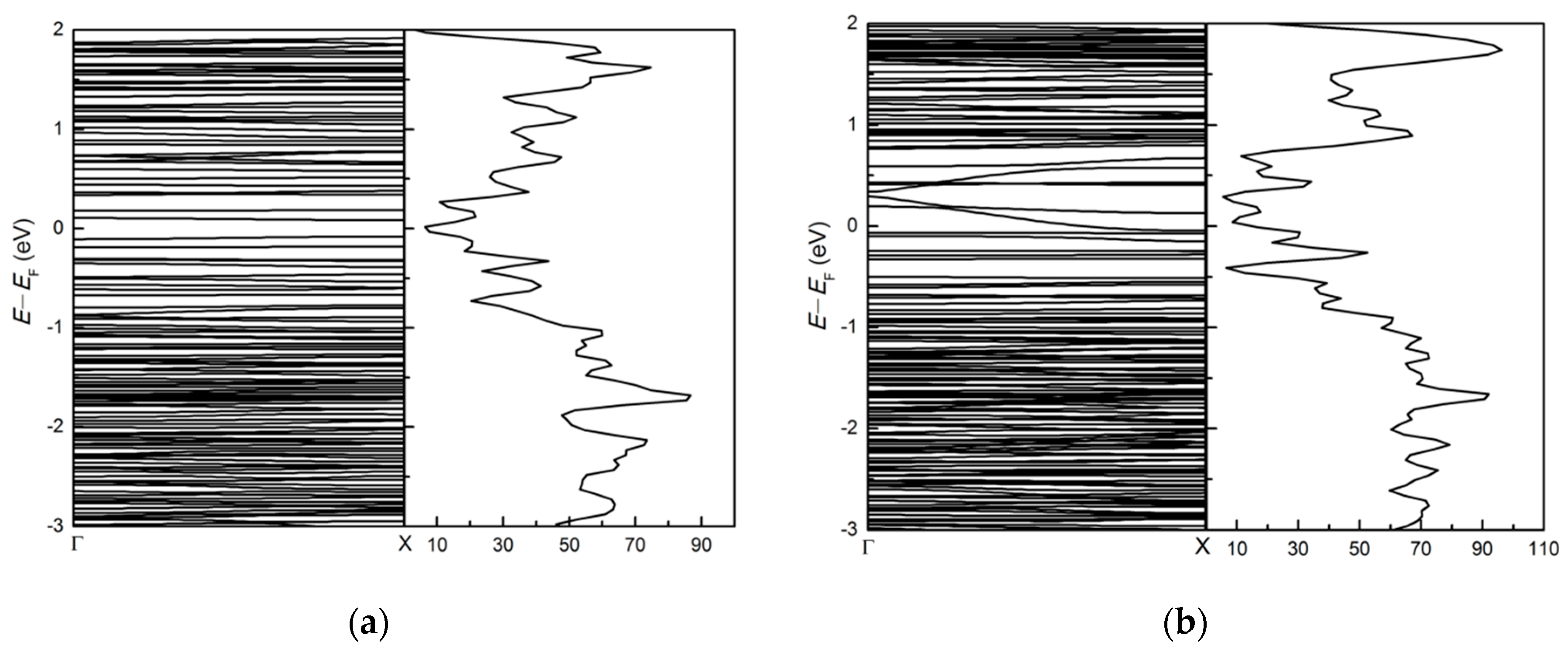
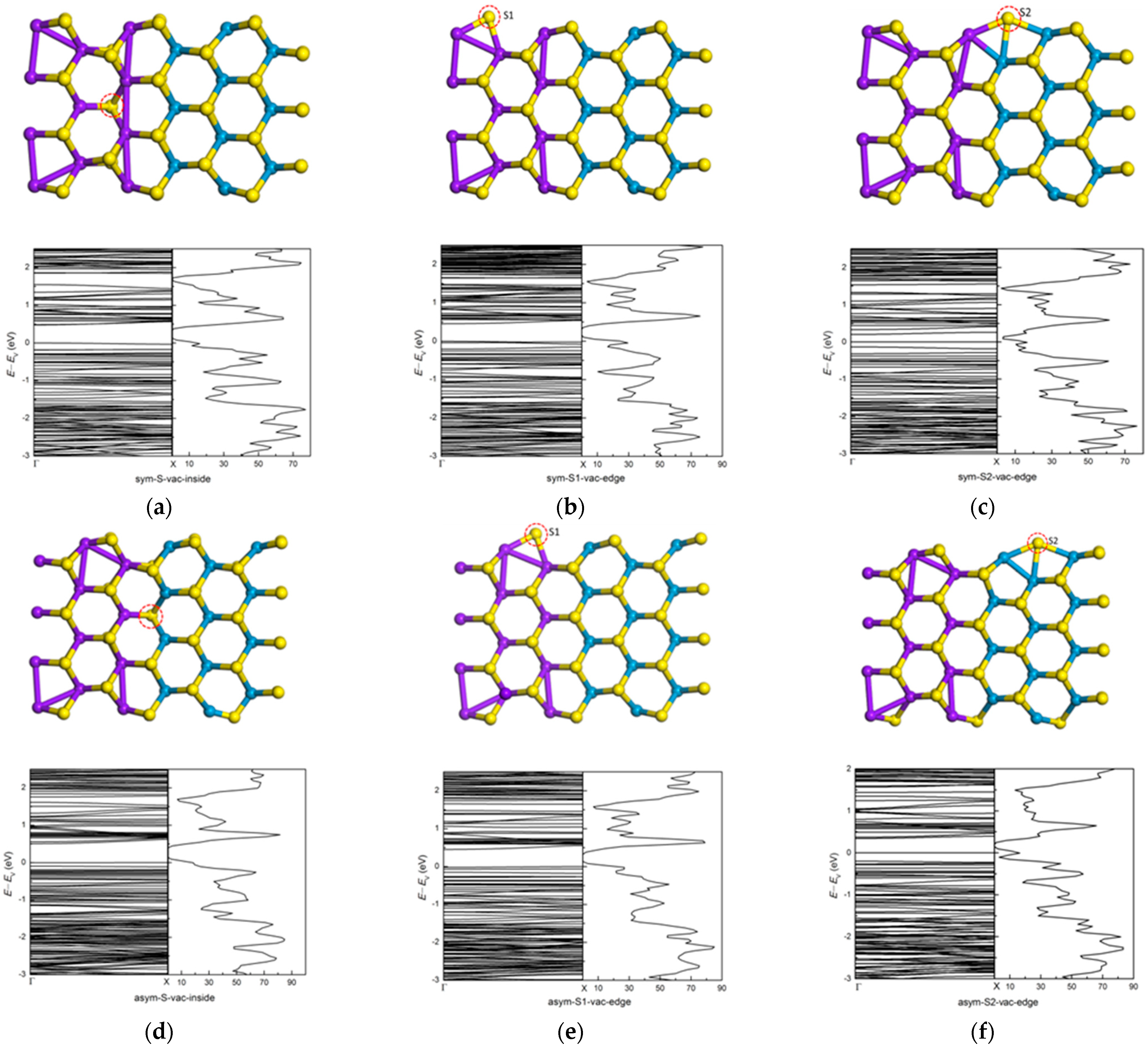
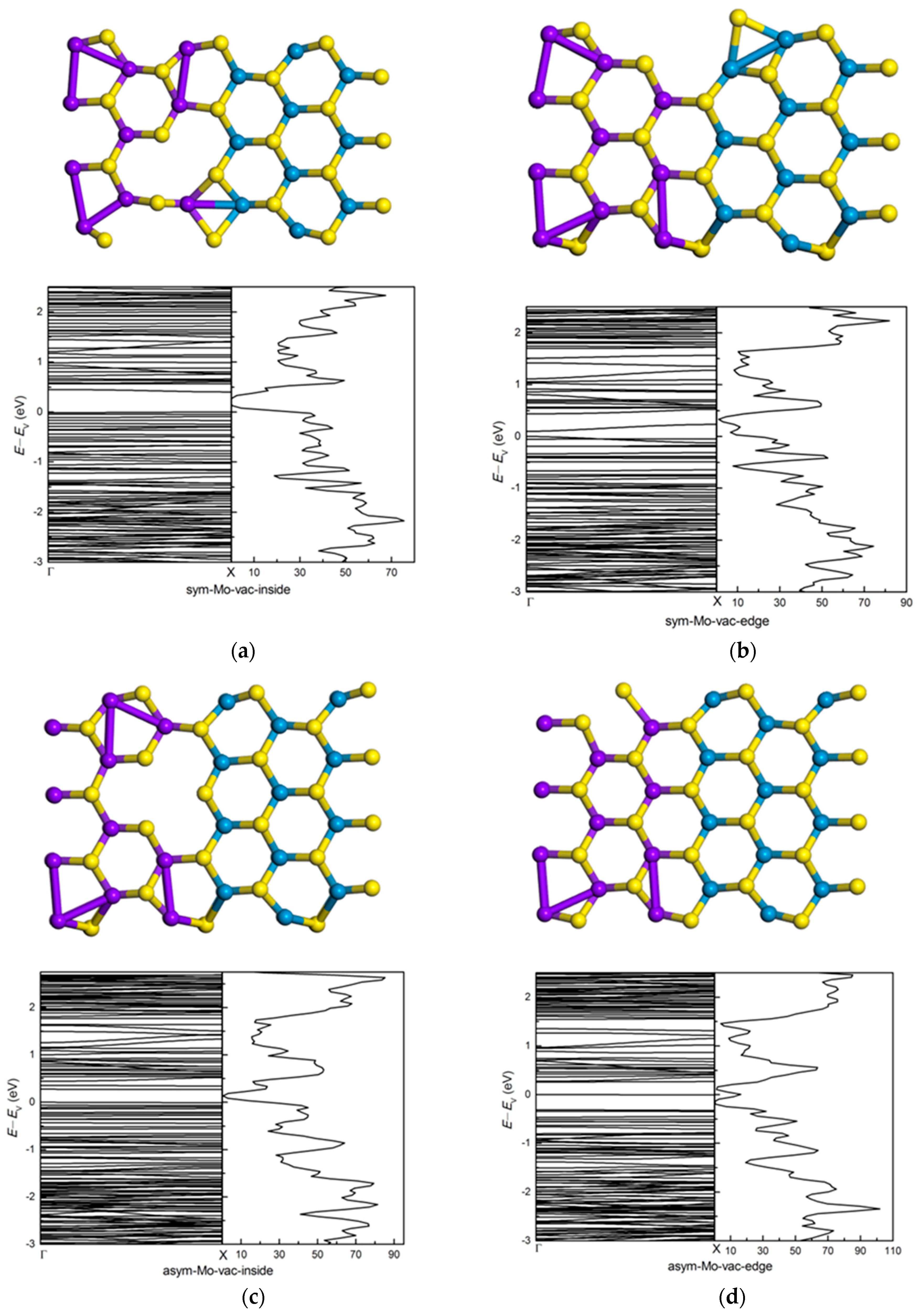
3.3. Effects of Atomic Vacancies in (MoS2)3/(WS2)3 Armchair Nanoribbon
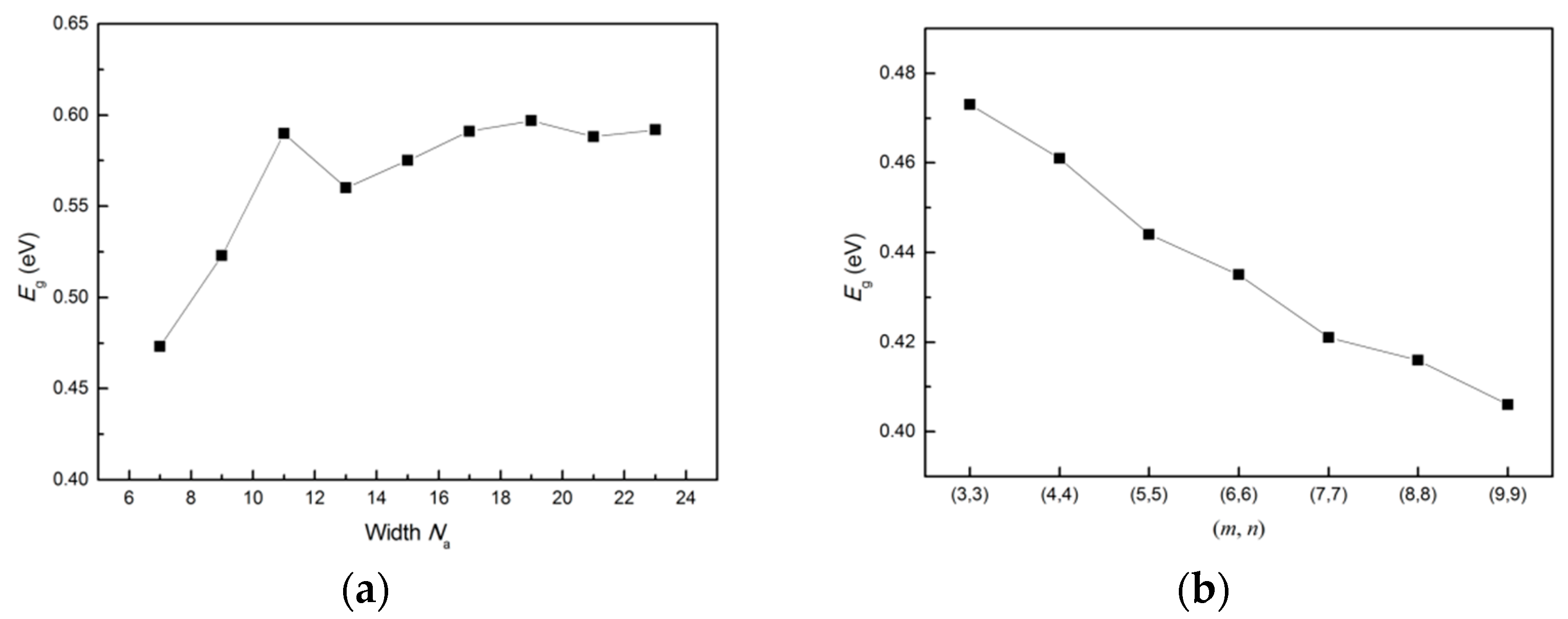
3.4. Band Gap Variation with Ribbon Width and Period
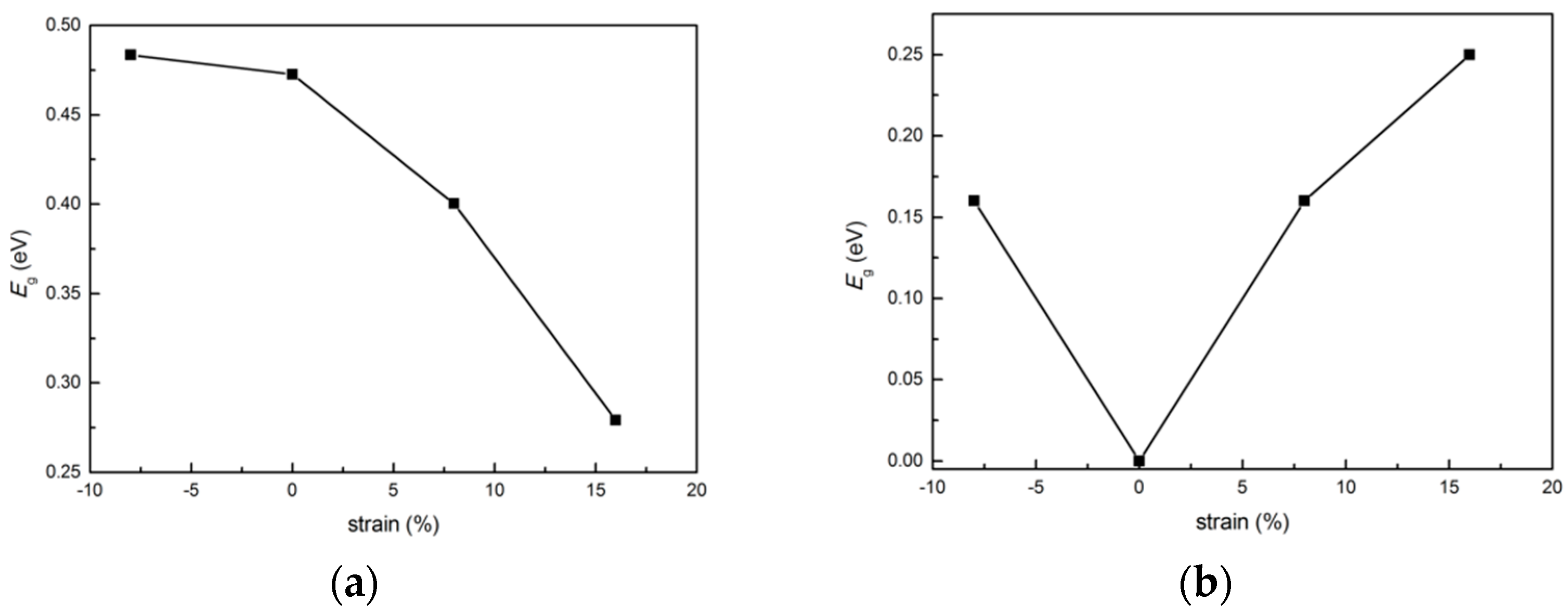
3.5. Bang Gap Variation with Biaxial Strain
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Matter. 2007, 6, 183–191. [Google Scholar] [CrossRef]
- Li, X.S.; Cai, W.W.; An, J.H.; Kim, S.; Nah, J.; Yang, D.X.; Piner, R.; Velamakanni, A.; Jung, I.; Tutuc, E.; et al. Large-Area Synthesis of High-Quality and Uniform Graphene Films on Copper foils. Science 2009, 324, 1312–1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oughaddou, H.; Enriquez, H.; Tchalala, M.R.; Yildirim, H.; Mayne, A.J.; Bendounan, A.; Dujardin, G.; Ali, M.A.; Kara, A. Silicene, a promising new 2D material. Prog. Surf. Sci. 2015, 90, 46–83. [Google Scholar]
- Song, L.; Ci, L.; Lu, H.; Sorokin, P.B.; Jin, C.; Ni, J.; Kvashnin, A.G.; Kvashnin, D.G.; Lou, J.; Yakobson, B.I.; et al. Large Scale Growth and Characterization of Atomic Hexagonal Boron Nitride Layers. Nano Lett. 2010, 10, 3209–3215. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Neal, A.T.; Zhu, Z.; Luo, Z.; Xu, X.; Tomnek, D.; Ye, P.D. Phosphorene: An unexplored 2D semiconductor with a high hole mobility. ACS Nano 2014, 8, 4033–4041. [Google Scholar] [CrossRef] [Green Version]
- Kotekar-Patil, D.; Deng, J.; Wong, S.L.; Lau, C.S.; Goh, K.E.J. Single layer MoS2 nanoribbon field effect transistor. Appl. Phys. Lett. 2019, 114, 013508. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, K.; Yamauchi, J. Electronic structure and scanning-tunneling- microscopy image of molybdenum dichalcogenide surfaces. Phys. Rev. B 1995, 51, 17085–17095. [Google Scholar] [CrossRef]
- Davelou, D.; Kopidakis, G.; Kaxiras, E.; Remediakis, I.N. Nanoribbon edges of transition-metal dichalcogenides: Stability and electronic properties. Phys. Rev. B 2017, 96, 165436. [Google Scholar] [CrossRef] [Green Version]
- Gusakova, J.; Wang, X.; Shiau, L.L.; Krivosheeva, A.; Shaposhnikov, V.; Borisenko, V.; Gusakov, V.; Tay, B.K. Electronic Properties of Bulk and Monolayer TMDs: Theoretical Study within DFT Framework (GVJ-2e Method). Phys. Stat. Sol. A 2017, 214, 1700218. [Google Scholar] [CrossRef]
- Butler, S.Z.; Hollen, S.M.; Cao, L.; Cui, Y.; Gupta, J.A.; Gutiérrez, H.R.; Heinz, T.F.; Hong, S.S.; Huang, J.; Ismach, A.F.; et al. Progress, challenges, and opportunities in two-dimensional materials beyond graphene. ACS Nano 2013, 7, 2898–2926. [Google Scholar] [CrossRef]
- Radisavljevic, B.; Radenovic, A.; Brivio, J.; Giacometti, V.; Kis, A. Single-layer MoS2 transistors. Nat. Nanotechnol. 2011, 6, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Mak, K.F.; Lee, C.; Hone, J.; Shan, J.; Heinz, T.F. Atomically Thin MoS2: A New Direct-Gap Semiconductor. Phys. Rev. Lett. 2010, 105, 136805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albe, K.; Klein, A. Density-functional-theory calculations of electronic band structure of single-crystal and single layer WS2. Phys. Rev. B 2002, 66, 73413. [Google Scholar] [CrossRef]
- Böker, T.; Severin, R.; Müller, A.; Janowitz, C.; Manzke, R. Band structure of MoS2, MoSe2, and α-MoTe2: Angle-resolved photoelectron spectroscopy and ab initio calculations. Phys. Rev. B 2001, 64, 235305. [Google Scholar] [CrossRef] [Green Version]
- Conley, H.J.; Wang, B.; Ziegler, J.I.; Haglund, R.F.; Pantelides, S.T.; Bolotin, K.I. Bandgap Engineering of Strained Monolayer and Bilayer MoS2. Nano Lett. 2013, 13, 3626–3630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kam, K.K.; Parkinson, B.A. Detailed Photocurrent Spectroscopy of the Semiconducting Group VI Transition Metal Dichalcogenides. J. Phys. Chem. 1982, 86, 463–467. [Google Scholar] [CrossRef]
- Duan, X.; Wang, C.; Shaw, J.C.; Cheng, R.; Chen, Y.; Li, H.; Wu, X.; Tang, Y.; Zhang, Q.; Pan, A.; et al. Lateral epitaxial growth of two-dimensional layered semiconductor heterojunctions. Nat. Nanotechnol. 2014, 9, 1024–1030. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, P.; Duan, X.; Zang, K.; Luo, J.; Duan, X. Robust epitaxial growth of two-dimensional heterostructures, multiheterostructures, and superlattices. Science 2017, 357, 788–792. [Google Scholar] [CrossRef] [Green Version]
- Oh, J.; Kim, Y.; Chung, S.; Kim, H.; Son, J.G. Fabrication of a MoS2/Graphene nanoribbon heterojunction network for improved thermoelectric properties. Adv. Mater. Interfaces 2019, 6, 1901333. [Google Scholar] [CrossRef]
- Xie, S.; Tu, L.; Han, Y.; Huang, L.; Kang, K.; Lao, K.U.; Poddar, P.; Park, C.; Muller, D.A.; DiStasio, R.A., Jr.; et al. Coherent, atomically thin transition-metal dichalcogenide superlattices with engineered strain. Science 2018, 359, 1131–1136. [Google Scholar] [CrossRef] [Green Version]
- Ding, G.; He, J.; Gao, G.Y.; Yao, K. Two-dimensional MoS2-MoSe2 lateral superlattice with minimized lattice thermal conductivity. J. Appl. Phys. 2018, 124, 165101. [Google Scholar] [CrossRef]
- Li, X.D.; Yu, S.; Wu, S.Q.; Wen, Y.H.; Zhou, S.; Zhu, Z.Z. Structural and Electronic Properties of Superlattice Composed of Graphene and Monolayer MoS2. J. Phys. Chem. C 2013, 117, 15347–15353. [Google Scholar] [CrossRef]
- Chen, Y.; Cui, P.; Ren, X.; Zhang, C.; Jin, C.; Zhang, Z.; Shih, C.K. Fabrication of MoSe2 nanoribbons via an unusual morphological phase transition. Nature Commun. 2017, 8, 15315. [Google Scholar] [CrossRef] [Green Version]
- Zan, W.; Zhang, Z.; Yang, Y.; Yao, X.; Li, S.; Yakobson, B.I. Width-dependent phase crossover in transition metal dichalcogenide nanoribbons. Nanotechnology 2019, 30, 075701. [Google Scholar] [CrossRef]
- Li, Q.; Newberg, J.T.; Walter, E.C.; Hemminger, J.C.; Penner, R.M. Polycrystalline Molybdenum Disulfide (2H-MoS2) Nano- and Microribbons by Electrochemical/Chemical Synthesis. Nano Lett. 2004, 4, 277–281. [Google Scholar] [CrossRef]
- Li, Y.F.; Zhou, Z.; Zhang, S.B.; Chen, Z.F. MoS2 Nanoribbons: High Stability and Unusual Electronic and Magnetic Properties. J. Am. Chem. Soc. 2008, 130, 16739–16744. [Google Scholar] [CrossRef]
- Ataca, C.; Sahin, H.; Akturk, E.; Ciraci, S. Mechanical and Electronic Properties of MoS2 Nanoribbons and Their Defects. J. Phys. Chem. C 2011, 115, 3934–3941. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, B.; Gupta, A. Effect of nanoribbon width and strain on the electronic properties of the WS2 nanoribbon. J. Electron. Devices 2014, 20, 1746–1754. [Google Scholar] [CrossRef] [Green Version]
- Erdogan, E.; Popov, I.H.; Enyashin, A.N.; Seifert, G. Transport properties of MoS2 nanoribbons: Edge priority. Eur. Phys. J. B 2012, 85, 33. [Google Scholar] [CrossRef]
- Fei, J.; Meng, L.; Nie, T.; Luan, Y. Electronic properties of 1T-MoS2 nanoribbon and its homojunction nanoribbon. Phys. Stat. Sol. B 2017, 254, 1600728. [Google Scholar] [CrossRef]
- López-Urías, F.; Elías, A.L.; Perea-López, N.; Gutiérrez, H.R.; Terrones, M.; Terrones, H. Electronic, magnetic, optical, and edge-reactivity properties of semiconducting and metallic WS2 nanoribbons. 2D Mater. 2015, 2, 015002. [Google Scholar] [CrossRef]
- Pan, H.; Zhang, Y.W. Edge-dependent structural, electronic and magnetic properties of MoS2 nanoribbons. J. Mater. Chem. 2012, 22, 7280. [Google Scholar] [CrossRef]
- Parija, A.; Choi, Y.H.; Liu, Z.; Andrews, J.L.; De Jesus, L.R.; Fakra, S.C.; Al-Hashimi, M.; Batteas, J.D.; Prendergast, D.; Banerjee, S. Mapping catalytically relevant edge electronic states of MoS2. ACS Cent. Sci. 2018, 4, 493–503. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wan, L.; Yu, Y.; Wang, B.; Xu, F.; Wei, Y.; Zhao, Y. Modulation of Electronic Structure of Armchair MoS2 Nanoribbon. J. Phys. Chem. C 2015, 119, 22164–22171. [Google Scholar] [CrossRef]
- Wang, R.; Zhou, X.; Xu, X.; Hu, J.; Pan, J. The indirect–direct band gap tuning in armchair MoS2 nanoribbon by edge passivation. J. Phys. D: Appl. Phys. 2017, 50, 095102. [Google Scholar] [CrossRef]
- DavoodianIdalik, M.; Kordbacheh, A.; Velashjerdi, F. Structural, electronic and transport properties of an edge terminated armchair MoS2 nanoribbon with N, O and F atoms. AIP Adv. 2019, 9, 035144. [Google Scholar] [CrossRef] [Green Version]
- Moghadasi, A.; Roknabadi, M.R.; Ghorbani, S.R.; Modarresi, M. Electronic and phononic modulation of MoS2 under biaxial strain. Physica B 2017, 526, 96–101. [Google Scholar] [CrossRef]
- Dong, L.; Namburu, R.R.; O’Regan, T.P.; Dubey, M.; Dongare, A.M. Theoretical study on strain-induced variations in electronic properties of monolayer MoS2. J. Mater. Sci. 2014, 49, 6762–6771. [Google Scholar] [CrossRef]
- Soni, H.R.; Fyta, M. Two-Dimensional Metallic/Semiconducting MoS2 under Biaxial Strain. ACS Appl. Nano Mater. 2018, 1, 5562–5570. [Google Scholar] [CrossRef]
- Chen, S.F.; Wu, Y.R. Electronic properties of MoS2 nanoribbon with strain using tight-binding method. Phys. Stat. Sol. B 2017, 254, 1600565. [Google Scholar] [CrossRef]
- Lloyd, D.; Liu, X.; Christopher, J.W.; Cantley, L.; Wadehra, A.; Kim, B.L.; Goldberg, B.B.; Swan, A.K.; Bunch, J.S. Band Gap Engineering with Ultralarge Biaxial Strains in Suspended Monolayer MoS2. Nano Lett. 2016, 16, 5836–5841. [Google Scholar] [CrossRef] [Green Version]
- Lu, P.; Wu, X.; Guo, W.; Zeng, X.C. Strain-dependent electronic and magnetic properties of MoS2 monolayer, bilayer, nanoribbons and nanotubes. Phys. Chem. Chem. Phys. 2012, 14, 13035–13040. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Ju, W.; Zhang, R.; Guo, C.; Zheng, J.; Yong, Y.; Li, X. Bandgap engineering of MoS2/MX2 (MX2 = WS2, MoSe2 and WSe2) heterobilayers subjected to biaxial strain and normal compressive strain. RSC Adv. 2016, 6, 18319. [Google Scholar] [CrossRef]
- Guo, H.; Lu, N.; Wang, L.; Wu, X.; Zeng, X.C. Tuning electronic and magnetic properties of early transition-metal dichalcogenides via tensile strain. J. Phys. Chem. C 2014, 118, 7242–7249. [Google Scholar] [CrossRef] [Green Version]
- Xi, J.; Zhao, T.; Wang, D.; Shuai, Z. Tunable electronic properties of two-dimensional transition metal dichalcogenide alloys: A First-Principles prediction. J. Phys. Chem. Lett. 2014, 5, 285–291. [Google Scholar] [CrossRef]
- Johari, P.; Shenoy, V.B. Tuning the electronic properties of semiconducting transition metal dichalcogenides by applying mechanical strains. ACS Nano 2012, 6, 5449–5456. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Poole, C.; Mak, K.F.; Shan, J. Experimental demonstration of continuous electronic structure tuning via strain in atomically thin MoS2. Nano Lett. 2013, 13, 2931–2936. [Google Scholar] [CrossRef] [Green Version]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Nardelli, M.B.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef] [Green Version]
- Rappe, A.M.; Rabe, K.M.; Kaxiras, E.; Joannopoulos, J.D. Optimized pseudopotentials. Phys. Rev. B 1990, 41, 1227. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.W.; Kwon, H.; Kim, S.K.; Ryu, S.; Yun, W.S.; Kim, D.H.; Hwang, J.H.; Kang, J.S.; Baik, J.; Shin, H.J.; et al. Band-gap Transition Induced by Interlayer Van der Waals Interaction in MoS2. Phys. Rev. B 2011, 84, 045409. [Google Scholar] [CrossRef]
- Lu, N.; Guo, H.; Wang, L.; Wu, X.; Zeng, X.C. van der Waals trilayers and superlattices: Modification of electronic structures of MoS2 by intercalation. Nanoscale 2014, 6, 4566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, G.P. Theoretical Modelling of Semiconductor Surfaces; World Scientific: Singapore, 1999. [Google Scholar]
- Li, X.M.; Long, M.Q.; Cui, L.L.; Yang, K.W.; Zhang, D.; Ding, J.F.; Xu, H. Effects of line defects on spin-dependent electronic transport of zigzag MoS2 nanoribbons. AIP Adv. 2016, 6, 015015. [Google Scholar] [CrossRef] [Green Version]
- Ataca, C.; Ciraci, S. Functionalization of Single-Layer MoS2 Honeycomb Structures. J. Phys. Chem. C 2011, 115, 13303–13311. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhang, K.; Xie, G. Remarkable suppression of thermal conductivity by point defects in MoS2 nanoribbons. Appl. Surf. Sci. 2016, 360, 107–112. [Google Scholar] [CrossRef]
- Han, Y.; Zhou, J.; Dong, J. Electronic and magnetic properties of MoS2 nanoribbons with sulfurline vacancy defects. Appl. Surf. Sci. 2015, 346, 470–476. [Google Scholar] [CrossRef]
- Xiao, S.L.; Yu, W.Z.; Gao, S.P. Edge preference and band gap characters of MoS2 and WS2 nanoribbons. Surf. Sci. 2016, 653, 107–112. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Edge Energy (eV) | Interface Energy (eV) | Passivation Energy (eV) |
|---|---|---|---|
| zz-(MoS2)3/(WS2)3-NR | +0.89 | −2.27 | |
| zz-(MoS2)3/(WS2)3-NR-H | −0.59 | ||
| 7-ac-(MoS2)3/(WS2)3-NR-sym | −0.26 | −2.28 | |
| 7-ac-(MoS2)3/(WS2)3-NR-sym-H | −8.84 | ||
| 7-ac-(MoS2)3/(WS2)3-NR-sym-H-S | −2.13 | ||
| 7-ac-(MoS2)3/(WS2)3-NR-sym-H-O | −3.86 | ||
| 8-ac-(MoS2)3/(WS2)3-NR-asym | +3.10 | −2.28 | |
| 8-ac-(MoS2)3/(WS2)3-NR-asym-H | −9.15 | ||
| 8-ac-(MoS2)3/(WS2)3-NR-asym-H-S | −2.44 | ||
| 8-ac-(MoS2)3/(WS2)3-NR-asym-H-O | −4.22 | ||
| zz-(MoS2)3/(MoTe2)3-NR | −3.09 | −5.89 | |
| zz-(MoS2)3/(MoTe2)3-NR-H | −0.34 | ||
| 7-ac-(MoS2)3/(MoTe 2)3-NR-sym | −1.89 | −3.48 | |
| 7-ac-(MoS2)3/(MoTe 2)3-NR-sym-H | −8.48 | ||
| 7-ac-(MoS2)3/(MoTe 2)3-NR-sym-H-S | −1.72 | ||
| 7-ac-(MoS2)3/(MoTe 2)3-NR-sym-H-O | −3.24 | ||
| 8-ac-(MoS2)3/(MoTe2)3-NR-asym | −2.70 | −4.08 | |
| 8-ac-(MoS2)3/(MoTe2)3-NR-asym-H | −8.26 | ||
| 8-ac-(MoS2)3/(MoTe2)3-NR-asym-H-S | −1.50 | ||
| 8-ac-(MoS2)3/(MoTe2)3-NR-asym-H-O | −3.10 |
| Systems | (Å) | (Å) | (Å) | (Å) | (Å) | (Å) | (Å) | ω1 | ω2 | ω3 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| (MoS2)3/(WS2)3 _ANR | sym | 0.266 | 0.271 | −0.074 (S-Mo) −0.086 (S-W) | 0.340 | 0.357 | 10° | 12° | |||
| asym | 0.275 | 0.277 | −0.077 (S-Mo) −0.088 (S-W) | 0.352 | 0.365 | 11° | 13° | ||||
| (MoS2)3/(MoTe2)3 _ANR | sym | 0.469 (Mo-S) 1.020 (Mo-Te) | 0.114 | −0.803 | 0.355 | 1.823 | 11° | 48° | |||
| asym | 0.509 (Mo-S) 0.969 (Mo-Te) | 0.057 | −0.869 | 0.452 | 1.838 | 16° | 50° | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Srivastava, G.P. Tunable Electronic Properties of Lateral Monolayer Transition Metal Dichalcogenide Superlattice Nanoribbons. Nanomaterials 2021, 11, 534. https://doi.org/10.3390/nano11020534
Wang J, Srivastava GP. Tunable Electronic Properties of Lateral Monolayer Transition Metal Dichalcogenide Superlattice Nanoribbons. Nanomaterials. 2021; 11(2):534. https://doi.org/10.3390/nano11020534
Chicago/Turabian StyleWang, Jinhua, and Gyaneshwar P. Srivastava. 2021. "Tunable Electronic Properties of Lateral Monolayer Transition Metal Dichalcogenide Superlattice Nanoribbons" Nanomaterials 11, no. 2: 534. https://doi.org/10.3390/nano11020534