
Bioconjugation of a PNA Probe to Zinc Oxide Nanowires for Label-Free Sensing

 ,
, 
 , , , , , ,
, , , , , ,  ,
,  and
and
Abstract
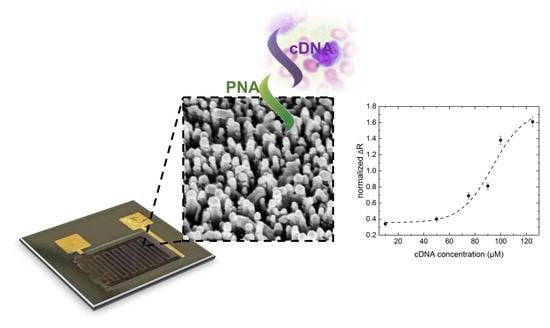
:
1. Introduction
2. Experimental Section
2.1. Chemicals and Reagents
2.2. PNA Synthesis and Analysis
2.3. DNA Synthesis and Analysis
2.4. Circular Dichroism (CD)
2.5. Non-Denaturing Polyacrylamide Gel Electrophoresis (PAGE)
2.6. Fabrication Process of Zinc Oxide Nanowires Electrical Sensor
2.7. Functionalization of ZnONWs Surface
2.8. Scanning Electron Microscopy
2.9. Hybridization Experiments
2.10. Photoluminescence Spectroscopy
2.11. Fluorescence Microscopy
2.12. Electrical Measurements
3. Results and Discussion
3.1. PNA/DNA Interaction in Solution Studies
3.2. The ZnONW Electrical Sensor: Geometry and Morphology
3.3. Bioconjugation of PNA to the ZnONW Surface
3.4. The Interaction between PNA-Modified ZnONWs and DNA
3.5. PNA-ZnONWs Transducer Specificity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arshavsky-Graham, S.; Massad-Ivanir, N.; Segal, E.; Weiss, S. Porous Silicon-Based Photonic Biosensors: Current Status and Emerging Applications. Anal. Chem. 2019, 91, 441–467. [Google Scholar] [CrossRef] [PubMed]
- Terracciano, M.; Rea, I.; Borbone, N.; Moretta, R.; Oliviero, G.; Piccialli, G.; De Stefano, L. Porous silicon-based aptasensors: The next generation of label-free devices for health monitoring. Molecules 2019, 24, 2216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bala, A.; Górski, Ł. Application of nucleic acid analogues as receptor layers for biosensors. Anal. Methods 2016, 8, 236–244. [Google Scholar] [CrossRef]
- Teles, F.R.R.; Fonseca, L.P. Trends in DNA biosensors. Talanta 2008, 77, 606–623. [Google Scholar] [CrossRef]
- Musumeci, D.; Platella, C.; Riccardi, C.; Moccia, F.; Montesarchio, D. Fluorescence Sensing Using DNA Aptamers in Cancer Research and Clinical Diagnostics. Cancers 2017, 9, 174. [Google Scholar] [CrossRef] [Green Version]
- Uhlmann, E.; Peyman, A.; Breipohl, G.; Will, D.W. PNA: Synthetic Polyamide Nucleic Acids with Unusual Binding Properties. Angew. Chemie Int. Ed. 1998, 37, 2796–2823. [Google Scholar] [CrossRef]
- Moretta, R.; Terracciano, M.; Borbone, N.; Oliviero, G.; Schiattarella, C.; Piccialli, G.; Falanga, A.P.; Marzano, M.; Dardano, P.; de Stefano, L.; et al. PNA-based graphene oxide/porous silicon hybrid biosensor: Towards a label-free optical assay for brugada syndrome. Nanomaterials 2020, 10, 2233. [Google Scholar] [CrossRef]
- Nielsen, P.E.; Egholm, M. An introduction to peptide nucleic acid. Curr. Issues Mol. Biol. 1999, 1, 89–104. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Mishra, A.; Puri, N. Peptide nucleic acids: Advanced tools for biomedical applications. J. Biotechnol. 2017, 259, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Sharma, C.; Awasthi, S.K. Versatility of peptide nucleic acids (PNAs): Role in chemical biology, drug discovery, and origins of life. Chem. Biol. Drug Des. 2017, 89, 16–37. [Google Scholar] [CrossRef]
- Amato, F.; Tomaiuolo, R.; Nici, F.; Borbone, N.; Elce, A.; Catalanotti, B.; D’Errico, S.; Morgillo, C.M.; de Rosa, G.; Mayol, L.; et al. Exploitation of a Very Small Peptide Nucleic Acid as a New Inhibitor of miR-509-3p Involved in the Regulation of Cystic Fibrosis Disease-Gene Expression. Biomed Res. Int. 2014, 2014, 610718. [Google Scholar] [CrossRef] [PubMed]
- Falanga, A.P.; Cerullo, V.; Marzano, M.; Feola, S.; Oliviero, G.; Piccialli, G.; Borbone, N. Peptide Nucleic Acid-Functionalized Adenoviral Vectors Targeting G-Quadruplexes in the P1 Promoter of Bcl-2 Proto-Oncogene: A New Tool for Gene Modulation in Anticancer Therapy. Bioconjug. Chem. 2019, 30, 572–582. [Google Scholar] [CrossRef] [PubMed]
- Amato, J.; Pagano, B.; Borbone, N.; Oliviero, G.; Gabelica, V.; de Pauw, E.; Derrico, S.; Piccialli, V.; Varra, M.; Giancola, C.; et al. Targeting G-Quadruplex structure in the human c-Kit promoter with short PNA sequences. Bioconjug. Chem. 2011, 22, 654–663. [Google Scholar] [CrossRef] [Green Version]
- Fabbri, E.; Tamanini, A.; Jakova, T.; Gasparello, J.; Manicardi, A.; Corradini, R.; Sabbioni, G.; Finotti, A.; Borgatti, M.; Lampronti, I.; et al. A peptide nucleic acid against microrna miR-145-5p enhances the expression of the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) in Calu-3 Cells. Molecules 2018, 23, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terracciano, M.; de Stefano, L.; Borbone, N.; Politi, J.; Oliviero, G.; Nici, F.; Casalino, M.; Piccialli, G.; Dardano, P.; Varra, M.; et al. Solid phase synthesis of a thrombin binding aptamer on macroporous silica for label free optical quantification of thrombin. RSC Adv. 2016, 6, 86762–86769. [Google Scholar] [CrossRef]
- Papari, G.P.; Silvestri, B.; Vitiello, G.; de Stefano, L.; Rea, I.; Luciani, G.; Aronne, A.; Andreone, A. Morphological, Structural, and Charge Transfer Properties of F-Doped ZnO: A Spectroscopic Investigation. J. Phys. Chem. C 2017, 121, 16012–16020. [Google Scholar] [CrossRef]
- Caicedo, N.; Leturcq, R.; Raskin, J.P.; Flandre, D.; Lenoble, D. Detection mechanism in highly sensitive ZnO nanowires network gas sensors. Sens. Actuators B 2019, 297, 126602. [Google Scholar] [CrossRef]
- Li, X.; Zhao, C.; Liu, X. A paper-based microfluidic biosensor integrating zinc oxide nanowires for electrochemical glucose detection. Microsys. Nanoeng. 2015, 1, 1. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wang, Y.H.; Zhao, C.; Liu, X. Paper-based piezoelectric touch pads with hydrothermally grown zinc oxide nanowires. ACS Appl. Mater. Interfaces 2014, 6, 22004–22012. [Google Scholar] [CrossRef]
- Ko, S.H.; Ko, S.H.; Lee, D.; Hotz, N.; Yeo, J.; Hong, S.; Nam, K.H.; Grigoropoulos, C.P. Digital selective growth of ZnO nanowire arrays from inkjet-printed nanoparticle seeds on a flexible substrate. Langmuir 2012, 28, 4787–4792. [Google Scholar] [CrossRef]
- Chen, J.; Ruther, R.E.; Tan, Y.; Bishop, L.M.; Hamers, R.J. Molecular Adsorption on ZnO(1010) Single-Crystal Surfaces: Morphology and Charge Transfer. Langmuir 2012, 28, 10437–10445. [Google Scholar] [CrossRef]
- Chianese, G.; Terracciano, M.; Moretta, R.; Cappiello, P.; Vitiello, G.; Aronne, A.; Schiattarella, C.; de Stefano, L.; Rea, I. Synthesis and Surface Modification of Nanostructured F-Doped ZnO: Toward a Transducer for Label-Free Optical Biosensing. Appl. Sci. 2019, 9, 3380. [Google Scholar] [CrossRef] [Green Version]
- Politi, J.; Rea, I.; Dardano, P.; de Stefano, L.; Gioffrè, M. Versatile synthesis of ZnO nanowires for quantitative optical sensing of molecular biorecognition. Sens. Actuators B 2015, 220, 705–711. [Google Scholar] [CrossRef]
- Egholm, M.; Buchardt, O.; Christensen, L.; Behrens, C.; Freier, S.M.; Driver, D.A.; Berg, R.H.; Kim, S.K.; Norden, B.; Nielsen, P.E. PNA hybridizes to complementary oligonucleotides obeying the Watson–Crick hydrogen-bonding rules. Nature 1993, 365, 566–568. [Google Scholar] [CrossRef]
- Zarrilli, F.; Amato, F.; Morgillo, C.M.; Pinto, B.; Santarpia, G.; Borbone, N.; Errico, S.D.; Catalanotti, B.; Piccialli, G.; Castaldo, G.; et al. Peptide Nucleic Acids as miRNA Target Protectors for the Treatment of Cystic Fibrosis. Molecules 2017, 22, 1144. [Google Scholar] [CrossRef] [Green Version]
- Syed, A.; Kalloudis, M.; Koutsos, V.; Mastropaolo, E. Controlled hydrothermal growth of vertically-aligned zinc oxide nanowires using silicon and polyimide substrates. Microelectron. Eng. 2015, 145, 86–90. [Google Scholar] [CrossRef] [Green Version]
- Tsay, C.Y.; Hsiao, I.P.; Chang, F.Y.; Hsu, C.L. Improving the photoelectrical characteristics of self-powered p-GaN film/n-ZnO nanowires heterojunction ultraviolet photodetectors through gallium and indium co-doping. Mater. Sci. Semicond. Process. 2021, 121, 105295. [Google Scholar] [CrossRef]
- Park, G.C.; Hwang, S.M.; Lee, S.M.; Choi, J.H.; Song, K.M.; Kim, H.Y.; Kim, H.S.; Eum, S.J.; Jung, S.B.; Lim, J.H.; et al. Hydrothermally grown in-doped zno nanorods on p-gan films for color-tunable heterojunction light-emitting-diodes. Sci. Rep. 2015, 5, 1–10. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Sequence |
|---|---|
| PNA | AcNH-tttctctcccaa-KK-NH2 (N to C) |
| PNA* | FITC-(AEEA)2-tttctctcccaa-KK-NH2 (N to C) |
| cDNA | TTGGGAGAGAAA (5′-3′) |
| cDNA* | 6FAM-TTGGGAGAGAAA (5′-3′) |
| ctrlDNA | CCTTTTTTTTTT (5′-3′) |
| ctrlDNA* | 6FAM-CCTTTTTTTTTT (5′-3′) |
| Functionalization Step | ΔRN |
|---|---|
| APTES | 0.47 ± 0.03 |
| BS3 | 0.52 ± 0.04 |
| PNA | 0.46 ± 0.04 |
| Passivated | 0.59 ± 0.04 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crisci, T.; Falanga, A.P.; Casalino, M.; Borbone, N.; Terracciano, M.; Chianese, G.; Gioffrè, M.; D’Errico, S.; Marzano, M.; Rea, I.; et al. Bioconjugation of a PNA Probe to Zinc Oxide Nanowires for Label-Free Sensing. Nanomaterials 2021, 11, 523. https://doi.org/10.3390/nano11020523
Crisci T, Falanga AP, Casalino M, Borbone N, Terracciano M, Chianese G, Gioffrè M, D’Errico S, Marzano M, Rea I, et al. Bioconjugation of a PNA Probe to Zinc Oxide Nanowires for Label-Free Sensing. Nanomaterials. 2021; 11(2):523. https://doi.org/10.3390/nano11020523
Chicago/Turabian StyleCrisci, Teresa, Andrea Patrizia Falanga, Maurizio Casalino, Nicola Borbone, Monica Terracciano, Giovanna Chianese, Mariano Gioffrè, Stefano D’Errico, Maria Marzano, Ilaria Rea, and et al. 2021. "Bioconjugation of a PNA Probe to Zinc Oxide Nanowires for Label-Free Sensing" Nanomaterials 11, no. 2: 523. https://doi.org/10.3390/nano11020523