The aim of this work is to evaluate the effect of the NC size and purity on the Pr3+ optical properties by analyzing in detail NCs prepared through five different Y2O3: Pr3+ synthesis procedures. Firstly, the crystalline structure and particle size is studied by PXRD and Raman spectroscopy, and the presence of Pr3+ is confirmed by reflectance spectroscopy. The influence of the synthesis method itself as well as diverse parameters, such as precursors ratio or calcination temperature, on the crystal size, size distribution, and particle aggregation has been investigated by TEM. Secondly, a thorough analysis of the Y2O3: Pr3+ NCs optical properties has been performed by means of emission and excitation spectra. Clearly, luminescence from Pr3+ ions in both C2 and S6 crystallographic sites is observed for all samples. Finally, the Pr3+ emission lifetime as a function of the NCs synthesis method is used as a figure of merit, as well as the dependence of the emission lifetime on Pr3+ concentration. The luminescence decay time is an appropriate parameter to calibrate the homogeneous distribution of Pr3+ and the presence of impurity traps acting as luminescence quenchers. Since all NCs have been synthesized with the same precursors, the only effect is due to the synthesis method and conditions.
3.1. Synthesis and Structural Characterization of Y2O3: Pr3+ Nanocrystals
All the so-obtained Y
2O
3: Pr
3+ NCs were initially studied by PXRD, and reflectance and Raman spectroscopy. The PXRD pattern analyses indicate that the five synthesis methods provide the same crystalline structure (
Figure 1 and
Figure S1, ESI). Indeed, all the Y
2O
3: Pr
3+ NCs samples show a pure cubic phase (space group
Ia) in which two different crystallographic sites, C
2 (75%) and S
6 (25%), can be occupied by Pr
3+ ions [
46]. In both sites, Pr
3+ is present in a six-fold coordination surrounded by oxygen ions [
15]. As the structure is body-centered, the unit cell contains twice the primitive cell. The Y
3+ ions occupy two types of Wyckoff positions, 8b and 24d, whereas all O ions occupy the 48e position.
From the crystal space group, 120 vibrational modes are possible. The irreducible representations for the optic and acoustic vibrational modes are [
47]
Among these 120 modes, there are 51 grouped into 17 infrared active modes of T
u symmetry and 54 into 22 Raman active modes (4A
g + 4E
g + 14T
g). Consequently, up to 22 lines would be expected in the Raman spectra. However, a smaller number of lines was experimentally observed, probably due to the superposition of different types of transitions. All the samples exhibit similar Raman spectra (
Figure 2 and
Figure S2, ESI). The Raman peaks observed in the 0–600 cm
−1 range were assigned according to refs. [
47,
48], being the most intense one located at around 376.4 cm
−1. This band corresponds to a T
g vibration type. On the other hand, the peaks observed in the region of the external lattice vibrations—i.e., below 200 cm
−1—are associated to pure Y
3+ vibrations [
47].
Table 1 shows the Raman frequencies, peak width (FWHM) and their symmetry assignment. The data were obtained by fitting to Lorentzian functions. No relationship between FWHM and nanocrystal size or synthesis method was found within the experimental resolution (0.6 cm
−1). According to dispersion curves [
49], a displacement of the most prominent band (
Figure S3, ESI), placed at
ca. 376.4 cm
−1, towards lower frequencies is expected upon decreasing the nanocrystal size. However, this peak does not present any displacement independently of the synthesis method or nanocrystal size, and thus there is no confinement effect due to the size.
In addition, the reflectance spectrum of all samples is dominated by a broad band centered below 400 nm (
Figure 3 and
Figure S4, ESI), which is associated with interconfigurational 4
f2 → 4
f15
d1 transitions of Pr
3+ ions hosted in the C-Y
2O
3 lattice. Low-intensity and sharp peaks are assigned to
f-f intraconfigurational transitions. The transition from the ground state to the
1G
4 excited state is weakly observed at
ca. 1000 nm. The absorption peaks detected from 1300 to 1800 nm correspond to transitions from the
3H
4 ground state to the
3F
J multiplets [
15]. There is a jump in absorbance intensity at 800 nm due to the change of detector and diffraction grating that is not corrected by the baseline correction.
Thus, PXRD as well as Raman and reflectance spectroscopy confirm that Y2O3 NCs doped with Pr3+ obtained via the five synthesis methods of choice present the same crystalline structure and vibrational peaks. It is worth mentioning that these synthesis methods produce NCs with sizes in the 20–60 nm range. This size range can minimize the surface effects due to Pr3+ ions located at the NCs surface. In the following sub-sections, an exhaustive analysis of particle size, size distribution, and aggregation of NCs is performed by means of TEM images analysis. This is very relevant to understand the optical properties of the as-prepared nanoparticles.
3.1.1. Combustion Method
The Y
2O
3: Pr
3+ NCs prepared by combustion method were calcined at 900 °C to improve the crystallinity. TEM images (
Figure 4) revealed the presence of highly agglomerated NCs with a grain size in the 10–40 nm range. This NC size is in good agreement with the one estimated by PXRD (35.7(5) nm), thus indicating single domain NCs. Additionally, TGA (
Figure S5a, ESI) showed a small weight loss of
ca. 1.5% from RT to 400 °C related to the presence of reagent traces.
3.1.2. Molten Salt Method
Y
2O
3: Pr
3+ NCs obtained via molten salt method look polyhedral and well dispersed, with an average size of 67(19) nm (
Figure 5 and
Figure S6, ESI). PXRD analysis (
Figure S1b, ESI) showed a good crystallinity grade and an average grain size of 77.7(4) nm, which was compatible with TEM observations. TG + DSC measurements (
Figure S5b, ESI) displayed several weight losses from RT to 800 °C that decreased with the increase in the number of washing cycles, and thus could be a sign of the presence of remaining NaNO
3 and KNO
3 salts.
3.1.3. Sol–Gel Pechini Method
Different parameters were modified to optimize the synthesis of Y
2O
3: Pr
3+ NCs by sol–gel Pechini method, specifically the type of hydroxyl carboxylic acid or chelating agent, as well as temperature and calcination time (
Table 2). Firstly, citric acid was selected as chelating agent (Entry 1). The formed gel was calcined at 800 °C for 16 h, leading to the formation of NCs surrounded by a presumably organic layer (
Figure S7, ESI). No changes were observed by TEM when the calcination time was increased up to 24 h (Entry 2 and
Figure S8, ESI). However, such a layer was removed by increasing the calcination temperature up to 900 °C (Entry 3), as confirmed by TEM and TG + DSC analyses (
Figure 6 and
Figures S9 and S5c, ESI). Under these conditions, the synthesized NCs are polyhedral crystallites (
Figure S1c, ESI) and show two populations of different size, 26(6) and 62(16) nm. Indeed, the average grain size determined by PXRD was 51(9) nm. Finally, EDTA was used instead of citric acid (Entry 4). The obtained NCs are more homogenous in size, 40(12) nm, but exhibit agglomeration (
Figure S10, ESI).
3.1.4. Homogeneous Precipitation Method
In general, the homogeneous precipitation method led to the generation of small crystalline NCs with grain size in the 20–30 nm range forming spherical nanoparticles with an excellent dispersion. Firstly, spheres of 260(26) nm (and a few of 106(14) nm) containing NCs with 20.2(2) nm in size (
Figure S11, ESI) were observed using the following reaction conditions: 360 mL of solvent, 0.485 mol of urea and 2 h as reaction time (
Table 3, entry 1). Subsequent synthesis reactions were carried out by modifying the water volume, which was increased up to 720 mL and reduced to 200 mL (Entries 2 and 3). In both cases, the nanospheres presented appropriate dispersion and well-defined spherical morphology, while the NC size distribution hardly changed and was 21.8(3) and 25.2(4) nm, respectively. In addition, the sphere size showed no appreciable differences, with an average size of 233(41) nm when H
2O volume was doubled and 229(20) nm when it was reduced to 200 mL (
Figures S12 and S13, ESI). Consequently, it seems that the water volume does not affect the size of NCs and nanospheres nor the dispersion. Similarly, the reaction time had no influence on these properties, since no changes were observed for reaction times of 45, 60, 90, 120, and 180 min.
Interestingly, a change in the urea content produced remarkable differences, not only in size but also in nanoparticle dispersion. A clear tendency was confirmed by PXRD analysis and TEM images, in which an increase in the amount of urea led to a reduction of nanoparticle size. Indeed, NCs synthesized using 0.166 mol of urea (Entry 4) showed a grain size of 26.9(3) nm and average sphere size of 338(35) nm (
Figure S14, ESI). In contrast, an average NC size of 22.2(2) nm and an average spherical nanoparticle size of 75(34) nm were observed with 1.415 mol of urea (Entry 5 and
Figure S15, ESI). In addition, these spherical nanoparticles exhibit a broad size distribution. An intermediate urea concentration (0.832 mol) provided NCs of 21.1(2) nm and spheres with 219(24) nm in size (Entry 6 and
Figure S16, ESI). On the other hand, the dispersion displayed an inverse trend; that is, samples prepared with low urea concentrations presented an excellent dispersion of nanospheres (
Figure S14, ESI), while high urea amounts led to slightly agglomerated nanoparticles (
Figure S15, ESI). In this sense, we were interested in finding the conditions that promote not only an appropriate size and good dispersion of the formed spheres but also a narrow particle size distribution, which will lead to a better understanding of the NCs optical performance. In this context, Y
2O
3: Pr
3+ NCs with a grain size of 22.4(3) nm, an average sphere size of 155(20) nm and excellent dispersion (
Figure 7 and
Figures S17 and S18, ESI) were obtained by the use of 200 mL of deionized H
2O and 0.832 mol of urea (Entry 7). Moreover, no organic or precursor traces were found by TG + DSC analysis (
Figure S5d, ESI).
3.1.5. Solvothermal Method
Finally, we synthesized Y
2O
3: Pr
3+ NCs through the solvothermal method. The synthesis process required heating the reaction mixture at 180 °C in a Teflon-lined stainless-steel autoclave. Then, the resulting sample was calcined at 800 °C for 4 h (
Table 4, entry 1). These conditions led to the formation of polyhedral NCs that exhibit some aggregation and 31(7) nm in size (
Figure S19, ESI), while the grain size determined by PXRD was 21.1(1) nm. Additionally, an organic layer was observed. This was confirmed by a TGA in which several weight losses were identified below 500 °C. Consequently, the calcination temperature was increased up to 900 °C in order to suppress this layer (Entry 2). The resulting NCs showed a polyhedral morphology and an increase in size up to 42(11) nm (
Figure S20, ESI), which was consistent with PXRD analysis (31.1(3) nm). Besides, the organic layer was successfully removed. The effect of additional parameters was also studied. The concentration of RE precursors was doubled (Entry 3), which resulted in a considerable increase in the NC aggregation, although the polyhedral morphology was not altered (
Figure S21, ESI). PXRD analysis showed a grain size of 43.1(3) nm, which was in good agreement with that observed by TEM (40(10) nm), indicating NCs with single domain structure. The effect of the employed alcohol was also examined, and thus EG was replaced by EtOH (Entry 4). TEM images showed NCs with an average size of 28(10) nm and remarkable aggregation. Furthermore, the presence of the above-mentioned organic layer was also detected (
Figure S22, ESI). On the other hand, the calcination time was increased up to 8 h (Entry 5), which led to a strong aggregation (
Figure S23, ESI). To sum up, high concentrations of precursors, the use of other alcohols and longer calcination times produce an increase in NC aggregation.
Finally, the influence of the temperature was also analyzed. An increase in both reaction and calcination temperatures generally leads to bigger NCs. Specifically, an increase in the autoclave temperature from 180 °C to 220 °C (Entry 6) provided NCs of 47(12) nm in size, while no organic layer was observed (
Figure S24, ESI). Next, maintaining the reaction temperature at 220 °C, the calcination temperature was increased up to 1000 °C (Entry 7). These conditions promoted a great increase in the average NC size up to 68(15) nm (
Figure 8 and
Figure S25, ESI), while that determined by PXRD was 52.8(5) nm. Additionally, TEM images (
Figure 8 and
Figure S25, ESI) and TG + DSC analysis (
Figure S5e, ESI) confirmed that no organic contamination was present.
3.2. Optical Properties of Synthesized Y2O3: Pr3+ Nanocrystals
To analyze the optical properties of the NCs prepared following the five aforementioned synthesis procedures, emission and excitation spectra were recorded for selected samples. RT emission spectra were studied upon direct excitation of Pr
3+ in the UV (at 292 and 330 nm) or 490 nm into the
3P
J multiplet (for both sites) (
Figure 9 and
Figures S26 and S27, ESI). The two UV excitation wavelengths allowed us to record two different emission spectra, which we associated to Pr
3+ placed at C
2 and S
6 sites, respectively. The intensity of the Pr
3+ emission spectrum at the C
2 site is about an order of magnitude more intense than the one corresponding to the S
6, in agreement with the 3:1 ratio of the two sites and the absence of inversion center for the C
2 site. All samples were analyzed under the same conditions in terms of experimental setup and geometry. The NCs prepared by five different synthesis procedures present similar emission spectra, with only slight differences regarding luminescence intensity. All emission spectra exhibit peaks between 600 and 670 nm which are characteristic of emission from
1D
2 to
3H
4 level, while peaks located in the range of 700–750 nm are assigned to the transition
1D
2 →
3H
5 (
Figure 10).
The differences observed in emission intensities for the NCs can be related to features such as Pr
3+ distribution, size, surface effects, and crystallinity. As an example, it is worth mentioning that the luminescence intensity shown by NCs prepared through Pechini method is about an order of magnitude more intense than that displayed by Y
2O
3: Pr
3+ NCs obtained via molten salt synthesis (
Figure S26 and S27, ESI). Nevertheless, the luminescence intensity is a difficult magnitude to compare among samples and therefore the luminescence lifetime (τ) was chosen as a more accurate parameter to perform a proper comparison of the optical properties, since it does not depend on the geometry, configuration of the experimental setup, and grain size.
The excitation spectra were also studied monitoring the emission from the
1D
2 multiplet to the
3H
4 ground state (λ
em = 717 and 603 nm for C
2 and S
6, respectively). All synthesized Pr
3+-doped Y
2O
3 NCs showed similar excitation spectra (
Figure 11 and
Figures S28 and S29, ESI), dominated by broad bands in the UV region resulting from the interconfigurational transition 4
f2 → 4
f15
d1 of Pr
3+, since yttria presents no absorption in this below-gap spectral range [
15]. According to Aumüller et al. [
14], this transition occurs at higher energies at C
2 sites than at S
6 sites for sesquioxides. Thus, the broad excitation band centred at 280 nm is assigned to Pr
3+ ions at C
2 whereas the peak at 317 nm is assigned to Pr
3+ ions at S
6 site. Additionally, the sharp lines identified in the 430–520 nm wavelength range are attributed to the intraconfigurational transitions from the ground state to the
3P
J +
1I
6 multiplets (
Figure 10).
3.3. Comparative Study of the Synthetic Methods Based on the 1D2 Emission Lifetime of Y2O3: Pr3+ Nanocrystals
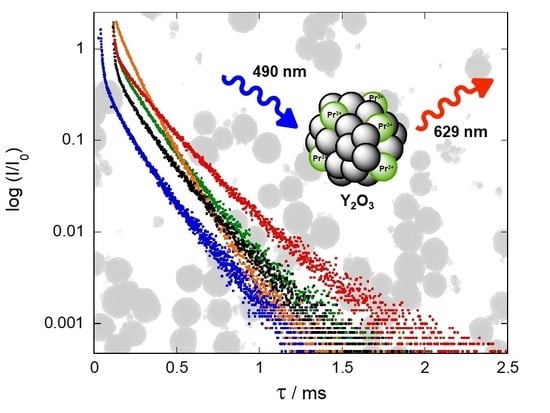
Luminescence lifetime (τ) measurements were performed to carry out a quantitative comparison of the luminescence efficiency of Y
2O
3: Pr
3+ NCs obtained by the different synthesis methods. The time evolution of
1D
2 →
3H
4 luminescence detected at 629 nm was recorded after direct excitation into
3P
J excited state at 429 nm for all synthesized samples. All luminescence decay curves were fitted to a double-exponential behavior that can be attributed to the excitation of both sites and the coincidence of peaks from both sites at this wavelength (
Figure 12). This fact, together with the probable energy transfer between both sites [
15], could be the reason of this bi-exponential behavior. Consequently, the shorter lifetime component could be assigned to Pr
3+ ions at the C
2 site, and the longer one, to Pr
3+ species at the S
6 site.
Firstly, diverse parameters were optimized for the five different synthesis procedures to maximize the luminescence lifetime. Assuming that all synthesis methods give rise to a similar occupancy of both crystallographic sites, the observed differences in the luminescence lifetime values can be attributed to parameters such as nanocrystal size, Pr
3+ distribution, crystallinity, and the presence of remaining reagent residues and molecules adsorbed at the NCs surface. In this context, Y
2O
3: Pr
3+ NCs synthesized by combustion method showed an increase in their average lifetime from 114 µs to 173 µs after the thermal treatment at 900 °C (
Figure 12 and
Figure S30, ESI). Firing the NCs at this temperature promoted not only a better crystallinity, but also the elimination of organic residues assumed to be the most important channel for non-radiative relaxation processes. This effect of remaining impurities on luminescence decay was clearly observed in NCs prepared via molten salt procedure. Indeed, the lifetime increased with the number of washing cycles. NCs that were washed-up 5, 7, and 10 times provided luminescence average lifetimes of 128, 138, and 160 µs, respectively (
Figure 12 and
Figures S31 and S32, ESI).
Samples prepared via Pechini method were also analyzed. As expected, an increase in calcination time and temperature (
Table 2, entries 1–3) had a positive effect on luminescence average lifetime, raising from 104 to 114 and 118 µs (
Figure 12 and
Figures S33 and S34, ESI), respectively. This may be attributed to the removal of the organic layer and a better crystallinity of the sample. In addition, the use of EDTA as chelating agent produced a slight improvement in lifetime up to 125 µs (
Figure S35, ESI), presumably caused by the monodisperse-size character of these NCs (
Table 2, entry 4). However, given that high aggregation was also promoted in this case, the enhancement in radiative lifetime was not remarkable enough to consider EDTA as a better chelating agent.
An increase in the luminescence lifetime was observed for Y
2O
3: Pr
3+ NCs synthetized through homogeneous precipitation method. Indeed, NCs prepared with the initial reaction conditions (
Table 5, entry 1) presented an average luminescence average lifetime of 152 µs (
Figure S36, ESI). The optimization of the different synthesis parameters not only provided better NCs in terms of size, dispersion, and crystallinity, but also improved the luminescence decay rate. For instance, the reduction in the amount of H
2O to 200 mL (Entry 2) led to an increase in the lifetime up to 161 µs (
Figure S37, ESI). In this line, 184 µs as a luminescence lifetime (
Figure 12) was observed under the optimized reaction conditions based on the use of 200 mL of deionized H
2O and 0.832 mol of urea (Entry 3).
Finally, we studied the Y
2O
3: Pr
3+ NCs synthesized via solvothermal method (
Table 6), which showed the longest luminescence lifetimes. A luminescence decay time of 198 μs (
Figure S38, ESI) was observed for NCs prepared at 180 °C and 800 °C as reaction and calcination temperatures, respectively (
Table 6, entry 1). The use of reaction conditions that produce a strong NC aggregation led to a decrease in the lifetime values (Entries 3–5 and
Figures S39–S41, ESI).
Conversely, an increase in the reaction and calcination temperatures improves the crystallinity, suppressing the observed organic layer, thus providing NCs with longer values for the luminescence lifetime (Entries 2, 6 and 7,
Figure 12, and
Figure S42, ESI). A radiative decay of 216 μs (
Figure 12) was measured using 220 °C and 1000 °C as reaction and calcination temperatures, respectively (Entry 7). Similarly, a subtle increase in the luminescence lifetime up to 223 μs (
Figure 12) was obtained with 220 °C and 900 °C (Entry 6).
The variations of luminescence lifetime attributed to non-radiative processes can be related to factors such as nanocrystal size, annealing temperature, impurities content, trap defects associated with the synthesis method, and small fluctuations in Pr
3+ concentration, to name a few. In fact, an increase in the nanocrystal size generally led to a reduction of surface effects and thus an improvement of luminescence properties, i.e., an increase of the luminescence lifetime (see
Table 5;
Table 6, entries 2, 6, and 7). On the other hand, it seems that an increase in the NC aggregation produces a decrease in the lifetime values (see
Table 6, entries 3–5). The NCs described in the present work are bigger than 20 nm, and the Pr
3+ concentration is low (0.1%). These features can minimize the surface effects mainly due to the small number of Pr
3+ ions located at the surface and the adsorption of molecules from the environment [
50,
51]. In this line, we have additionally studied the dependence of the emission lifetime on Pr
3+ concentration for NCs obtained through solvothermal, precipitation, and sol–gel Pechini methods (
Figure 13). As expected, a decrease in the luminescence decay rate values with an increase in the Pr
3+ concentration was observed, thus confirming that 0.1% is the optimal Pr
3+ content, regardless the synthesis procedure.
All things considered, the optimization of the different synthesis parameters for each discussed method has allowed to improve the optical properties of Y
2O
3: Pr
3+ NCs, therefore achieving longer luminescence lifetimes for the visible red Pr
3+ emission (
1D
2 →
3H
4).
Table 7 summarizes the optimized lifetimes for the NCs prepared via the five synthesis procedures. Some of the presented methods have proven to produce NCs with superior optical properties. Among the different procedures, the solvothermal synthesis provided the best results. This method is not only reproducible and time-efficient, but also produces NCs with appropriate size and dispersion. In addition, these Y
2O
3: Pr
3+ NCs showed one of the most intense emission spectra together with the longest luminescence lifetime. While the luminescence lifetime value for bulk 0.1% Pr
3+-doped Y
2O
3 is reported to be 124 µs [
32], some authors have described emission decay rates between 110–115 µs for Y
2O
3: Pr
3+ (0.1%) NCs [
17,
52]. To the best of our knowledge, 180 µs is the longest luminescence emission lifetime reported to date for Pr
3+-doped yttria NCs [
53]. Consequently, it is worth noting that three of the optimized methods described herein provide Y
2O
3: Pr
3+ NCs with the longest luminescence lifetime values described so far.

 ,
, 














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}