
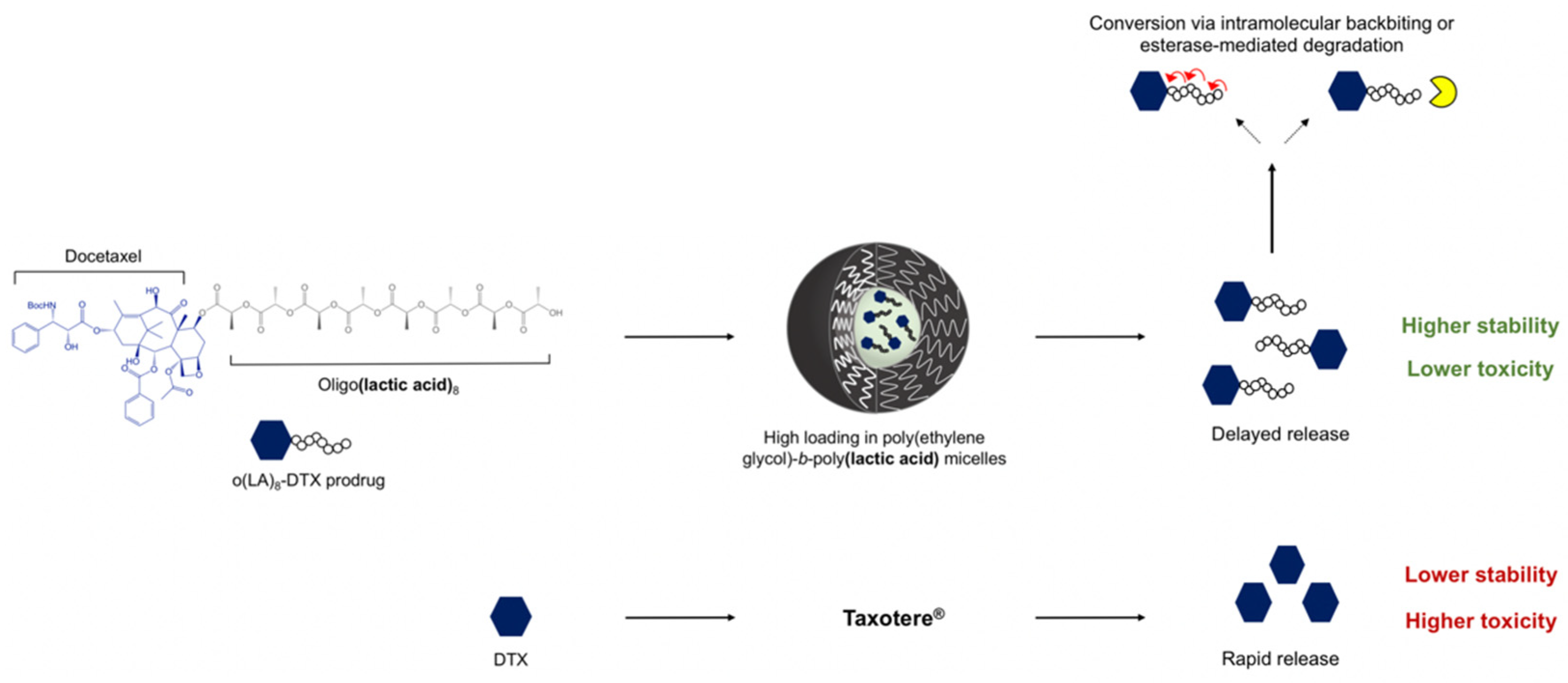
Oligo(Lactic Acid)8-Docetaxel Prodrug-Loaded PEG-b-PLA Micelles for Prostate Cancer

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
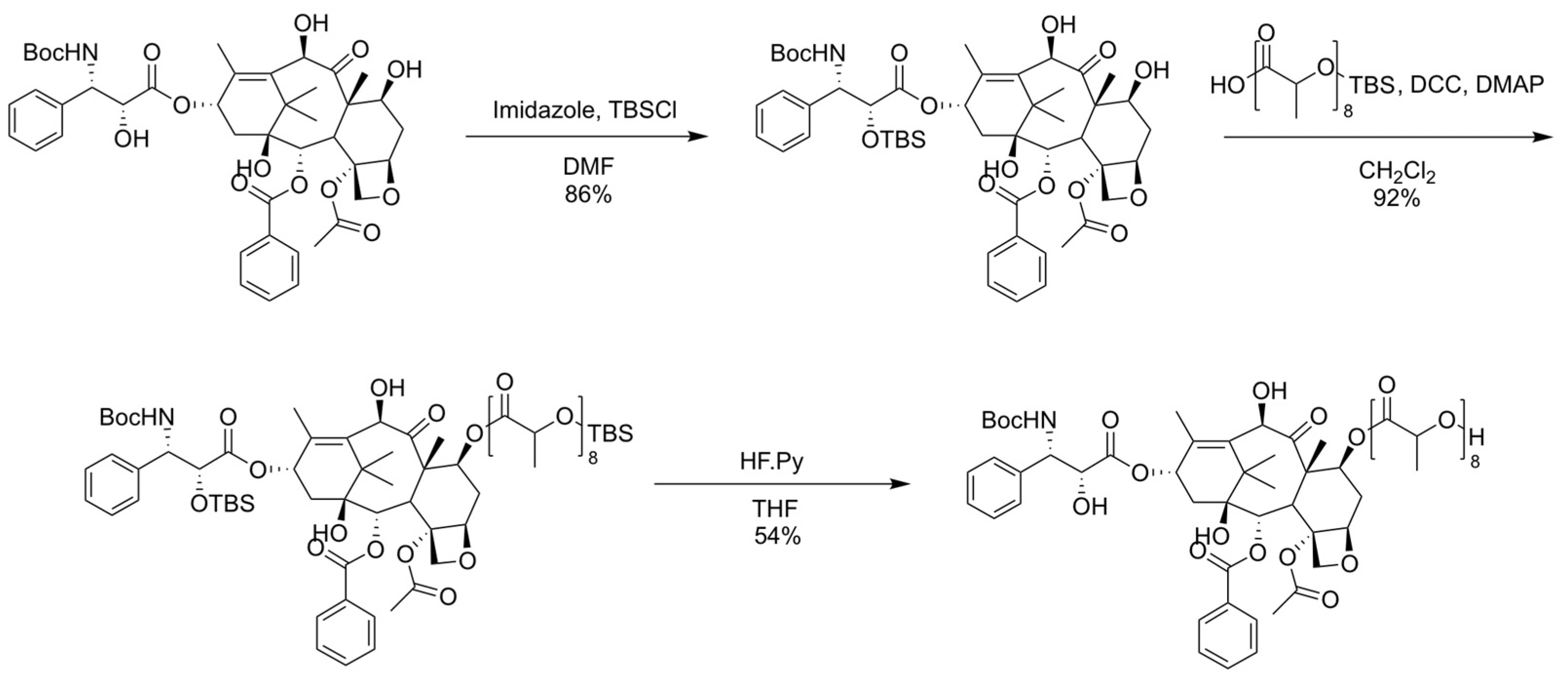
2.2. Synthesis, Purification, and Characterization of Oligo(Lactic Acid)8-Docetaxel Prodrug (o(LA)8-DTX)
2.3. Preparation and Characterization of o(LA)8-DTX-Loaded PEG4k-b-PLA2.2k Micelles
2.4. In Vitro Drug or Prodrug Release from Micelles
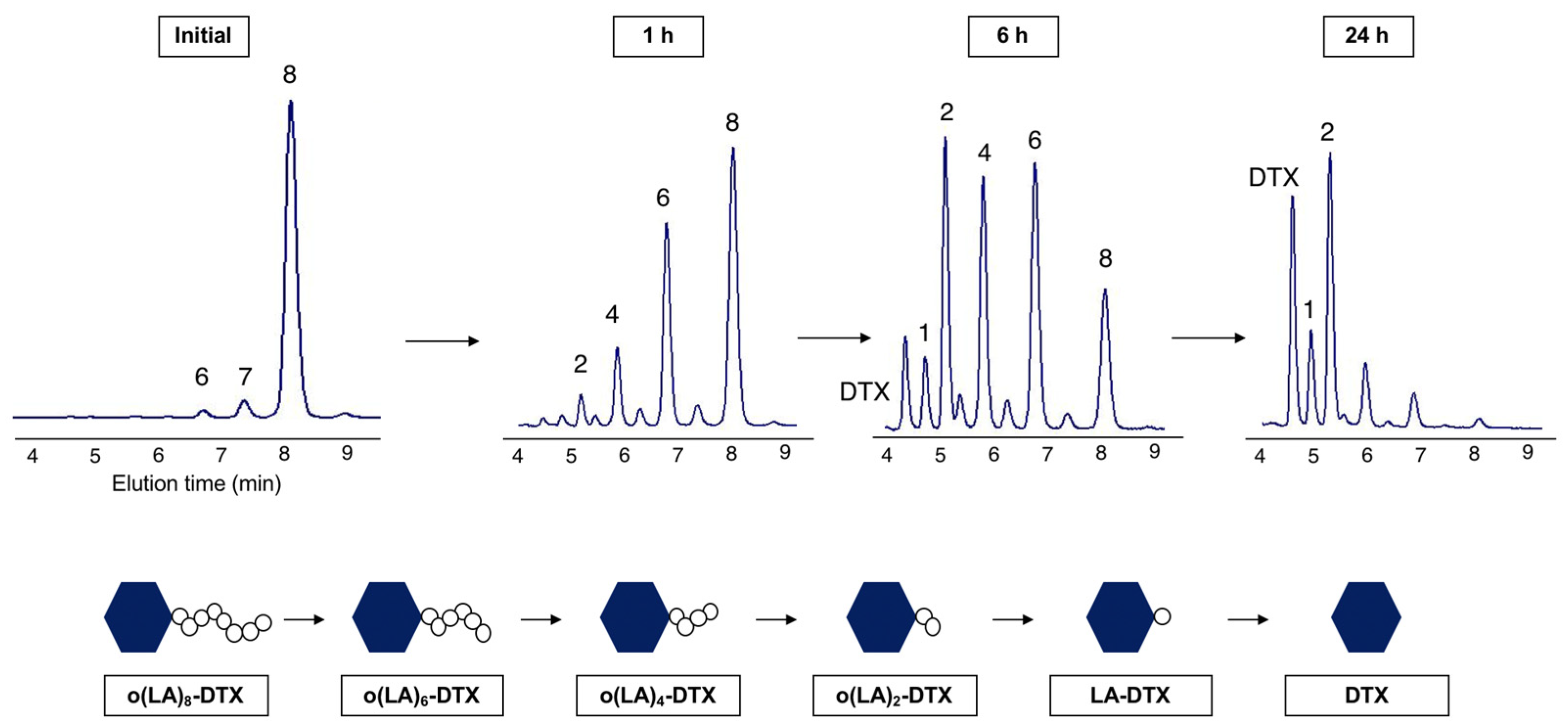
2.5. Backbiting Conversion of o(LA)8-DTX
2.6. In Vitro Plasma Stability of o(LA)8-DTX
2.7. In Vitro Cytotoxicity of o(LA)8-DTX
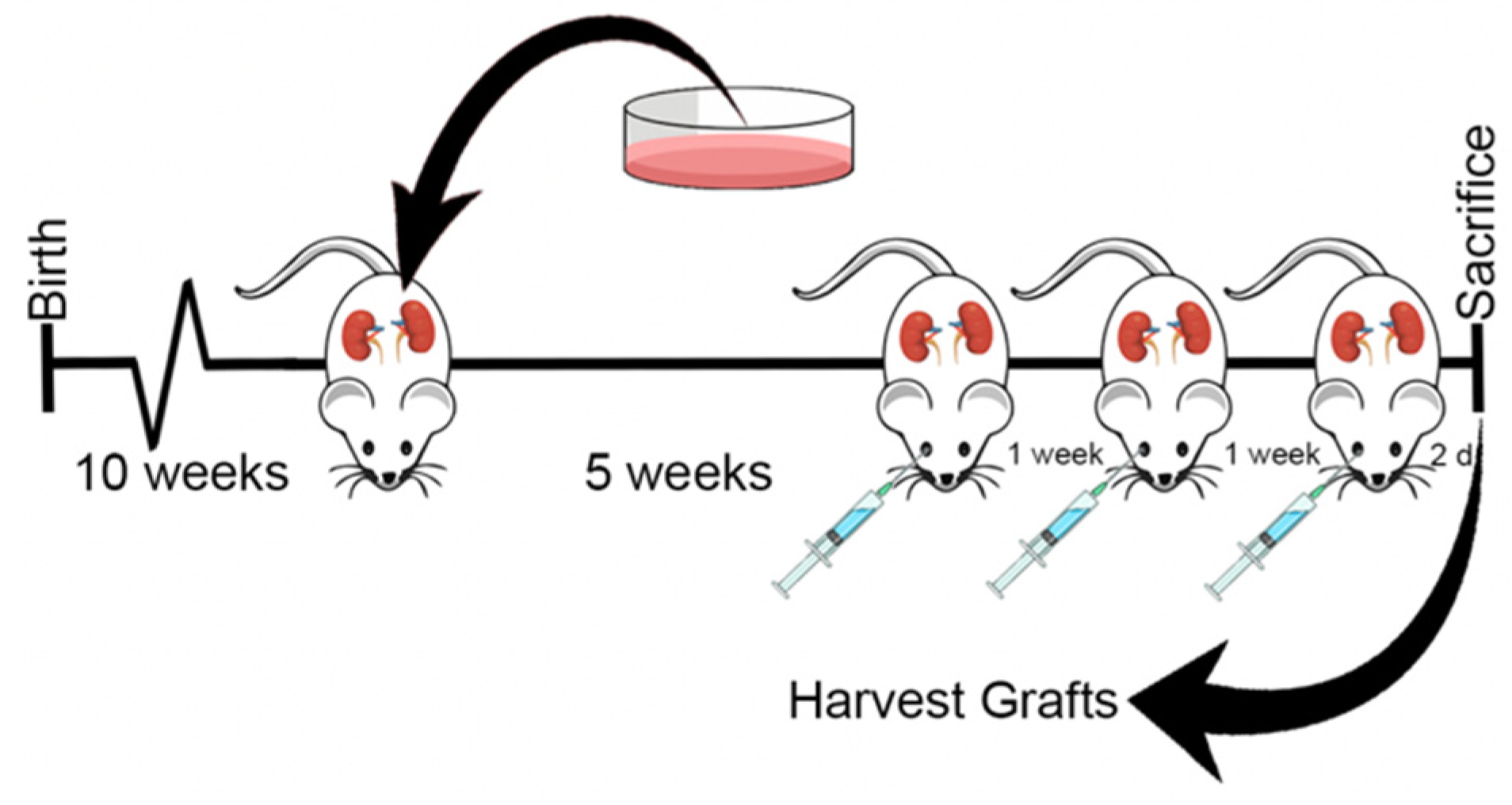
2.8. In Vivo Antitumor Efficacy in Prostate Cancer Xenograft Model
3. Results and Discussion
3.1. Synthesis and Characterization of o(LA)8-DTX
3.2. Loading and Characterization of o(LA)8-DTX-Loaded PEG4k-b-PLA2.2k Micelles
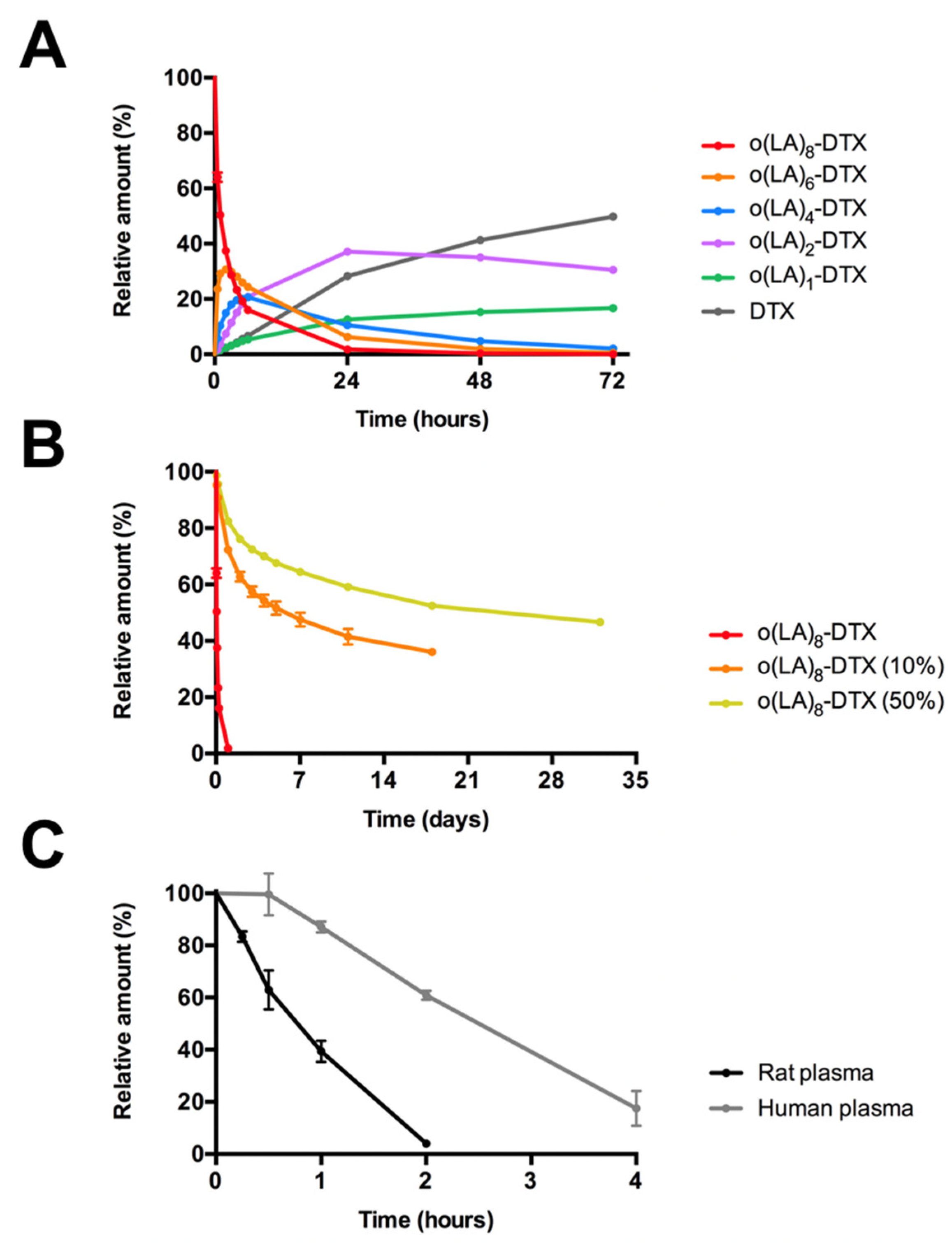
3.3. In Vitro Drug or Prodrug Release from Micelles
3.4. Backbiting Conversion of o(LA)8-DTX
3.5. In Vitro Plasma Stability of o(LA)8-DTX
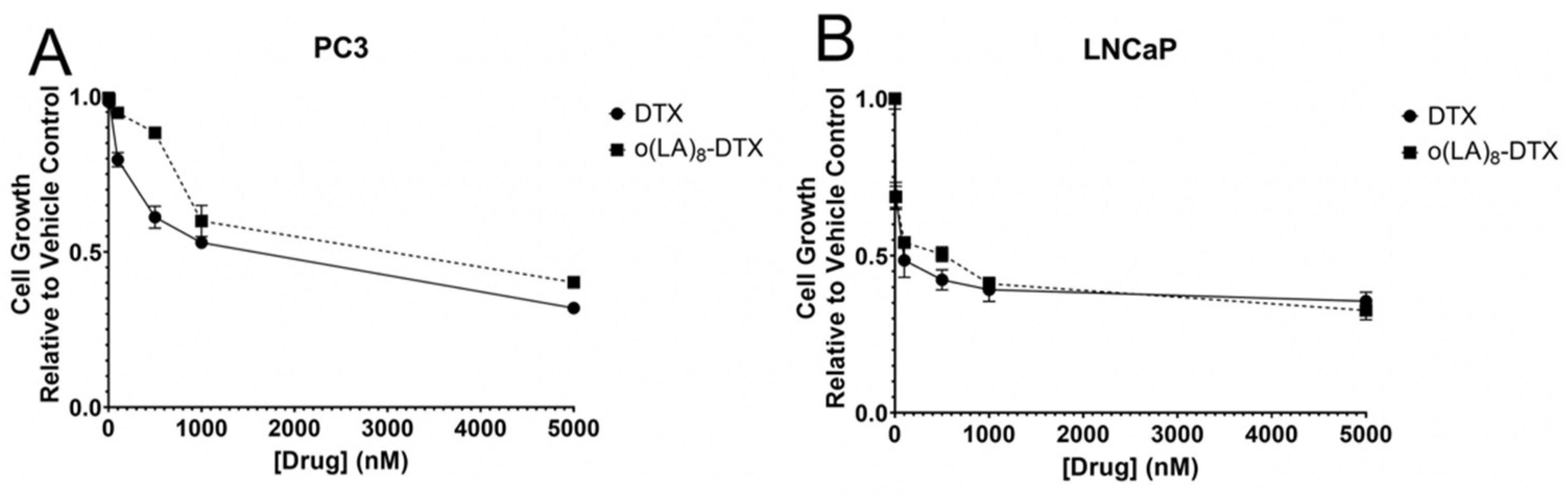
3.6. In Vitro Cytotoxicity of o(LA)8-DTX
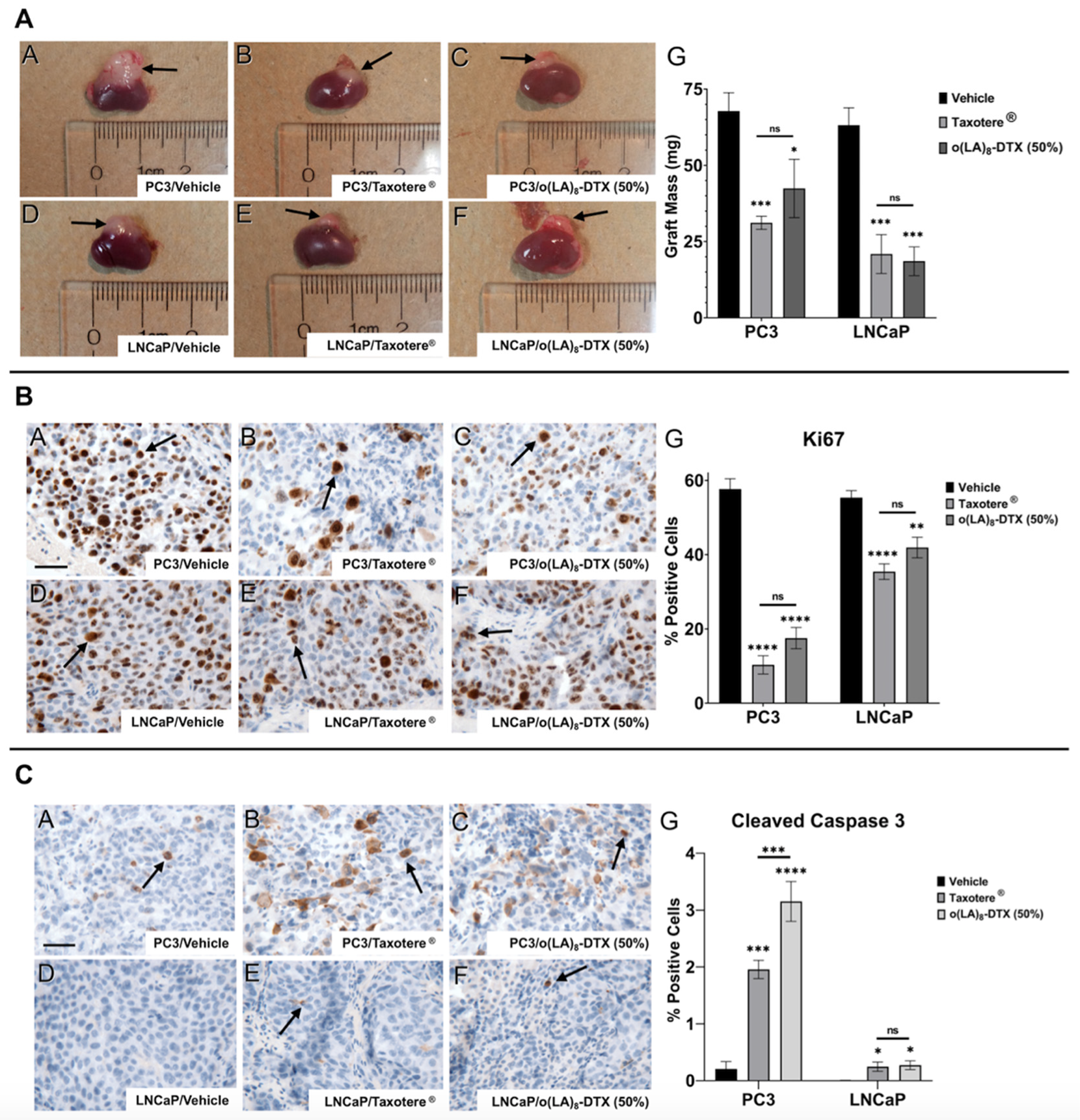
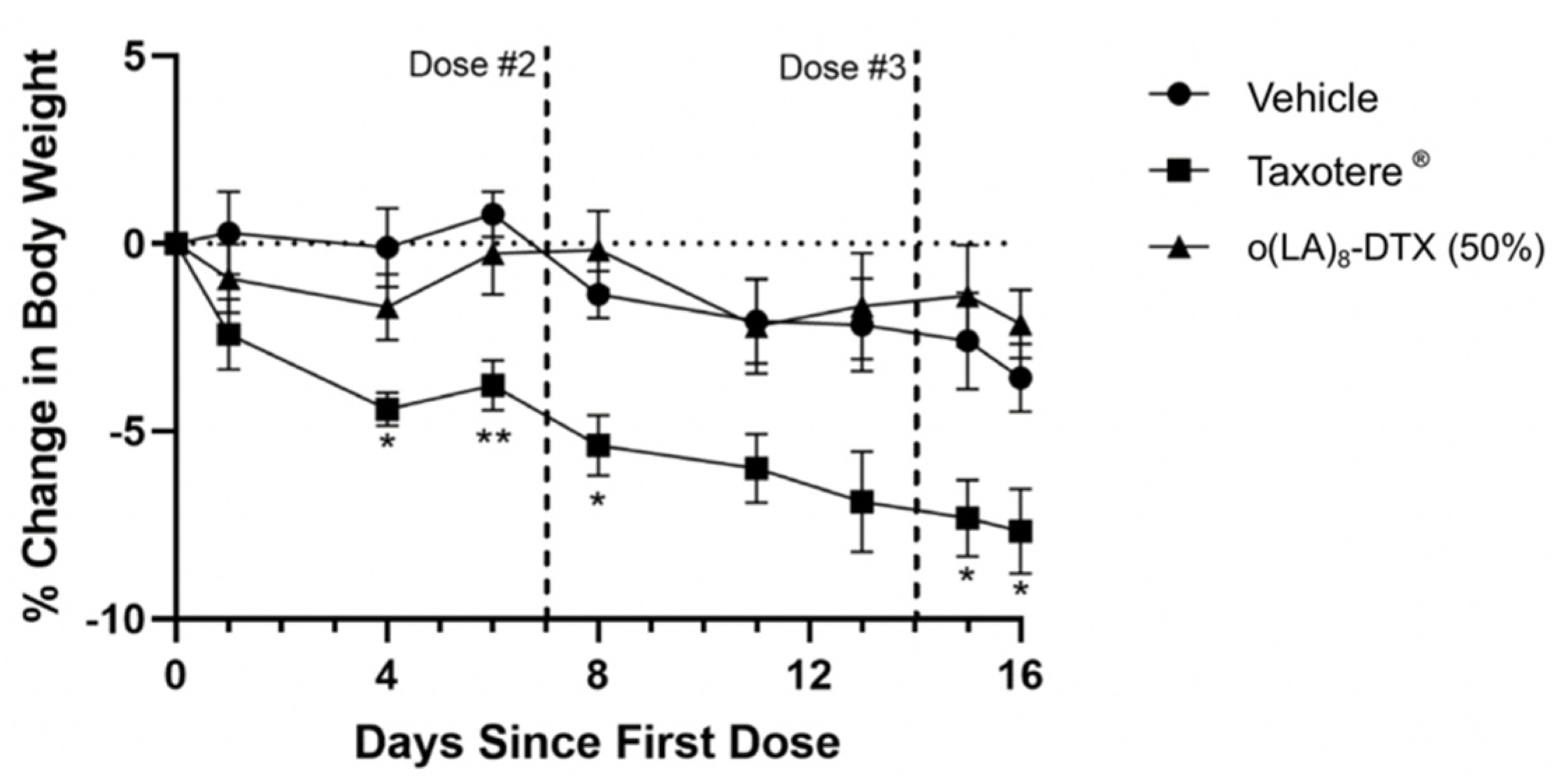
3.7. In Vivo Antitumor Efficacy in Prostate CANCER Model
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Hennenfent, K.L.; Govindan, R. Novel formulations of taxanes: A review. Old wine in a new bottle? Ann. Oncol. 2006, 17, 735–749. [Google Scholar] [CrossRef]
- Zhang, N.; Zhang, L. How nanotechnology can enhance docetaxel therapy. Int. J. Nanomed. 2013, 8, 2927–2941. [Google Scholar] [CrossRef] [Green Version]
- Imran, M.; Saleem, S.; Chaudhuri, A.; Ali, J.; Baboota, S. Docetaxel: An update on its molecular mechanisms, therapeutic trajectory and nanotechnology in the treatment of breast, lung and prostate cancer. J. Drug Deliv. Sci. Technol. 2020, 60, 101959. [Google Scholar] [CrossRef]
- Ganju, A.; Yallapu, M.M.; Khan, S.; Behrman, S.W.; Chauhan, S.C.; Jaggi, M. Nanoways to overcome docetaxel resistance in prostate cancer. Drug Resist. Updat. 2014, 17, 13–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartzberg, L.S.; Navari, R.M. Safety of Polysorbate 80 in the Oncology Setting. Adv. Ther. 2018, 35, 754–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, L.; Peng, J.; Zhao, Q.; Zhang, L.; Tang, X.; Chen, L.; Lei, M.; Qian, Z. A Novel MPEG-PDLLA-PLL Copolymer for Docetaxel Delivery in Breast Cancer Therapy. Theranostics 2017, 7, 2652–2672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Li, S.; Han, Y.; Guan, J.; Chung, S.; Wang, C.; Li, D. Poly (Ethylene Glycol)–Polylactide Micelles for Cancer Therapy. Front. Pharmacol. 2018, 9, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croy, S.; Kwon, S.R.C.A.G.S. Polymeric Micelles for Drug Delivery. Curr. Pharm. Des. 2006, 12, 4669–4684. [Google Scholar] [CrossRef]
- Owen, S.C.; Chan, D.P.; Shoichet, M.S. Polymeric micelle stability. Nano Today 2012, 7, 53–65. [Google Scholar] [CrossRef]
- Shin, D.H.; Tam, Y.T.; Kwon, G.S. Polymeric micelle nanocarriers in cancer research. Front. Chem. Sci. Eng. 2016, 10, 348–359. [Google Scholar] [CrossRef]
- Cabral, H.; Miyata, K.; Osada, K.; Kataoka, K. Block Copolymer Micelles in Nanomedicine Applications. Chem. Rev. 2018, 118, 6844–6892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ernsting, M.J.; Tang, W.-L.; MacCallum, N.W.; Li, S.-D. Preclinical pharmacokinetic, biodistribution, and anti-cancer efficacy studies of a docetaxel-carboxymethylcellulose nanoparticle in mouse models. Biomaterials 2012, 33, 1445–1454. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jin, M.; Shao, S.; Huang, W.; Yang, F.; Chen, W.; Zhang, S.; Xia, G.; Gao, Z. Small-sized polymeric micelles incorporating docetaxel suppress distant metastases in the clinically-relevant 4T1 mouse breast cancer model. BMC Cancer 2014, 14, 329. [Google Scholar] [CrossRef] [Green Version]
- Hu, Q.; Rijcken, C.J.; Bansal, R.; Hennink, W.E.; Storm, G.; Prakash, J. Complete regression of breast tumour with a single dose of docetaxel-entrapped core-cross-linked polymeric micelles. Biomaterials 2015, 53, 370–378. [Google Scholar] [CrossRef]
- Bowerman, C.J.; Byrne, J.D.; Chu, K.S.; Schorzman, A.N.; Keeler, A.W.; Sherwood, C.A.; Perry, J.L.; Luft, J.C.; Darr, D.B.; Deal, A.M.; et al. Docetaxel-Loaded PLGA Nanoparticles Improve Efficacy in Taxane-Resistant Triple-Negative Breast Cancer. Nano Lett. 2017, 17, 242–248. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.-B.; Zeng, S.; Zhao, W. Highly Stable PEGylated Poly (lactic-co-glycolic acid) (PLGA) Nanoparticles for the Effective Delivery of Docetaxel in Prostate Cancers. Nanoscale Res. Lett. 2016, 11, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-W.; Yun, M.-H.; Jeong, S.W.; In, C.-H.; Kim, J.-Y.; Seo, M.-H.; Pai, C.-M.; Kim, S.-O. Development of docetaxel-loaded intravenous formulation, Nanoxel-PM™ using polymer-based delivery system. J. Control. Release 2011, 155, 262–271. [Google Scholar] [CrossRef]
- Liang, C.; Bai, X.; Qi, C.; Sun, Q.; Han, X.; Lan, T.; Zhang, H.; Zheng, X.; Liang, R.; Jiao, J.; et al. Π electron-stabilized polymeric micelles potentiate docetaxel therapy in advanced-stage gastrointestinal cancer. Biomaterials 2021, 266, 120432. [Google Scholar] [CrossRef]
- Jun, Y.J.; Park, J.H.; Avaji, P.G.; Park, K.S.; Lee, K.E.; Lee, H.J.; Sohn, Y.S. Design of theranostic nanomedicine (II): Synthesis and physicochemical properties of a biocompatible polyphosphazene–docetaxel conjugate. Int. J. Nanomed. 2017, 12, 5373–5386. [Google Scholar] [CrossRef] [Green Version]
- Atrafi, F.; Dumez, H.; Mathijssen, R.H.; Oordt, C.W.M.V.D.H.V.; Rijcken, C.J.; Hanssen, R.; Eskens, F.A.; Schöffski, P. A phase I dose-escalation and pharmacokinetic study of a micellar nanoparticle with entrapped docetaxel (CPC634) in patients with advanced solid tumours. J. Control. Release 2020, 325, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Houdaihed, L.; Evans, J.C.; Allen, C. Overcoming the Road Blocks: Advancement of Block Copolymer Micelles for Cancer Therapy in the Clinic. Mol. Pharm. 2017, 14, 2503–2517. [Google Scholar] [CrossRef] [PubMed]
- Huynh, L.; Leroux, J.-C.; Allen, C. Enhancement of docetaxel solubility via conjugation of formulation-compatible moieties. Org. Biomol. Chem. 2009, 7, 3437–3446. [Google Scholar] [CrossRef] [PubMed]
- Repp, L.; Rasoulianboroujeni, M.; Lee, H.J.; Kwon, G.S. Acyl and oligo (lactic acid) prodrugs for PEG-b-PLA and PEG-b-PCL nano-assemblies for injection. J. Control. Release 2021, 330, 1004–1015. [Google Scholar] [CrossRef]
- Tam, Y.T.; Shin, D.H.; Chen, K.E.; Kwon, G.S. Poly (ethylene glycol)-block-poly (d,l-lactic acid) micelles containing oligo(lactic acid)8-paclitaxel prodrug: In Vivo conversion and antitumor efficacy. J. Control. Release 2019, 298, 186–193. [Google Scholar] [CrossRef]
- Tam, Y.T.; Gao, J.; Kwon, G.S. Oligo (lactic acid) n-Paclitaxel Prodrugs for Poly (ethylene glycol)-block-poly (lactic acid) Micelles: Loading, Release, and Backbiting Conversion for Anticancer Activity. J. Am. Chem. Soc. 2016, 138, 8674–8677. [Google Scholar] [CrossRef] [Green Version]
- Guenard, D.; Gueritte-Voegelein, F.; Potier, P. Taxol and taxotere: Discovery, chemistry, and structure-activity relationships. Acc. Chem. Res. 1993, 26, 160–167. [Google Scholar] [CrossRef]
- Skoczen, S.; McNeil, S.E.; Stern, S.T. Stable isotope method to measure drug release from nanomedicines. J. Control. Release 2015, 220, 169–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, K.S.; Schorzman, A.N.; Finniss, M.C.; Bowerman, C.J.; Peng, L.; Luft, J.C.; Madden, A.J.; Wang, A.; Zamboni, W.C.; DeSimone, J.M. Nanoparticle drug loading as a design parameter to improve docetaxel pharmacokinetics and efficacy. Biomaterials 2013, 34, 8424–8429. [Google Scholar] [CrossRef] [Green Version]
- de Jong, S.; Arias, E.; Rijkers, D.T.; van Nostrum, C.; Bosch, J.K.-V.D.; Hennink, W. New insights into the hydrolytic degradation of poly (lactic acid): Participation of the alcohol terminus. Polymer 2001, 42, 2795–2802. [Google Scholar] [CrossRef]
- Liederer, B.M.; Borchardt, R.T. Enzymes involved in the bioconversion of ester-based prodrugs. J. Pharm. Sci. 2006, 95, 1177–1195. [Google Scholar] [CrossRef] [PubMed]
- Komura, K.; Jeong, S.H.; Hinohara, K.; Qu, F.; Wang, X.; Hiraki, M.; Azuma, H.; Lee, G.-S.M.; Kantoff, P.W.; Sweeney, C.J. Resistance to docetaxel in prostate cancer is associated with androgen receptor activation and loss of KDM5D expression. Proc. Natl. Acad. Sci. USA 2016, 113, 6259–6264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, A.J.; Pilon, A.V.; MacDowell, R.T. Evaluation of growth and histology of human tumor xenografts implanted under the renal capsule of immunocompetent and immunodeficient mice. Cancer Res. 1985, 45, 4963–4969. [Google Scholar] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug/Prodrug (Target wt Loading %) | Actual wt Loading (%) | Encapsulation Efficiency (%) | Z-Average Particle Size (nm) | PDI | Room Temperature Stability | Release t1/2 (h) |
|---|---|---|---|---|---|---|
| DTX (10%) | 9.7 ± 0.1 | 97.2 ± 2.5 | 30.6 ± 1.7 | 0.18 ± 0.03 | <24 h | 3.0 |
| o(LA)8-DTX (10%) | 9.8 ± 0.6 | 98.2 ± 5.9 | 31.2 ± 0.5 | 0.07 ± 0.02 | >3 months | 16.1 |
| o(LA)8-DTX (50%) | 49.0 ± 0.8 | 98.1 ± 1.7 | 103.4 ± 10.9 | 0.10 ± 0.02 | >3 months | 39.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Repp, L.; Unterberger, C.J.; Ye, Z.; Feltenberger, J.B.; Swanson, S.M.; Marker, P.C.; Kwon, G.S. Oligo(Lactic Acid)8-Docetaxel Prodrug-Loaded PEG-b-PLA Micelles for Prostate Cancer. Nanomaterials 2021, 11, 2745. https://doi.org/10.3390/nano11102745
Repp L, Unterberger CJ, Ye Z, Feltenberger JB, Swanson SM, Marker PC, Kwon GS. Oligo(Lactic Acid)8-Docetaxel Prodrug-Loaded PEG-b-PLA Micelles for Prostate Cancer. Nanomaterials. 2021; 11(10):2745. https://doi.org/10.3390/nano11102745
Chicago/Turabian StyleRepp, Lauren, Christopher J. Unterberger, Zhengqing Ye, John B. Feltenberger, Steven M. Swanson, Paul C. Marker, and Glen S. Kwon. 2021. "Oligo(Lactic Acid)8-Docetaxel Prodrug-Loaded PEG-b-PLA Micelles for Prostate Cancer" Nanomaterials 11, no. 10: 2745. https://doi.org/10.3390/nano11102745