
NanoTraPPED—A New Method for Determining the Surface Energy of Nanoparticles via Pickering Emulsion Polymerization


Abstract
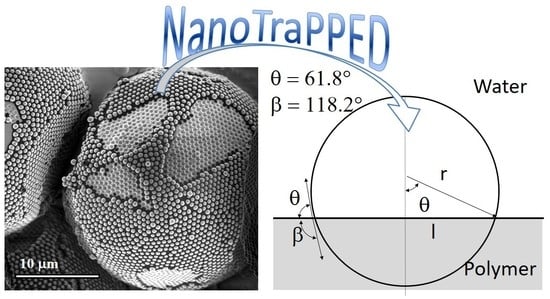
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis and Characterization of Silica Nanoparticles
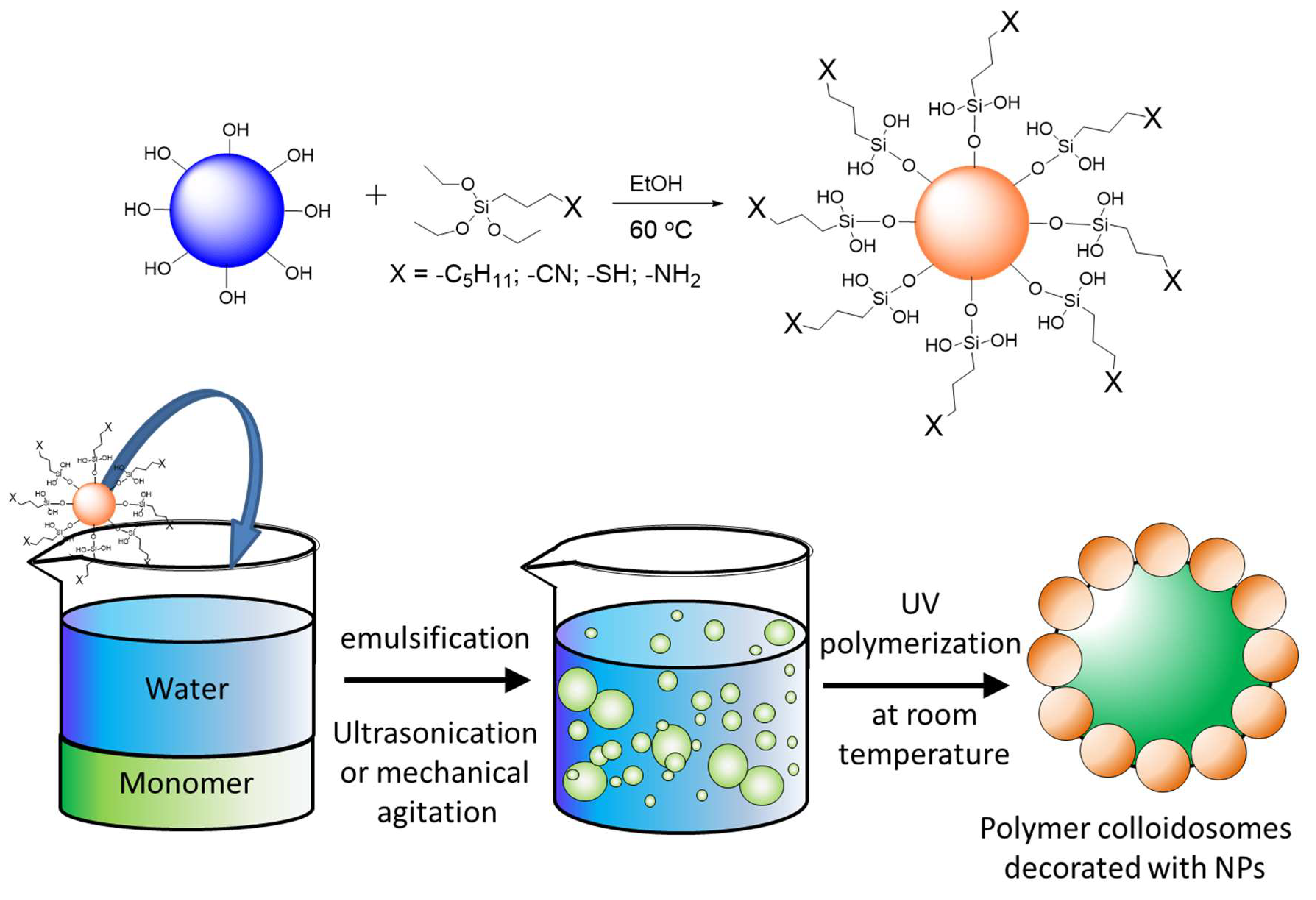
2.3. Surface Functionalization of Silica Nanoparticles
2.4. Pickering Emulsion Preparation and Polymerization
2.5. Measurement of the Contact Angle
3. Results
3.1. Synthesis of Silica Nanoparticles
3.2. Functionalization of Silica Nanoparticles
3.3. Generation of Pickering Emulsions Choice of Monomers
3.4. Pickering Emulsion Polymerization
4. Discussion
4.1. Measuring the Contact Angle of Nanoparticles at the Polymer-Water Interface
4.2. Adsorption Energy of the Nanoparticles
4.3. Determining the Surface Energy of Nanoparticles—The Polar and Disperse Components
4.4. Sources of Error, Limitations of the NanoTraPPED Method and Future Studies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Good, R.J. Contact Angle, Wetting, and Adhesion: A Critical Review. J. Adhes. Sci. Technol. 1993, 6, 3–36. [Google Scholar] [CrossRef]
- Owens, D.K.; Wendt, R.C. Estimation of the Surface Free Energy of Polymers. J. Appl. Polym. Sci. 1969, 13, 1741–1747. [Google Scholar] [CrossRef]
- Rabel, W. Einige Aspekte Der Benetzungstheorie Und Ihre Anwendung Auf Die Untersuchung Und Veränderung Der Oberflächeneigenschaften von Polymeren. Farbe Und Lack 1971, 77, 997–1005. [Google Scholar]
- Honciuc, A. Chemistry of Functional Materials Surfaces and Interfaces: Fundamentals and Applications, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2021; ISBN 978-0-12-821059-8. [Google Scholar]
- Fowkes, F.M. Determination of Interfacial Tensions, Contact Angles, and Dispersion Forces in Surfaces by Assuming Additivity of Intermolecular Interactions in Surfaces. J. Phys. Chem. 1962, 66, 382. [Google Scholar] [CrossRef]
- Olusanmi, D.; Jayawickrama, D.; Bu, D.; McGeorge, G.; Sailes, H.; Kelleher, J.; Gamble, J.F.; Shah, U.V.; Tobyn, M. A Control Strategy for Bioavailability Enhancement by Size Reduction: Effect of Micronization Conditions on the Bulk, Surface and Blending Characteristics of an Active Pharmaceutical Ingredient. Powder Technol. 2014, 258, 222–233. [Google Scholar] [CrossRef]
- Gigliobianco, M.; Casadidio, C.; Censi, R.; Di Martino, P. Nanocrystals of Poorly Soluble Drugs: Drug Bioavailability and Physicochemical Stability. Pharmaceutics 2018, 10, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karde, V.; Ghoroi, C. Fine Powder Flow under Humid Environmental Conditions from the Perspective of Surface Energy. Int. J. Pharm. 2015, 485, 192–201. [Google Scholar] [CrossRef]
- Parent, J.S.; Mrkoci, M.I.; Hennigar, S.L. Silica Agglomeration and Elastomer Reinforcement: Influence of Surface Modifications. Plast. Rubber Compos. 2003, 32, 114–121. [Google Scholar] [CrossRef]
- Lee, A.F.; Wilson, K. Recent Developments in Heterogeneous Catalysis for the Sustainable Production of Biodiesel. Catal. Today 2015, 242, 3–18. [Google Scholar] [CrossRef]
- Díaz, E.; Ordóñez, S.; Vega, A.; Coca, J. Characterization of Co, Fe and Mn-Exchanged Zeolites by Inverse Gas Chromatography. J. Chromatogr. A 2004, 1049, 161–169. [Google Scholar] [CrossRef]
- van der Zon, M.; Thoolen, H.; Hamersma, P.J.; Poels, E.K.; Bliek, A. Agglomeration and Adhesion of Catalyst Particles in Gas–Liquid Reactors. Catal. Today 2001, 66, 263–270. [Google Scholar] [CrossRef]
- Gromov, A.A.; Strokova, Y.I.; Teipel, U. Stabilization of Metal Nanoparticles—A Chemical Approach. Chem. Eng. Technol. 2009, 32, 1049–1060. [Google Scholar] [CrossRef]
- Shah, U.V.; Olusanmi, D.; Narang, A.S.; Hussain, M.A.; Tobyn, M.J.; Heng, J.Y.Y. Decoupling the Contribution of Dispersive and Acid-Base Components of Surface Energy on the Cohesion of Pharmaceutical Powders. Int. J. Pharm. 2014, 475, 592–596. [Google Scholar] [CrossRef]
- Karagüzel, C.; Can, M.F.; Sönmez, E.; Çelik, M.S. Effect of Electrolyte on Surface Free Energy Components of Feldspar Minerals Using Thin-Layer Wicking Method. J. Colloid Interface Sci. 2005, 285, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, M.; Hartmann, R. Specific Surface Free Energy Component Distributions and Flotabilities of Mineral Microparticles in Flotation—An Inverse Gas Chromatography Study. Colloids Surf. A Physicochem. Eng. Asp. 2017, 513, 380–388. [Google Scholar] [CrossRef]
- Zou, W.; Cao, Y.; Sun, C. Adsorption of Anionic Polyacrylamide onto Coal and Kaolinite: Changes of Surface Free Energy Components. Part. Sci. Technol. 2017, 35, 233–238. [Google Scholar] [CrossRef]
- Wu, D.; Binks, B.P.; Honciuc, A. Modeling the Interfacial Energy of Surfactant-Free Amphiphilic Janus Nanoparticles from Phase Inversion in Pickering Emulsions. Langmuir 2018, 34, 1225–1233. [Google Scholar] [CrossRef]
- Mihali, V.; Honciuc, A. Semiconductive Materials with Tunable Electrical Resistance and Surface Polarity Obtained by Asymmetric Functionalization of Janus Nanoparticles. Adv. Mater. Interfaces 2017, 4, 1700914. [Google Scholar] [CrossRef]
- Kawai, A.; Nagata, H.; Abe, H.; Takata, M. Adhesion between Photoresist and Inorganic Substrate. Jpn. J. Appl. Phys. 1991, 30, 121–125. [Google Scholar] [CrossRef]
- Levitin, G.; Timmons, C.; Hess, D.W. Photoresist and Etch Residue Removal. J. Electrochem. Soc. 2006, 153, G712. [Google Scholar] [CrossRef]
- Islam, M.S.; Tong, L.; Falzon, P.J. Influence of Metal Surface Preparation on Its Surface Profile, Contact Angle, Surface Energy and Adhesion with Glass Fibre Prepreg. Int. J. Adhes. Adhes. 2014, 51, 32–41. [Google Scholar] [CrossRef]
- Jamil, M.I.; Ali, A.; Haq, F.; Zhang, Q.; Zhan, X.; Chen, F. Icephobic Strategies and Materials with Superwettability: Design Principles and Mechanism. Langmuir 2018, 34, 15425–15444. [Google Scholar] [CrossRef]
- Wang, L.; McCarthy, T.J. Covalently Attached Liquids: Instant Omniphobic Surfaces with Unprecedented Repellency. Angew. Chem. Int. Ed. 2016, 55, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.-S.; Kang, S.H.; Tang, S.K.Y.; Smythe, E.J.; Hatton, B.D.; Grinthal, A.; Aizenberg, J. Bioinspired Self-Repairing Slippery Surfaces with Pressure-Stable Omniphobicity. Nature 2011, 477, 443–447. [Google Scholar] [CrossRef]
- Galet, L.; Patry, S.; Dodds, J. Determination of the Wettability of Powders by the Washburn Capillary Rise Method with Bed Preparation by a Centrifugal Packing Technique. J. Colloid Interface Sci. 2010, 346, 470–475. [Google Scholar] [CrossRef] [Green Version]
- Cui, Z.-G.; Binks, B.P.; Clint, J.H. Determination of Contact Angles on Microporous Particles Using the Thin-Layer Wicking Technique. Langmuir 2005, 21, 8319–8325. [Google Scholar] [CrossRef]
- Chibowski, E.; Gonzalez-Caballero, F. Theory and Practice of Thin-Layer Wicking. Langmuir 1993, 9, 330–340. [Google Scholar] [CrossRef]
- Preuss, M.; Butt, H.-J. Measuring the Contact Angle of Individual Colloidal Particles. J. Colloid Interface Sci. 1998, 208, 468–477. [Google Scholar] [CrossRef]
- Paunov, V.N. Novel Method for Determining the Three-Phase Contact Angle of Colloid Particles Adsorbed at Air−Water and Oil−Water Interfaces. Langmuir 2003, 19, 7970–7976. [Google Scholar] [CrossRef]
- Isa, L.; Lucas, F.; Wepf, R.; Reimhult, E. Measuring Single-Nanoparticle Wetting Properties by Freeze-Fracture Shadow-Casting Cryo-Scanning Electron Microscopy. Nat. Commun. 2011, 2, 438. [Google Scholar] [CrossRef]
- Wu, D.; Honciuc, A. Design of Janus Nanoparticles with PH-Triggered Switchable Amphiphilicity for Interfacial Applications. ACS Appl. Nano Mater. 2018, 1, 471–482. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Granick, S. Controlling the Geometry (Janus Balance) of Amphiphilic Colloidal Particles. Langmuir 2008, 24, 2438–2445. [Google Scholar] [CrossRef] [PubMed]
- Honciuc, A. Amphiphilic Janus Particles at Interfaces. In Flowing Matter; Toschi, F., Sega, M., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 95–136. ISBN 978-3-030-23369-3. [Google Scholar]
- Finkle, P.; Draper, H.D.; Hildebrand, J.H. The Theory of Emulsification. J. Am. Chem. Soc. 1923, 45, 2780–2788. [Google Scholar] [CrossRef]
- Bancroft, W.D. Applied Colloid Chemistry; General Theory; McGraw-Hill Book Company, Inc.: New York, NY, USA, 1921. [Google Scholar]
- Wu, D.; Honciuc, A. Contrasting Mechanisms of Spontaneous Adsorption at Liquid–Liquid Interfaces of Nanoparticles Constituted of and Grafted with PH-Responsive Polymers. Langmuir 2018, 34, 6170–6182. [Google Scholar] [CrossRef] [PubMed]
- Shi, B.; Peng, P.; Jia, L. Relationship Between Total Surface Tension of Monomer and Its Homopolymer. J. Macromol. Sci. Part B 2011, 50, 952–955. [Google Scholar] [CrossRef]
- Pieranski, P. Two-Dimensional Interfacial Colloidal Crystals. Phys. Rev. Lett. 1980, 45, 569–572. [Google Scholar] [CrossRef]
- Binks, B.P.; Lumsdon, S.O. Influence of Particle Wettability on the Type and Stability of Surfactant-Free Emulsions†. Langmuir 2000, 16, 8622–8631. [Google Scholar] [CrossRef]
- Du, K.; Glogowski, E.; Emrick, T.; Russell, T.P.; Dinsmore, A.D. Adsorption Energy of Nano- and Microparticles at Liquid−Liquid Interfaces. Langmuir 2010, 26, 12518–12522. [Google Scholar] [CrossRef]
- Wu, D.; Mihali, V.; Honciuc, A. PH-Responsive Pickering Foams Generated by Surfactant-Free Soft Hydrogel Particles. Langmuir 2019, 35, 212–221. [Google Scholar] [CrossRef]
- Luck, R.M.; Sadhir, R.K. Shrinkage in Conventional Monomers during Polymerization. In Expanding Monomers; Synthesis, Characterization and Applications; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 1992; pp. 1–17. [Google Scholar]
- Bonaccorso, F.; Succi, S.; Lauricella, M.; Montessori, A.; Tiribocchi, A.; Luo, K.H. Shear Dynamics of Confined Bijels. AIP Adv. 2020, 10, 095304. [Google Scholar] [CrossRef]
- Bonaccorso, F.; Montessori, A.; Tiribocchi, A.; Amati, G.; Bernaschi, M.; Lauricella, M.; Succi, S. LBsoft: A Parallel Open-Source Software for Simulation of Colloidal Systems. Comput. Phys. Commun. 2020, 256, 107455. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NP | Polymer | γP/W (mJ/m2) | Radius (nm) | Cosβ | ΔGdesorbtion (KJ/mol) |
|---|---|---|---|---|---|
| NP-OH | PSty | 35.48 | 250.34 | −0.65 | 849.8 |
| PMM | 20.82 | −0.54 | 850.6 | ||
| PDAEMA | 3.52 | −0.28 | 354.7 | ||
| PBM | 41.07 | −0.67 | 860.4 | ||
| PVAc | 26.47 | −0.64 | 677.7 | ||
| NP-C8 | PSty | 35.48 | 254.72 | 0.39 | 13,881.7 |
| PBM | 41.07 | 0.79 | 26,915.7 | ||
| PtBA | 1.05 | 0.43 | 438.9 | ||
| PVAc | 26.47 | 0.64 | 14,564.2 | ||
| PEM | 32.03 | 0.50 | 14,761.1 | ||
| NP-CN | PMM | 20.82 | 252.50 | −0.31 | 1980.2 |
| PDAEMA | 3.52 | −0.66 | 82.1 | ||
| PBM | 41.07 | −0.41 | 2836.5 | ||
| PtBA | 1.05 | −0.49 | 54.7 | ||
| PEM | 32.03 | −0.41 | 2212.1 | ||
| NP-SH | PMM | 20.82 | 249.67 | −0.28 | 2138.4 |
| PBM | 41.07 | −0.47 | 2234.2 | ||
| PtBA | 1.05 | −0.43 | 66.2 | ||
| PEM | 32.03 | −0.39 | 2340.1 | ||
| NP-NH2 | PSty | 35.48 | 249.43 | −0.85 | 148.9 |
| PDAEMA | 3.52 | −0.88 | 10.5 | ||
| PBM | 41.07 | −0.89 | 101.8 | ||
| PEM | 32.03 | −0.90 | 65.7 |
| Nanoparticle | γdNP/water (mN/m) | γpNP/water (mN/m) | γNP/water (mN/m) |
|---|---|---|---|
| NP-OH | 0.00 | 1.06 | 1.06 |
| NP-C8 | 111.65 | 9.79 | 121.44 |
| NP-CN | 0.00 | 2.77 | 2.77 |
| NP-NH2 | 0.00 | 0.14 | 0.14 |
| NP-SH | 0.21 | 3.10 | 3.10 |
| Nanoparticle | γdNP (mN/m) | Xd | γpNP (mN/m) | Xp | γNP (mN/m) |
|---|---|---|---|---|---|
| NP-OH | 25.40 | 0.43 | 33.92 | 0.57 | 59.32 |
| NP-C8 | 30.54 | 0.69 | 13.89 | 10.31 | 44.43 |
| NP-CN | 25.40 | 0.49 | 26.94 | 0.51 | 52.34 |
| NP-NH2 | 25.40 | 0.38 | 41.96 | 0.62 | 67.36 |
| NP-SH | 20.96 | 0.44 | 26.60 | 0.56 | 47.56 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Honciuc, A.; Negru, O.-I. NanoTraPPED—A New Method for Determining the Surface Energy of Nanoparticles via Pickering Emulsion Polymerization. Nanomaterials 2021, 11, 3200. https://doi.org/10.3390/nano11123200
Honciuc A, Negru O-I. NanoTraPPED—A New Method for Determining the Surface Energy of Nanoparticles via Pickering Emulsion Polymerization. Nanomaterials. 2021; 11(12):3200. https://doi.org/10.3390/nano11123200
Chicago/Turabian StyleHonciuc, Andrei, and Oana-Iuliana Negru. 2021. "NanoTraPPED—A New Method for Determining the Surface Energy of Nanoparticles via Pickering Emulsion Polymerization" Nanomaterials 11, no. 12: 3200. https://doi.org/10.3390/nano11123200