
Small-Sized Co-Polymers for Targeted Delivery of Multiple Imaging and Therapeutic Agents


Abstract
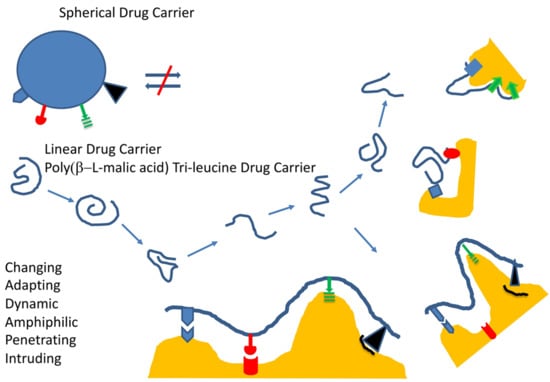
:
1. Introduction
1.1. Mini-Nanodrugs
1.2. Criteria Ruling the Design of Mini-Nano Carriers
1.2.1. General Structure, Function, and Desired Effects
1.2.2. Example of Mini-Nano Carriers, Composition, and Outstanding Properties
1.2.3. Permeation through Barriers by Spontaneous Diffusion or Receptor-Gated Access
Extravasation
After Extravasation: Several Multiple Cellular and Extracellular Hurdles
Passive (Diffusive) Pathways
“Active” Delivery Pathways
Transcytosis Pathways, Vectors
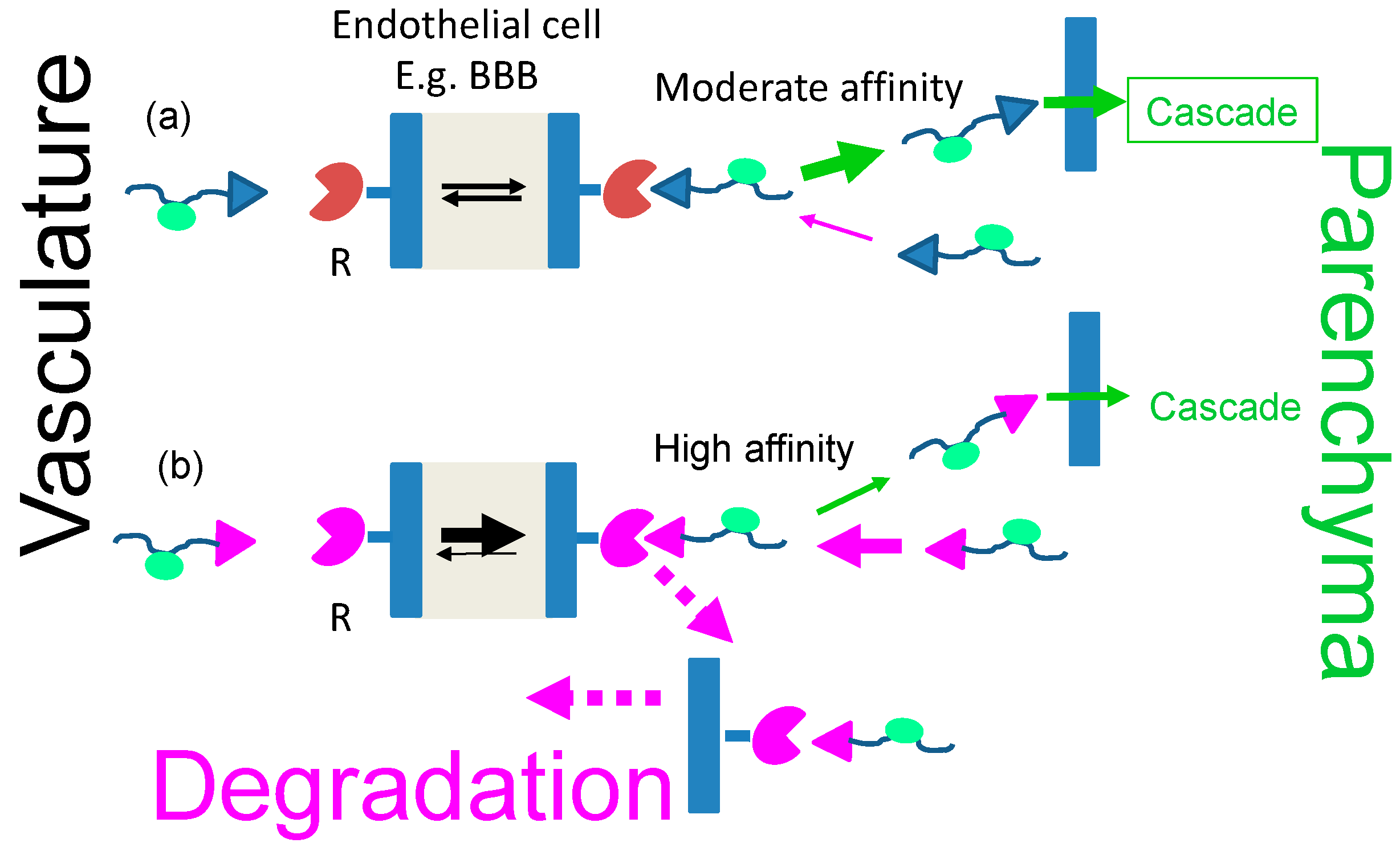
Receptor-Driven Permeation through Cascades of Gated Barriers
Consideration of Size and Shape Effects on BBB Permeation
Transcytosis from Blood to Brain
2. Favorable Reasons to Use Mini Nano Vehicles for Delivery into Brain
2.1. Semiquantitative Description of Cross-BBB Delivery
Simplified Transcytosis Model
2.2. Calculation of Approximate BBB Permeation Efficacies Using Quasi Equilibrium and Other Approximations
2.3. Effects on Transcytosis Efficacy at Selected Concentrations of Receptor and Ligand
2.4. The Dissociation Rate of the Ligand–Receptor Complex Is Coupled with the Affinity
2.4.1. The Vector Part of the Ligands Matters
2.4.2. Polymalic Acid Tri-Leucine Group “Boosts” the Function of the Vector Group
2.5. The Observed Impact of Vector–Receptor Affinity on Pharmaceutical Delivery
The History of Drug Delivery to Brain
2.6. Transcytosis and Cascade Reactions
2.6.1. How to Optimize the Flow through Cascade Barriers
2.6.2. Polymalic Acid Conjugates as Outstanding Candidates for Borderline Nanosized Drug Delivery Systems
Structural, Chemical, and Physical Background for PMLA-Based Mini-Nanodrugs
Molecular Weight
The Linear Structure of the Polymeric Platform
The Chemical Attachment of Ligands
2.6.3. Why Peptides Instead of Antibodies?
3. Examples of Mini-Nano Devices
3.1. Example 1: PMLA-Based Mini-Nano Carriers (MNCs) for Delivery across the BBB
Kinetics and Efficacy of Mini-Nanocarriers’ Permeation through BBB
3.2. Example 2: PMLA-Based Mini-Nano Imaging Agents (MNIAs) for Deep Brain Tumor Imaging by MRI Analysis and Near Infra-Red Fluorescence-Guided Tumor Resectinon
Mini MRI-Contrast Agents
3.3. Example 3: Image-Guided Resection of Glioblastoma
3.4. Example 4: PMLA-Based Mini-Nano Drugs for the Treatment of HER-Positive Breast Cancer
Conventional Cancer Targeting by Antibodies
4. Summary of Distinguished Features of PMLA-Based Mini-Nano Devices
4.1. Multifunctionality of the Mini-Nano Device
4.2. Cascade Targeting Affording Attachment of Several Peptides per MNC
4.3. Optimal Settings of Mini Nano Devices (MNDs)
5. Comparison with Non-PMLA Types of Mini-Nano Devices
6. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
Model Calculations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case # | Kd (nM) | [R]o (nM) | [L]o (nM) | [LR] (nM) | [R] (nM) | [R]/[R]o (%) |
|---|---|---|---|---|---|---|
| 1 | 1 | 1 | 10 | 0.909 | 0.091 | 9.0 |
| 2 | 1 | 2 | 10 | 1.818 | 0.182 | 9.1 |
| 3 | 1 | 1 | 20 | 0.952 | 0.048 | 4.8 |
| 4 | 1 | 2 | 20 | 1.905 | 0.095 | 4.8 |
| 5 | 10 | 1 | 10 | 0.5 | 0.5 | 50 |
| 6 | 10 | 2 | 10 | 1.0 | 1.0 | 50 |
| 7 | 10 | 1 | 20 | 0.667 | 0.333 | 33 |
| 8 | 10 | 2 | 20 | 1.333 | 0.667 | 33 |
| 9 | 10 | 4 | 100 | 3.696 | 0.364 | 9.1 |
| 10 | 50 | 4 | 100 | 2.667 | 1.333 | 33 |
| 11 | 100 | 4 | 100 | 2.0 | 2.0 | 50 |
| 12 | 100 | 1 | 10 | 0.091 | 0.909 | 91 |
References
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2021, 20, 102–124. [Google Scholar] [CrossRef]
- Wais, U.; Jackson, A.W.; He, T.; Zhang, H. Nanoformulation and encapsulation approaches for poorly water-soluble drug nanoparticles. Nanoscale 2016, 8, 1746–1769. [Google Scholar] [CrossRef] [PubMed]
- Farokhzad, O.C.; Langer, R. Impact of nanotechnology on drug delivery. Acs Nano 2009, 3, 16–20. [Google Scholar] [CrossRef]
- Rabanel, J.M.; Aoun, V.; Elkin, I.; Mokhtar, M.; Hildgen, P. Drug-loaded nanocarriers: Passive targeting and crossing of biological barriers. Curr. Med. Chem. 2012, 19, 3070–3102. [Google Scholar] [CrossRef] [PubMed]
- Ljubimova, J.Y.; Sun, T.; Mashouf, L.; Ljubimov, A.V.; Israel, L.L.; Ljubimov, V.A.; Falahatian, V.; Holler, E. Covalent nano delivery systems for selective imaging and treatment of brain tumors. Adv. Drug Deliv. Rev. 2017, 113, 177–200. [Google Scholar] [CrossRef] [PubMed]
- Mundargi, R.C.; Babu, V.R.; Rangaswamy, V.; Patel, P.; Aminabhavi, T.M. Nano/micro technologies for delivering macromolecular therapeutics using poly (d, l-lactide-co-glycolide) and its derivatives. J. Control. Release 2008, 125, 193–209. [Google Scholar] [CrossRef]
- Hosta-Rigau, L.; Schattling, P.; Teo, B.M.; Lynge, M.E.; Städler, B. Recent progress of liposomes in nanomedicine. J. Mater. Chem. B 2014, 2, 6686–6691. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Shi, J. MSN anti-cancer nanomedicines: Chemotherapy enhancement, overcoming of drug resistance, and metastasis inhibition. Adv. Mater. 2014, 26, 391–411. [Google Scholar] [CrossRef]
- Firme, C.P., III; Bandaru, P.R. Toxicity issues in the application of carbon nanotubes to biological systems. Nanomed. Nanotechnol. Biol. Med. 2010, 6, 245–256. [Google Scholar] [CrossRef]
- Schmid, G.; Kreyling, W.G.; Simon, U. Toxic effects and biodistribution of ultrasmall gold nanoparticles. Arch. Toxicol. 2017, 91, 3011–3037. [Google Scholar] [CrossRef] [Green Version]
- Ren, H.; Huang, X. Polyacrylate nanoparticles: Toxicity or new nanomedicine? Eur. Respir. J. 2010, 36, 218–221. [Google Scholar] [CrossRef] [Green Version]
- Hofmann-Amtenbrink, M.; Grainger, D.W.; Hofmann, H. Nanoparticles in medicine: Current challenges facing inorganic nanoparticle toxicity assessments and standardizations. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 1689–1694. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Liu, D.; Yang, Y. Anti-cancer peptides: Classification, mechanism of action, reconstruction and modification. Open Biol. 2020, 10, 200004. [Google Scholar] [CrossRef]
- Ljubimova, J.Y.; Portilla-Arias, J.; Patil, R.; Ding, H.; Inoue, S.; Markman, J.L.; Rekechenetskiy, A.; Konda, B.; Gangalum, P.R.; Chesnokova, A.; et al. Toxicity and efficacy evaluation of multiple targeted polymalic acid conjugates for triple-negative breast cancer treatment. J. Drug Target. 2013, 21, 956–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Göttler, T.; Holler, E. Screening for β-poly (l-malate) binding proteins by affinity chromatography. Biochem. Biophys. Res. Commun. 2006, 341, 1119–1127. [Google Scholar] [CrossRef]
- Angerer, B.; Holler, E. Large Complexes of. β-Poly (l-malate) with DNA Polymerase. alpha, Histones, and Other Proteins in Nuclei of Growing Plasmodia of Physarum polycephalum. Biochemistry 1995, 34, 14741–14751. [Google Scholar] [CrossRef] [PubMed]
- Israel, L.L.; Braubach, O.; Galstyan, A.; Chiechi, A.; Shatalova, E.S.; Grodzinski, Z.; Ding, H.; Black, K.L.; Ljubimova, J.Y.; Holler, E. A Combination of Tri-Leucine and Angiopep-2 Drives a Poly-Anionic Polymalic Acid Nanodrug Platform Across the Blood-Brain Barrier. ACS Nano 2019, 13, 1253–1271. [Google Scholar] [CrossRef] [PubMed]
- Stylianopoulos, T.; Munn, L.L.; Jain, R.K. Reengineering the Physical Microenvironment of Tumors to Improve Drug Delivery and Efficacy: From Mathematical Modeling to Bench to Bedside. Trends Cancer 2018, 4, 292–319. [Google Scholar] [CrossRef] [Green Version]
- Bien-Ly, N.; Yu, Y.J.; Bumbaca, D.; Elstrott, J.; Boswell, C.A.; Zhang, Y.; Luk, W.; Lu, Y.; Dennis, M.S.; Weimer, R.M. Transferrin receptor (TfR) trafficking determines brain uptake of TfR antibody affinity variants. J. Exp. Med. 2014, 211, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Li, R.; Deng, H.; Li, Y.; Cui, Y.; Zhang, H.; Dai, W.; He, B.; Zheng, Y.; Wang, X.; et al. Receptor mediated transcytosis in biological barrier: The influence of receptor character and their ligand density on the transmembrane pathway of active-targeting nanocarriers. Biomaterials 2018, 180, 78–90. [Google Scholar] [CrossRef]
- Tian, X.; Leite, D.M.; Scarpa, E.; Nyberg, S.; Fullstone, G.; Forth, J.; Matias, D.L.; Apriceno, A.; Poma, A.; Duro-Castano, A.; et al. On the shuttling across the blood-brain barrier via tubules formation: Mechanism and cargo avidity bias. Sci. Adv. 2020, 6, eabc4397. [Google Scholar] [CrossRef]
- Dai, Q.; Wilhelm, S.; Ding, D.; Syed, A.M.; Sindhwani, S.; Zhang, Y.; Chen, Y.Y.; MacMillan, P.; Chan, W.C. Quantifying the ligand-coated nanoparticle delivery to cancer cells in solid tumors. ACS Nano 2018, 12, 8423–8435. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Gangalum, P.R.; Galstyan, A.; Fox, I.; Patil, R.; Hubbard, P.; Murali, R.; Ljubimova, J.Y.; Holler, E. HER2-positive breast cancer targeting and treatment by a peptide-conjugated mini nanodrug. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 631–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, H.; Inoue, S.; Ljubimov, A.V.; Patil, R.; Portilla-Arias, J.; Hu, J.; Konda, B.; Wawrowsky, K.A.; Fujita, M.; Karabalin, N.; et al. Inhibition of brain tumor growth by intravenous poly(β-L-malic acid) nanobioconjugate with pH-dependent drug release. Proc. Natl. Acad. Sci. USA 2010, 107, 18143–18148. [Google Scholar] [CrossRef] [Green Version]
- Ljubimova, J.Y.; Ding, H.; Portilla-Arias, J.; Patil, R.; Gangalum, P.R.; Chesnokova, A.; Inoue, S.; Rekechenetskiy, A.; Nassoura, T.; Black, K.L.; et al. Polymalic Acid-based nano Biopolymers for Targeting of Multiple Tumor Markers: An Opportunity for Personalized Medicine? J. Vis. Exp. (JoVE) 2014, 88, e50668. [Google Scholar] [CrossRef] [Green Version]
- Rathberger, K.; Reisner, H.; Willibald, B.; Molitoris, H.-P.; Holler, E. Comparative synthesis and hydrolytic degradation of poly (L-malate) by myxomycetes and fungi. Mycol. Res. 1999, 103, 513–520. [Google Scholar] [CrossRef]
- Fischer, H.; Erdmann, S.; Holler, E. An unusual polyanion from Physarum polycephalum that inhibits homologous DNA-polymerase. alpha. in vitro. Biochemistry 1989, 28, 5219–5226. [Google Scholar] [CrossRef]
- Lee, B.-S.; Vert, M.; Holler, E. Water-soluble aliphatic polyesters: Poly(malic acid)s. In Biopolymers; Doi, Y., Steinbüchel, A., Eds.; Wiley-VCH: Weinheim, Germany, 2002; pp. 75–103. [Google Scholar]
- Cammas, S.; Guerin, P.; Girault, J.; Holler, E.; Gache, Y.; Vert, M. Natural poly (l-malic acid): NMR shows a poly (3-hydroxy acid)-type structure. Macromolecules 1993, 26, 4681–4684. [Google Scholar] [CrossRef]
- Vert, M.; Fournier, P.; Boustta, M.; Domurado, D.; Guérin, P.; Braud, C.; Holler, H. Poly (β-malic acid) and tailor-made derivatives: Fate in vivo. Macromol. Rep. 1994, 31, 723–730. [Google Scholar]
- Zeng, W.; Zhang, B.; Liu, Q.; Chen, G.; Liang, Z. Analysis of the L-malate biosynthesis pathway involved in poly (β-L-malic acid) production in Aureobasidium melanogenum GXZ-6 by addition of metabolic intermediates and inhibitors. J. Microbiol. 2019, 57, 281–287. [Google Scholar] [CrossRef]
- Regina, A.; Demeule, M.; Che, C.; Lavallee, I.; Poirier, J.; Gabathuler, R.; Beliveau, R.; Castaigne, J.P. Antitumour activity of ANG1005, a conjugate between paclitaxel and the new brain delivery vector Angiopep-2. Br. J. Pharmacol. 2008, 155, 185–197. [Google Scholar] [CrossRef]
- Bertrand, Y.; Currie, J.-C.; Demeule, M.; Régina, A.; Ché, C.; Abulrob, A.; Fatehi, D.; Sartelet, H.; Gabathuler, R.; Castaigne, J.-P.; et al. Transport characteristics of a novel peptide platform for CNS therapeutics. J. Cell. Mol. Med. 2010, 14, 2827–2839. [Google Scholar] [CrossRef] [Green Version]
- Patil, R.; Galstyan, A.; Sun, T.; Shatalova, E.S.; Butte, P.; Mamelak, A.N.; Carico, C.; Kittle, D.S.; Grodzinski, Z.B.; Chiechi, A. Polymalic acid chlorotoxin nanoconjugate for near-infrared fluorescence guided resection of glioblastoma multiforme. Biomaterials 2019, 206, 146–159. [Google Scholar] [CrossRef] [PubMed]
- Patil, R.; Galstyan, A.; Grodzinski, Z.B.; Shatalova, E.S.; Wagner, S.; Israel, L.L.; Ding, H.; Black, K.L.; Ljubimova, J.Y.; Holler, E. Single-and Multi-Arm Gadolinium MRI Contrast Agents for Targeted Imaging of Glioblastoma. Int. J. Nanomed. 2020, 15, 3057–3070. [Google Scholar] [CrossRef]
- Staquicini, F.I.; Ozawa, M.G.; Moya, C.A.; Driessen, W.H.; Barbu, E.M.; Nishimori, H.; Soghomonyan, S.; Flores, L.G., 2nd; Liang, X.; Paolillo, V. Systemic combinatorial peptide selection yields a non-canonical iron-mimicry mechanism for targeting tumors in a mouse model of human glioblastoma. J. Clin. Investig. 2011, 121, 161. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.; Jiang, M.; Jiang, D.; Feng, X.; Yao, J.; Song, Q.; Chen, H.; Gao, X.; Chen, J. Enhancing Glioblastoma-Specific Penetration by Functionalization of Nanoparticles with an Iron-Mimic Peptide Targeting Transferrin/Transferrin Receptor Complex. Mol. Pharm. 2015, 12, 2947–2961. [Google Scholar] [CrossRef]
- Huang, N.; Lu, S.; Liu, X.-G.; Zhu, J.; Wang, Y.-J.; Liu, R.-T. PLGA nanoparticles modified with a BBB-penetrating peptide co-delivering Aβ generation inhibitor and curcumin attenuate memory deficits and neuropathology in Alzheimer’s disease mice. Oncotarget 2017, 8, 81001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, H.; Anderson, B.; Mao, Q.; Davidson, B.L. Recombinant human adenovirus: Targeting to the human transferrin receptor improves gene transfer to brain microcapillary endothelium. J. Virol. 2000, 74, 11359–11366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, T.; Yang, L.; Liu, Y.; Zhou, X.; Sun, J.; Liu, J. Sialic acid (SA)-modified selenium nanoparticles coated with a high blood–brain barrier permeability peptide-B6 peptide for potential use in Alzheimer’s disease. Acta Biomater. 2015, 25, 172–183. [Google Scholar] [CrossRef]
- Liu, Z.; Gao, X.; Kang, T.; Jiang, M.; Miao, D.; Gu, G.; Hu, Q.; Song, Q.; Yao, L.; Tu, Y. B6 peptide-modified PEG-PLA nanoparticles for enhanced brain delivery of neuroprotective peptide. Bioconjugate Chem. 2013, 24, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Oller-Salvia, B.; Sánchez-Navarro, M.; Ciudad, S.; Guiu, M.; Arranz-Gibert, P.; Garcia, C.; Gomis, R.R.; Cecchelli, R.; García, J.; Giralt, E. MiniAp-4: A venom-inspired peptidomimetic for brain delivery. Angew. Chem. Int. Ed. 2016, 55, 572–575. [Google Scholar] [CrossRef] [Green Version]
- Soroceanu, L.; Gillespie, Y.; Khazaeli, M.; Sontheimer, H. Use of chlorotoxin for targeting of primary brain tumors. Cancer Res. 1998, 58, 4871–4879. [Google Scholar] [PubMed]
- Veiseh, M.; Gabikian, P.; Bahrami, S.B.; Veiseh, O.; Zhang, M.; Hackman, R.C.; Ravanpay, A.C.; Stroud, M.R.; Kusuma, Y.; Hansen, S.J.; et al. Tumor Paint: A Chlorotoxin:Cy5.5 Bioconjugate for Intraoperative Visualization of Cancer Foci. Cancer Res. 2007, 67, 6882. [Google Scholar] [CrossRef] [Green Version]
- Deshane, J.; Garner, C.C.; Sontheimer, H. Chlorotoxin inhibits glioma cell invasion via matrix metalloproteinase-2. J. Biol. Chem. 2003, 278, 4135–4144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, B.-W.; Zhang, H.-T.; Wu, C.; Berezov, A.; Zhang, X.; Dua, R.; Wang, Q.; Kao, G.; O’Rourke, D.M.; Greene, M.I. Rationally designed anti-HER2/neu peptide mimetic disables p185 HER2/neu tyrosine kinases in vitro and in vivo. Nat. Biotechnol. 2000, 18, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Berezov, A.; Zhang, H.-T.; Greene, M.I.; Murali, R. Disabling erbB receptors with rationally designed exocyclic mimetics of antibodies: Structure− function analysis. J. Med. Chem. 2001, 44, 2565–2574. [Google Scholar] [CrossRef]
- Briand, J.P.; Muller, S.; Van Regenmortel, M.H. Synthetic peptides as antigens: Pitfalls of conjugation methods. J. Immunol. Methods 1985, 78, 59–69. [Google Scholar] [CrossRef]
- Fernandez, L.; Bustos, R.; Zapata, C.; Garcia, J.; Jauregui, E.; Ashraf, G. Immunogenicity in protein and peptide based-therapeutics: An overview. Curr. Protein Pept. Sci. 2018, 19, 958–971. [Google Scholar] [CrossRef]
- Sela-Culang, I.; Kunik, V.; Ofran, Y. The structural basis of antibody-antigen recognition. Front. Immunol. 2013, 4, 302. [Google Scholar] [CrossRef] [Green Version]
- Burkovitz, A.; Leiderman, O.; Sela-Culang, I.; Byk, G.; Ofran, Y. Computational identification of antigen-binding antibody fragments. J. Immunol. 2013, 190, 2327–2334. [Google Scholar] [CrossRef] [Green Version]
- Niewoehner, J.; Bohrmann, B.; Collin, L.; Urich, E.; Sade, H.; Maier, P.; Rueger, P.; Stracke, J.O.; Lau, W.; Tissot, A.C. Increased brain penetration and potency of a therapeutic antibody using a monovalent molecular shuttle. Neuron 2014, 81, 49–60. [Google Scholar] [CrossRef] [Green Version]
- McCully, M.; Sanchez-Navarro, M.; Teixido, M.; Giralt, E. Peptide Mediated Brain Delivery of Nano-and Submicroparticles: A Synergistic Approach. Curr. Pharm. Des. 2018, 24, 1366–1376. [Google Scholar] [CrossRef] [PubMed]
- Sarin, H.; Kanevsky, A.S.; Wu, H.; Brimacombe, K.R.; Fung, S.H.; Sousa, A.A.; Auh, S.; Wilson, C.M.; Sharma, K.; Aronova, M.A. Effective transvascular delivery of nanoparticles across the blood-brain tumor barrier into malignant glioma cells. J. Transl. Med. 2008, 6, 80. [Google Scholar] [CrossRef] [Green Version]
- Tosi, G.; Fano, R.A.; Bondioli, L.; Badiali, L.; Benassi, R.; Rivasi, F.; Ruozi, B.; Forni, F.; Vandelli, M.A. Investigation on mechanisms of glycopeptide nanoparticles for drug delivery across the blood–brain barrier. Nanomedicine 2011, 6, 423–436. [Google Scholar] [CrossRef]
- Shamloo, A.; GhafarZadeh, E.; Alasty, A. Modeling and simulation of crossing magnetic nanoparticles through Blood Brain Barrier (BBB). In Proceedings of the 2014 36th Annual International Conference of the IEEE Engineering in Medicine and Biology Society, Chicago, IL, USA, 26–30 August 2014; pp. 5280–5283. [Google Scholar]
- Lee, K.L.; Hubbard, L.C.; Hern, S.; Yildiz, I.; Gratzl, M.; Steinmetz, N.F. Shape matters: The diffusion rates of TMV rods and CPMV icosahedrons in a spheroid model of extracellular matrix are distinct. Biomater. Sci. 2013, 1, 581–588. [Google Scholar] [CrossRef] [Green Version]
- Betzer, O.; Shilo, M.; Opochinsky, R.; Barnoy, E.; Motiei, M.; Okun, E.; Yadid, G.; Popovtzer, R. The effect of nanoparticle size on the ability to cross the blood–brain barrier: An in vivo study. Nanomedicine 2017, 12, 1533–1546. [Google Scholar] [CrossRef]
- Liu, X.; Sui, B.; Sun, J. Size-and shape-dependent effects of titanium dioxide nanoparticles on the permeabilization of the blood–brain barrier. J. Mater. Chem. B 2017, 5, 9558–9570. [Google Scholar] [CrossRef] [PubMed]
- Israel, L.L.; Galstyan, A.; Holler, E.; Ljubimova, J.Y. Magnetic iron oxide nanoparticles for imaging, targeting and treatment of primary and metastatic tumors of the brain. J. Control. Release 2020, 320, 45–62. [Google Scholar] [CrossRef]
- Maeda, H.; Fang, J.; Inutsuka, T.; Kitamoto, Y. Vascular permeability enhancement in solid tumor: Various factors, mechanisms involved and its implications. Int. Immunopharmacol. 2003, 3, 319–328. [Google Scholar] [CrossRef]
- Arvanitis, C.D.; Ferraro, G.B.; Jain, R.K. The blood–brain barrier and blood–tumour barrier in brain tumours and metastases. Nat. Rev. Cancer 2020, 20, 26–41. [Google Scholar] [CrossRef]
- Jolliet-Riant, P.; Tillement, J.P. Drug transfer across the blood-brain barrier and improvement of brain delivery. Fundam. Clin. Pharmacol. 1999, 13, 16–26. [Google Scholar] [CrossRef]
- Pardridge, W.M. Delivery of biologics across the blood–brain barrier with molecular Trojan horse technology. BioDrugs 2017, 31, 503–519. [Google Scholar] [CrossRef]
- Pardridge, W.M. Drug and gene targeting to the brain with molecular Trojan horses. Nat. Rev. Drug Discov. 2002, 1, 131–139. [Google Scholar] [CrossRef]
- Pardridge, W.M.; Boado, R.J.; Giugliani, R.; Schmidt, M. Plasma pharmacokinetics of valanafusp alpha, a human insulin receptor antibody-iduronidase fusion protein, in patients with mucopolysaccharidosis type I. BioDrugs 2018, 32, 169–176. [Google Scholar] [CrossRef]
- Chang, R.; Al Maghribi, A.; Vanderpoel, V.; Vasilevko, V.; Cribbs, D.H.; Boado, R.; Pardridge, W.M.; Sumbria, R.K. Brain Penetrating Bifunctional Erythropoietin–Transferrin Receptor Antibody Fusion Protein for Alzheimer’s Disease. Mol. Pharm. 2018, 15, 4963–4973. [Google Scholar] [CrossRef]
- Yu, Y.J.; Atwal, J.K.; Zhang, Y.; Tong, R.K.; Wildsmith, K.R.; Tan, C.; Bien-Ly, N.; Hersom, M.; Maloney, J.A.; Meilandt, W.J. Therapeutic bispecific antibodies cross the blood-brain barrier in nonhuman primates. Sci. Transl. Med. 2014, 6, 261ra154. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.J.; Zhang, Y.; Kenrick, M.; Hoyte, K.; Luk, W.; Lu, Y.; Atwal, J.; Elliott, J.M.; Prabhu, S.; Watts, R.J. Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci. Transl. Med. 2011, 3, 84ra44. [Google Scholar] [CrossRef] [PubMed]
- Georgieva, J.V.; Hoekstra, D.; Zuhorn, I.S. Smuggling drugs into the brain: An overview of ligands targeting transcytosis for drug delivery across the blood–brain barrier. Pharmaceutics 2014, 6, 557–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dehouck, B.; Fenart, L.; Dehouck, M.-P.; Pierce, A.; Torpier, G.; Cecchelli, R. A new function for the LDL receptor: Transcytosis of LDL across the blood–brain barrier. J. Cell Biol. 1997, 138, 877–889. [Google Scholar] [CrossRef] [PubMed]
- Demeule, M.; Currie, J.C.; Bertrand, Y.; Che, C.; Nguyen, T.; Regina, A.; Gabathuler, R.; Castaigne, J.P.; Beliveau, R. Involvement of the low-density lipoprotein receptor-related protein in the transcytosis of the brain delivery vector Angiopep-2. J. Neurochem. 2008, 106, 1534–1544. [Google Scholar] [CrossRef]
- Demeule, M.; Regina, A.; Che, C.; Poirier, J.; Nguyen, T.; Gabathuler, R.; Castaigne, J.-P.; Beliveau, R. Identification and design of peptides as a new drug delivery system for the brain. J. Pharmacol. Exp. Ther. 2008, 324, 1064–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, X.; Nyberg, S.; Sharp, P.S.; Madsen, J.; Daneshpour, N.; Armes, S.P.; Berwick, J.; Azzouz, M.; Shaw, P.; Abbott, N.J.; et al. LRP-1-mediated intracellular antibody delivery to the Central Nervous System. Sci. Rep. 2015, 5, 11990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, W.; Xiong, C.; Zhang, R.; Shi, L.; Huang, M.; Zhang, G.; Song, S.; Huang, Q.; Liu, G.-y.; Li, C. Receptor-mediated transcytosis: A mechanism for active extravascular transport of nanoparticles in solid tumors. J. Control. Release 2012, 161, 959–966. [Google Scholar] [CrossRef] [Green Version]
- Preston, J.E.; Abbott, N.J.; Begley, D.J. Transcytosis of macromolecules at the blood–brain barrier. In Advances in Pharmacology; Elsevier: Amsterdam, The Netherlands, 2014; Volume 71, pp. 147–163. [Google Scholar]
- Pardridge, W.M. Vector-mediated drug delivery to the brain. Adv. Drug Deliv. Rev. 1999, 36, 299–321. [Google Scholar] [CrossRef]
- Scheltens, P.; Blennow, K.; Breteler, M.M.B.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Perry, J.L.; Reuter, K.G.; Luft, J.C.; Pecot, C.V.; Zamboni, W.; DeSimone, J.M. Mediating passive tumor accumulation through particle size, tumor type, and location. Nano Lett. 2017, 17, 2879–2886. [Google Scholar] [CrossRef] [Green Version]
- Patil, R.; Ljubimov, A.V.; Gangalum, P.R.; Ding, H.; Portilla-Arias, J.; Wagner, S.; Inoue, S.; Konda, B.; Rekechenetskiy, A.; Chesnokova, A.; et al. MRI Virtual Biopsy and Treatment of Brain Metastatic Tumors with Targeted Nanobioconjugates: Nanoclinic in the Brain. ACS Nano 2015, 9, 5594–5608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, F.; Rojas, O.J.; Genzer, J.; Gurgel, P.V.; Carbonell, R.G. Affinity interactions of human immunoglobulin G with short peptides: Role of ligand spacer on binding, kinetics, and mass transfer. Anal. Bioanal. Chem. 2016, 408, 1829–1841. [Google Scholar] [CrossRef]
- Pope, M.E.; Soste, M.V.; Eyford, B.A.; Anderson, N.L.; Pearson, T.W. Anti-peptide antibody screening: Selection of high affinity monoclonal reagents by a refined surface plasmon resonance technique. J. Immunol. Methods 2009, 341, 86–96. [Google Scholar] [CrossRef]
- Katsamba, P.S.; Navratilova, I.; Calderon-Cacia, M.; Fan, L.; Thornton, K.; Zhu, M.; Bos, T.V.; Forte, C.; Friend, D.; Laird-Offringa, I. Kinetic analysis of a high-affinity antibody/antigen interaction performed by multiple Biacore users. Anal. Biochem. 2006, 352, 208–221. [Google Scholar] [CrossRef] [PubMed]
- Canovi, M.; Markoutsa, E.; Lazar, A.N.; Pampalakis, G.; Clemente, C.; Re, F.; Sesana, S.; Masserini, M.; Salmona, M.; Duyckaerts, C. The binding affinity of anti-Aβ1-42 MAb-decorated nanoliposomes to Aβ1-42 peptides in vitro and to amyloid deposits in post-mortem tissue. Biomaterials 2011, 32, 5489–5497. [Google Scholar] [CrossRef]
- Yang, D.; Singh, A.; Wu, H.; Kroe-Barrett, R. Comparison of biosensor platforms in the evaluation of high affinity antibody-antigen binding kinetics. Anal. Biochem. 2016, 508, 78–96. [Google Scholar] [CrossRef] [Green Version]
- Canziani, G.A.; Klakamp, S.; Myszka, D.G. Kinetic screening of antibodies from crude hybridoma samples using Biacore. Anal. Biochem. 2004, 325, 301–307. [Google Scholar] [CrossRef]
- Benveniste, M.; Mayer, M.L. Structure-activity analysis of binding kinetics for NMDA receptor competitive antagonists: The influence of conformational restriction. Br. J. Pharmacol. 1991, 104, 207–221. [Google Scholar] [CrossRef] [Green Version]
- Geng, L.; Wang, Z.; Yang, X.; Li, D.; Lian, W.; Xiang, Z.; Wang, W.; Bu, X.; Lai, W.; Hu, Z. Structure-based design of peptides with high affinity and specificity to HER2 positive tumors. Theranostics 2015, 5, 1154. [Google Scholar] [CrossRef] [Green Version]
- Nederpelt, I.; Bunnik, J.; IJzerman, A.P.; Heitman, L.H. Kinetic Profile of Neuropeptide–Receptor Interactions. Trends Neurosci. 2016, 39, 830–839. [Google Scholar] [CrossRef]
- Lee, S.-M.; Booe, J.M.; Gingell, J.J.; Sjoelund, V.; Hay, D.L.; Pioszak, A.A. N-Glycosylation of asparagine 130 in the extracellular domain of the human calcitonin receptor significantly increases peptide hormone affinity. Biochemistry 2017, 56, 3380–3393. [Google Scholar] [CrossRef] [PubMed]
- Oller-Salvia, B.; Sánchez-Navarro, M.; Giralt, E.; Teixidó, M. Blood–brain barrier shuttle peptides: An emerging paradigm for brain delivery. Chem. Soc. Rev. 2016, 45, 4690–4707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, H.; Portilla-Arias, J.; Patil, R.; Black, K.L.; Ljubimova, J.Y.; Holler, E. Distinct mechanisms of membrane permeation induced by two polymalic acid copolymers. Biomaterials 2013, 34, 217–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, H.; Portilla-Arias, J.; Patil, R.; Black, K.L.; Ljubimova, J.Y.; Holler, E. The optimization of polymalic acid peptide copolymers for endosomolytic drug delivery. Biomaterials 2011, 32, 5269–5278. [Google Scholar] [CrossRef] [Green Version]
- Inoue, S.; Ding, H.; Portilla-Arias, J.; Hu, J.; Konda, B.; Fujita, M.; Espinoza, A.; Suhane, S.; Riley, M.; Gates, M.; et al. Polymalic acid–based nanobiopolymer provides efficient systemic breast cancer treatment by inhibiting both HER2/neu receptor synthesis and activity. Cancer Res. 2011, 71, 1454–1464. [Google Scholar] [CrossRef] [Green Version]
- Patil, R.; Portilla-Arias, J.; Ding, H.; Konda, B.; Rekechenetskiy, A.; Inoue, S.; Black, K.L.; Holler, E.; Ljubimova, J.Y. Cellular delivery of doxorubicin via pH-controlled hydrazone linkage using multifunctional nano vehicle based on poly (β-L-malic acid). Int. J. Mol. Sci. 2012, 13, 11681–11693. [Google Scholar] [CrossRef]
- Lee, B.-S.; Fujita, M.; Khazenzon, N.M.; Wawrowsky, K.A.; Wachsmann-Hogiu, S.; Farkas, D.L.; Black, K.L.; Ljubimova, J.Y.; Holler, E. Polycefin, a New Prototype of a Multifunctional Nanoconjugate Based on Poly(β-l-malic acid) for Drug Delivery. Bioconjugate Chem. 2006, 17, 317–326. [Google Scholar] [CrossRef] [Green Version]
- Patil, R.; Portilla-Arias, J.; Ding, H.; Inoue, S.; Konda, B.; Hu, J.; Wawrowsky, K.A.; Shin, P.K.; Black, K.L.; Holler, E.; et al. Temozolomide Delivery to Tumor Cells by a Multifunctional Nano Vehicle Based on Poly(β-l-malic acid). Pharm. Res. 2010, 27, 2317–2329. [Google Scholar] [CrossRef] [Green Version]
- Schönenbrücher, H.; Adhikary, R.; Mukherjee, P.; Casey, T.A.; Rasmussen, M.A.; Maistrovich, F.D.; Hamir, A.N.; Kehrli, M.E.; Richt, J.A.; Petrich, J.W. Fluorescence-Based Method, Exploiting Lipofuscin, for Real-Time Detection of Central Nervous System Tissues on Bovine Carcasses. J. Agric. Food Chem. 2008, 56, 6220–6226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinohara, M.; Tachibana, M.; Kanekiyo, T.; Bu, G. Role of LRP1 in the pathogenesis of Alzheimer’s disease: Evidence from clinical and preclinical studies. J. Lipid Res. 2017, 58, 1267–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zheng, M.; Shimoni, O.; Banks, W.A.; Bush, A.I.; Gamble, J.R.; Shi, B. Development of Novel Therapeutics Targeting the Blood-Brain Barrier: From Barrier to Carrier. Adv. Sci. 2021, 8, 2101090. [Google Scholar] [CrossRef] [PubMed]
- Ljubimova, Y.J.; Black, K.L.; Ljubimov, A.V.; Holler, E. Biodegradable multitargeting nanoconjugates for drug delivery. In Multifunctional Pharmaceutical Nanocarriers; Buolanwini, J.K., Ed.; Springer: Berlin, Germany, 2008; pp. 233–262. [Google Scholar]
- Choudhury, H.; Pandey, M.; Chin, P.X.; Phang, Y.L.; Cheah, J.Y.; Ooi, S.C.; Mak, K.-K.; Pichika, M.R.; Kesharwani, P.; Hussain, Z. Transferrin receptors-targeting nanocarriers for efficient targeted delivery and transcytosis of drugs into the brain tumors: A review of recent advancements and emerging trends. Drug Deliv. Transl. Res. 2018, 8, 1545–1563. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Lv, Q.; Lu, J.; Yao, H.; Lv, X.; Jiang, F.; Lu, A.; Zhang, G. Prodrug strategies for paclitaxel. Int. J. Mol. Sci. 2016, 17, 796. [Google Scholar] [CrossRef] [Green Version]
- Helms, H.C.; Abbott, N.J.; Burek, M.; Cecchelli, R.; Couraud, P.-O.; Deli, M.A.; Förster, C.; Galla, H.J.; Romero, I.A.; Shusta, E.V.; et al. In vitro models of the blood–brain barrier: An overview of commonly used brain endothelial cell culture models and guidelines for their use. J. Cereb. Blood Flow Metab. 2016, 36, 862–890. [Google Scholar] [CrossRef]
- Cho, C.F.; Wolfe, J.M.; Fadzen, C.M.; Calligaris, D.; Hornburg, K.; Chiocca, E.A.; Agar, N.Y.R.; Pentelute, B.L.; Lawler, S.E. Blood-brain-barrier spheroids as an in vitro screening platform for brain-penetrating agents. Nat. Commun. 2017, 8, 15623. [Google Scholar] [CrossRef]
- Lidický, O.; Šírová, M.; Etrych, T. HPMA copolymer-based polymer conjugates for the delivery and controlled release of retinoids. Physiol. Res. 2016, 65, S233–S241. [Google Scholar] [CrossRef] [PubMed]
- Zarschler, K.; Rocks, L.; Licciardello, N.; Boselli, L.; Polo, E.; Garcia, K.P.; De Cola, L.; Stephan, H.; Dawson, K.A. Ultrasmall inorganic nanoparticles: State-of-the-art and perspectives for biomedical applications. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 1663–1701. [Google Scholar] [CrossRef] [PubMed]
- Boselli, L.; Polo, E.; Castagnola, V.; Dawson, K.A. Regimes of biomolecular ultrasmall nanoparticle interactions. Angew. Chem. Int. Ed. 2017, 56, 4215–4218. [Google Scholar] [CrossRef] [Green Version]
- Nemmar, A.; Beegam, S.; Yuvaraju, P.; Yasin, J.; Tariq, S.; Attoub, S.; Ali, B.H. Ultrasmall superparamagnetic iron oxide nanoparticles acutely promote thrombosis and cardiac oxidative stress and DNA damage in mice. Part. Fibre Toxicol. 2015, 13, 22. [Google Scholar] [CrossRef] [Green Version]
- Sousa, A.A.; Hassan, S.A.; Knittel, L.L.; Balbo, A.; Aronova, M.A.; Brown, P.H.; Schuck, P.; Leapman, R.D. Biointeractions of ultrasmall glutathione-coated gold nanoparticles: Effect of small size variations. Nanoscale 2016, 8, 6577–6588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassano, D.; Pocoví-Martínez, S.; Voliani, V. Ultrasmall-in-Nano Approach: Enabling the Translation of Metal Nanomaterials to Clinics. Bioconjug. Chem. 2018, 29, 4–16. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, D.; Liang, G.; Xu, W.; Tao, Z. Biosynthetic Polymalic Acid as a Delivery Nanoplatform for Translational Cancer Medicine. Trends Biochem. Sci. March 2021, 46, 213–224. [Google Scholar] [CrossRef] [PubMed]












| Feature | Verification | Effect |
|---|---|---|
| Size (volume) | Low mass, hydrodynamic diameter < 10 nm | Fast and deep penetration |
| Shape | High axial (high aspect) ratio | Fast and deep penetration assisted by geometry |
| Platform molecularity | Single molecule | Stability against spontaneous disassembly |
| Functionality | Multiple ligands | Multiple targeting and delivery |
| Ligand attachment | Covalent | Controlled assembly |
| Drug attachment | Reversible if prodrug | Controlled release |
| Targeting | Multiple targeting through gated bio barriers | Tuned affinity-gating receptors regulate movement through cascades |
| High affinity targeting Low affinity targeting | Affinity locked ligand-receptor Speedy delivery via ligand-receptor | Lock-in for antigen-antibody capture Transport through multiple junctions |
| Mini-Nano Device (MNC, MNIA, MND) and Peptide Vector | Formula a | MW (g/mol) | Size b (nm) | ζ-Potential (mV) | Kdiss c (μM) | Dose d (µmol/kg) | Serum t½ (h) e | Site t½ (h) f |
|---|---|---|---|---|---|---|---|---|
| Platform | poly (β-l-malic acid) (PMLA) | 50 × 103 | 3.3 ± 1.5 | −16 ± 0.9 | - | - | - | - |
| Angiopep-2 (AP2) Vector | TFFYGGSRGKRNNFKTEEYC [32,33] | 2404 | - | - | 0.33 [32,33] | 12–30 [32,33] | - | - |
| MNC [34] | P(50 kDa)/LLL(40%) /PEG3400-AP2(2%) /rh(1%) [17] | 165 × 103 | 4.5 ± 1.5 | −11.6 ± 1.8 | - | 0.068–0.548 [17] | 1.2 [17] | 2–3 [17] |
| MNIA (MRI contrast) | P(60 kDa)/PEG600(Gd-DOTA)3(10%)/AP2(1%) /rh(0.5%) [35] | 270 × 103 | 9.4 ± 1.6 | −8.2 ± 1.72 | - | - | - | - |
| Fe-mimetic Vector: cTfRL | CRTIGPSVC (S-S disulfide bridge) [36] | 932 | - | - | - | 5–40 [37,38] | - | - |
| MNC [17] | P(50 kDa)/LLL(40%) /PEG2000cTfRL(2%)/rh(1%) [17] | 142 × 103 | - | −9.58 ± 1.1 | - | 0.068–0.548 [17] | - | - |
| TfR-mimetic Vector: B6 | CGHKAKGPRK [39,40,41] | - | - | - | - | - | - | - |
| MNC [17] | P(50 kDa/LLL(40%) /PEG2000B6(2%)/rh (1%) [17] | 153 × 103 | - | −6.1 | - | 0.068–0.548 [17] | - | - |
| MiniAp-4 Vector M4 | H-[Dap]KAPETALD-NH2 (Dap-d lactam bridge) | 911 | - | −10.4 ± 1.3 | - | 0.2–1.04 [42] | - | - |
| MNC [17] | P(50 kDa)/LLL(40%) /PEG2000-M4(2%)/rh (1%) [15] | 139 × 103 | - | - | - | 0.068–0.548 [17] | - | - |
| Chlorotoxin Vector (to glioma) CTX | MCMPCFTTDHQMARKCDDCCGGKGRGKCYGPQCLCR; (CTX) [43,44,45] | 3996 | - | - | 0.66 [23] | 0.05–0.15 [44] | - | - |
| MNIA (fluorescence) | P(60 kDa)/LLL(40%) /PEG2000-CTX(1.5%) /ICG(2%) [34] | 160 × 103 | 11.8 ± 16 | −20.5 ± 1.8 | - | - | 1.5 [34] | 9.5 [34] |
| Vector to HER2 (HER2-mimetic) [23] AHNP | YCDGFYACYMDV-NH2 (S-S disulfide bridge) | 1647 | - | - | 0.52 [23,46,47] | - | - | - |
| MND [23] | P(50 kDa)/LLL(40%) /StarPEG(PEG200 (AHNP)2(2%) /AON(1.5%) | 331 × 103 | 7.8 ± 2.1 | −13.8 ± 1.3 | 4.6 [23] | 0.75 | - | 10 |
| MND | P(50 kDa)/LLL(40%) /StarPEG(PEG200 (AHNP)2(2%) /DTX(5%) | 274 × 103 | - | - | - | 5.0 | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ljubimova, J.Y.; Ramesh, A.; Israel, L.L.; Holler, E. Small-Sized Co-Polymers for Targeted Delivery of Multiple Imaging and Therapeutic Agents. Nanomaterials 2021, 11, 2996. https://doi.org/10.3390/nano11112996
Ljubimova JY, Ramesh A, Israel LL, Holler E. Small-Sized Co-Polymers for Targeted Delivery of Multiple Imaging and Therapeutic Agents. Nanomaterials. 2021; 11(11):2996. https://doi.org/10.3390/nano11112996
Chicago/Turabian StyleLjubimova, Julia Y., Arshia Ramesh, Liron L. Israel, and Eggehard Holler. 2021. "Small-Sized Co-Polymers for Targeted Delivery of Multiple Imaging and Therapeutic Agents" Nanomaterials 11, no. 11: 2996. https://doi.org/10.3390/nano11112996