Lipid-Based Drug Delivery Nanoplatforms for Colorectal Cancer Therapy



Abstract
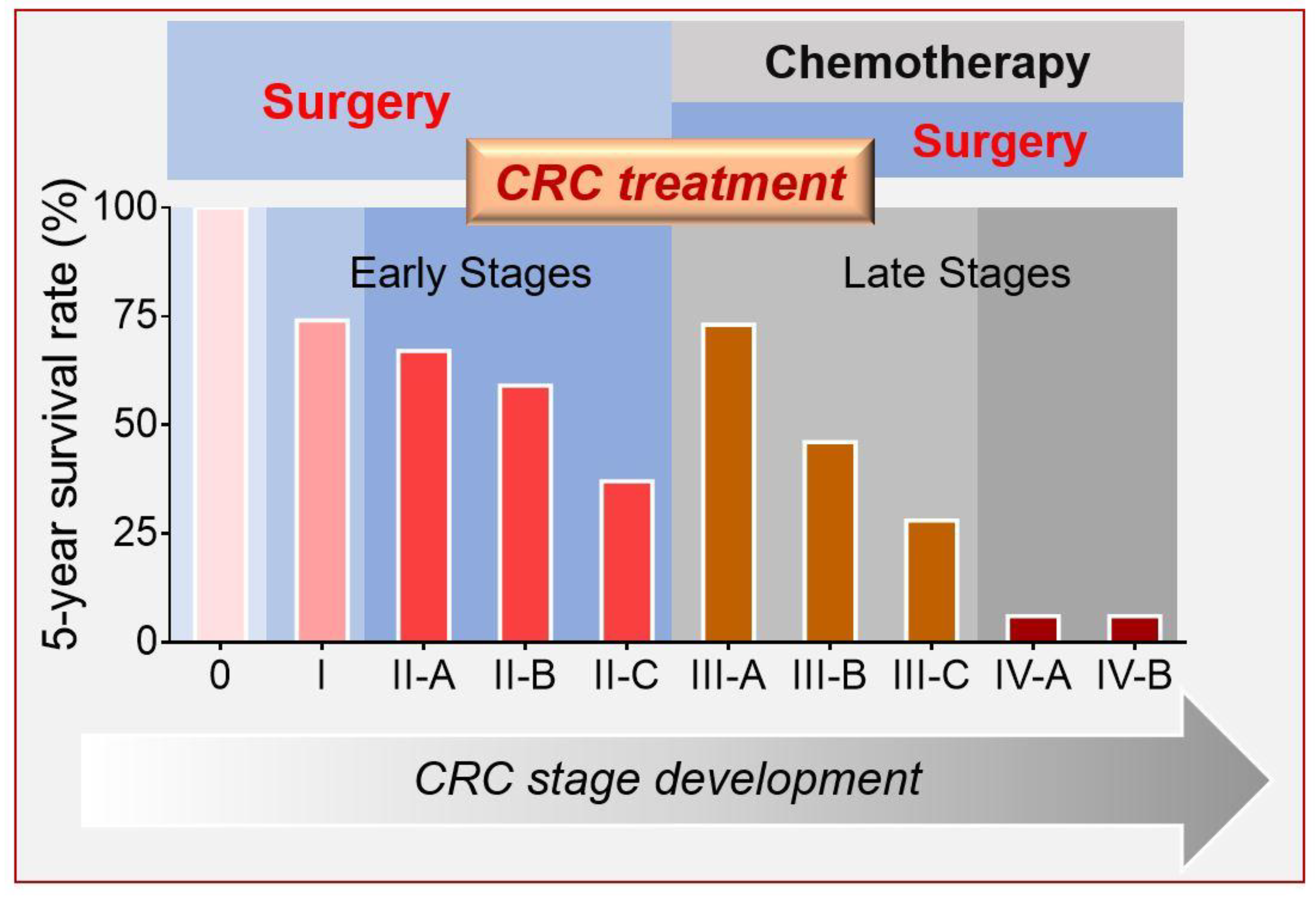
:1. Introduction
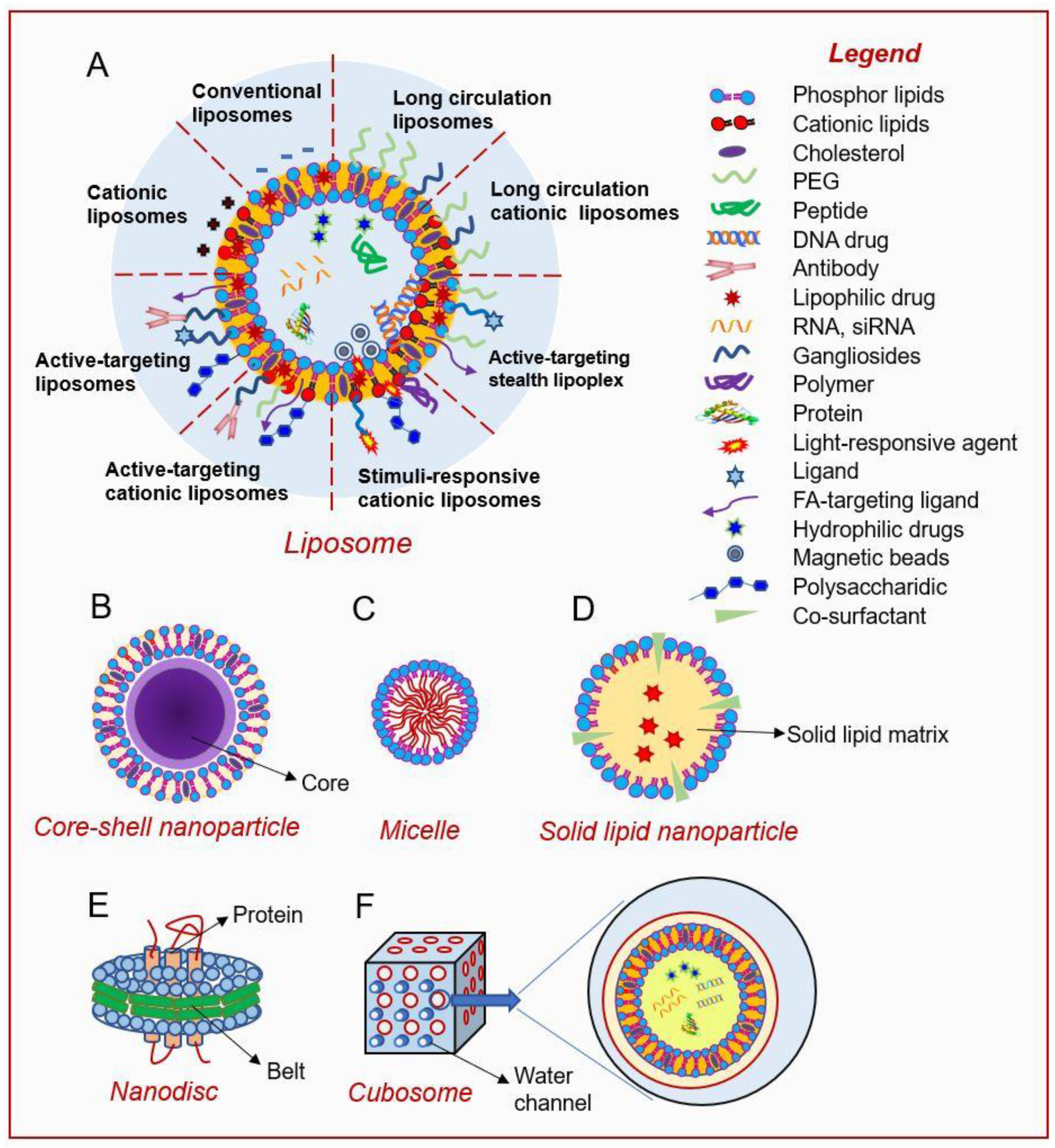
2. Current Lipid-Based Nanoplatforms
2.1. Liposomal Nanoparticles
2.1.1. Conventional Liposomes
2.1.2. Long-Circulating Liposomes (LCLs)
2.1.3. Active-Targeting Liposomes
2.1.4. Stimuli-Sensitive Liposomes
2.1.5. Cationic Liposomes
2.2. Variants of Liposomes
2.2.1. Prodrug Approach
2.2.2. Core-Shell Lipid Nanoparticles
2.3. Lipid Micelles
2.4. Solid Lipid Nanoparticles (SLNs)
2.5. Lipid Nanodiscs
2.6. Nano-Cubosomes
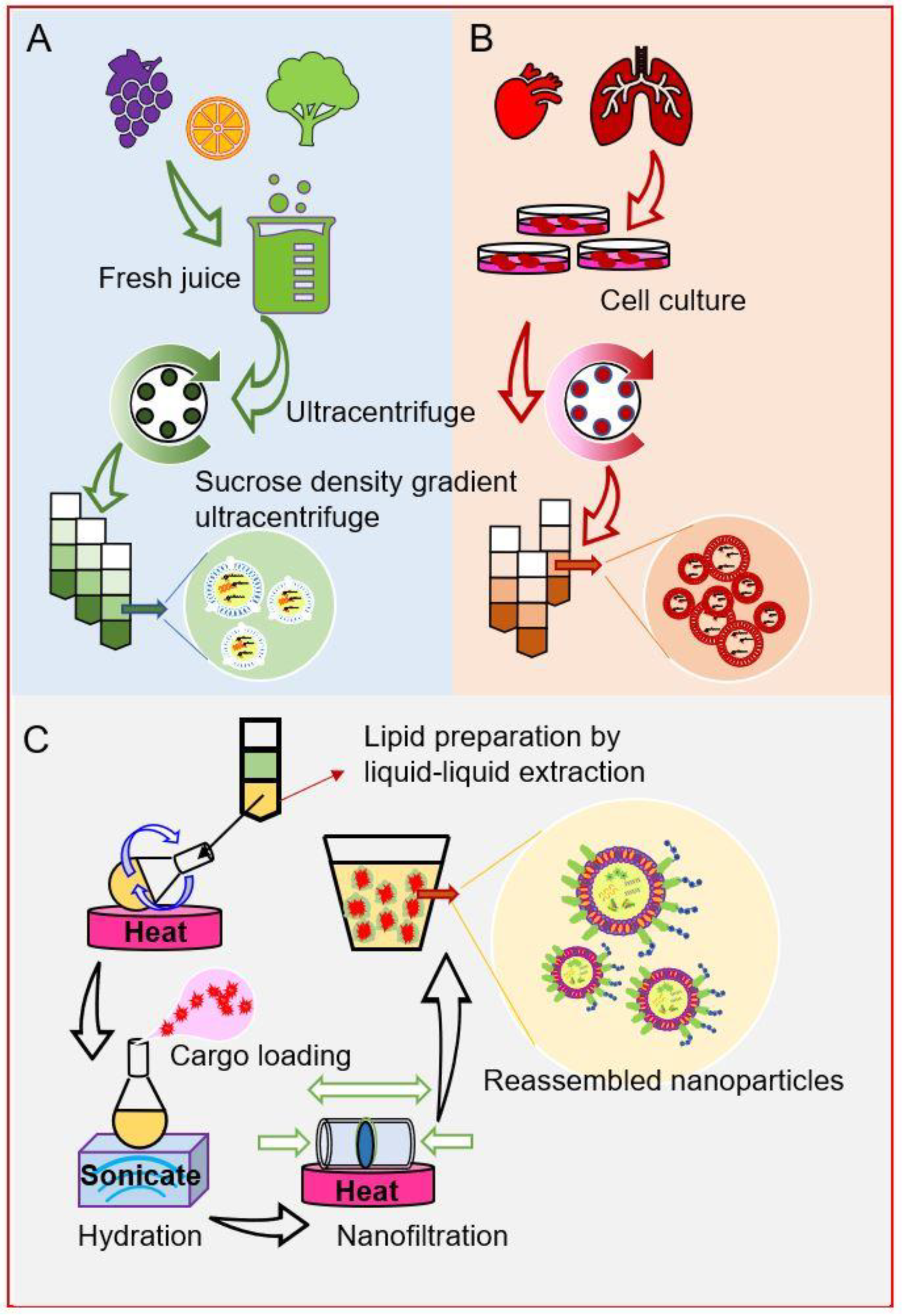
2.7. Plant-Derived Lipid Nanoparticles (PDLNPs)
2.8. Exosomes from Mammalian Cells
3. Deliverable Targets for CRC Treatment
3.1. Receptors and Transporters on CRC Cells
3.2. Target the Colon Cancer Stem Cells (CCSCs)
3.3. CRC Microenvironment
3.4. Target the CRC Metastatic Liver Cancer
3.5. Lipid Nanoparticles Mediated Gene Therapy
3.6. Multiple Targeting Strategies
4. Delivery Routes of Lipid-Based Nanoparticles in CRC Treatment
4.1. Oral Administration
4.2. Injections and IV Delivery
4.3. Other Delivery Routes
5. Conclusions and Prospects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wheat, C.L.; Clark-Snustad, K.; Devine, B.; Grembowski, D.; Thornton, T.A.; Ko, C.W. Worldwide incidence of colorectal cancer, leukemia, and lymphoma in inflammatory bowel disease: An updated systematic review and meta-analysis. Gastroenterol. Res. Pract. 2016, 2016, 1632439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favoriti, P.; Carbone, G.; Greco, M.; Pirozzi, F.; Pirozzi, R.E.; Corcione, F. Worldwide burden of colorectal cancer: A review. Updates Surg. 2016, 68, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA A Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Fedewa, S.A.; Ahnen, D.J.; Meester, R.G.S.; Barzi, A.; Jemal, A. Colorectal cancer statistics, 2017. CA A Cancer J. Clin. 2017, 67, 177–193. [Google Scholar] [CrossRef]
- Skyrud, K.D.; Myklebust, T.A.; Bray, F.; Eriksen, M.T.; de Lange, T.; Larsen, I.K.; Moller, B. How many deaths from colorectal cancer can be prevented by 2030? A scenario-based quantification of risk factor modification, screening, and treatment in Norway. Cancer Epidemiol. Biomark. 2017, 26, 1420–1426. [Google Scholar] [CrossRef] [Green Version]
- Ye, P.; Xi, Y.; Huang, Z.; Xu, P. Linking obesity with colorectal cancer: Epidemiology and mechanistic insights. Cancers 2020, 12, 1408. [Google Scholar] [CrossRef]
- Karin, M.; Bogut, A.; Hojsak, I.; Babic, E.; Volaric, M.; Bevanda, M. Nutritional status and its effect on complications in patients with colorectal cancer. Wien. Klin. Wochenschr. 2020. [Google Scholar] [CrossRef]
- Lin, T.C.; Chien, W.C.; Hu, J.M.; Tzeng, N.S.; Chung, C.H.; Pu, T.W.; Hsiao, C.W.; Chen, C.Y. Risk of colorectal cancer in patients with alcoholism: A nationwide, population-based nested case-control study. PLoS ONE 2020, 15, e0232740. [Google Scholar] [CrossRef]
- Tao, H.; O′Neil, A.; Choi, Y.; Wang, W.; Wang, J.; Wang, Y.; Jia, Y.; Chen, X. Pre- and post-diagnosis diabetes as a risk factor for all-cause and cancer-specific mortality in breast, prostate, and colorectal cancer survivors: A prospective cohort study. Front. Endocrinol. 2020, 11, 60. [Google Scholar] [CrossRef] [Green Version]
- Li, J.B.; Luo, S.; Wong, M.C.S.; Li, C.; Feng, L.F.; Peng, J.H.; Li, J.H.; Zhang, X. Longitudinal associations between BMI change and the risks of colorectal cancer incidence, cancer-relate and all-cause mortality among 81,388 older adults: BMI change and the risks of colorectal cancer incidence and mortality. BMC Cancer 2019, 19, 1082. [Google Scholar] [CrossRef]
- Ji, H.; Lu, L.; Huang, J.; Liu, Y.; Zhang, B.; Tang, H.; Sun, D.; Zhang, Y.; Shang, H.; Li, Y.; et al. IL1A polymorphisms is a risk factor for colorectal cancer in Chinese Han population: A case control study. BMC Cancer 2019, 19, 181. [Google Scholar] [CrossRef] [PubMed]
- Schoormans, D.; Husson, O.; Denollet, J.; Mols, F. Is Type D personality a risk factor for all-cause mortality? A prospective population-based study among 2625 colorectal cancer survivors from the PROFILES registry. J. Psychosom. Res. 2017, 96, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Baghad, I.; Erguibi, D.; Chehab, F.; Nadifi, S. Risk of colorectal cancer and clotting factor gene polymorphisms in Moroccan Population. Int. J. Adv. Res. 2017, 5, 1141–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.M.; MacKinlay, A.; Semira, C.; Schieber, C.; Jimeno Yepes, A.J.; Lee, B.; Wong, R.; Hettiarachchige, C.K.H.; Gunn, N.; Tie, J.; et al. Stage-based variation in the effect of primary tumor side on all stages of colorectal cancer recurrence and survival. Clin. Colorectal Cancer 2018, 17, e569–e577. [Google Scholar] [CrossRef]
- Weiser, M.R. AJCC 8th edition: Colorectal cancer. Ann. Surg. Oncol. 2018, 25, 1454–1455. [Google Scholar] [CrossRef] [Green Version]
- Tian, Q.; Liu, Y.; Zhang, Y.; Song, Z.; Yang, J.; Zhang, J.; Guo, T.; Gao, W.; Dai, F.; He, C. THBS2 is a biomarker for AJCC stages and a strong prognostic indicator in colorectal cancer. J. B.U.ON. Off. J. Balk. Union Oncol. 2018, 23, 1331–1336. [Google Scholar]
- Bennedsgaard, K.; Ventzel, L.; Themistocleous, A.C.; Bennett, D.L.; Jensen, A.B.; Jensen, A.R.; Andersen, N.T.; Jensen, T.S.; Tankisi, H.; Finnerup, N.B. Long-term symptoms of polyneuropathy in breast and colorectal cancer patients treated with and without adjuvant chemotherapy. Cancer Med. 2020, 9, 5114–5123. [Google Scholar] [CrossRef]
- Lee, C.S.; Murphy, D.J.; McMahon, C.; Nolan, B.; Cullen, G.; Mulcahy, H.; Sheahan, K.; Barnes, E.; Fennelly, D.; Ryan, E.J.; et al. Visceral adiposity is a risk factor for poor prognosis in colorectal cancer patients receiving adjuvant chemotherapy. J. Gastrointest. Cancer 2015, 46, 243–250. [Google Scholar] [CrossRef]
- He, Y.; Liu, P.; Zhang, Y.; Deng, X.; Meng, W.; Wei, M.; Yang, T.; Wang, Z.; Qiu, M. Low-dose capecitabine adjuvant chemotherapy in elderly stage II/III colorectal cancer patients (LC-ACEC): Study protocol for a randomized controlled trial. Trials 2015, 16, 238. [Google Scholar] [CrossRef] [Green Version]
- Nordlinger, B.; Rougier, P.; Arnaud, J.P.; Debois, M.; Wils, J.; Ollier, J.C.; Grobost, O.; Lasser, P.; Wals, J.; Lacourt, J.; et al. Adjuvant regional chemotherapy and systemic chemotherapy versus systemic chemotherapy alone in patients with stage II-III colorectal cancer: A multicentre randomised controlled phase III trial. Lancet Oncol. 2005, 6, 459–468. [Google Scholar] [CrossRef]
- Duran, G.; Cruz, R.; Simoes, A.R.; Barros, F.; Balboa, E.; Giraldez, J.M.; Bernardez, B.; Anido, U.; Candamio, S.; Lopez-Lopez, R.; et al. Efficacy and toxicity of adjuvant chemotherapy on colorectal cancer patients: How much influence from the genetics? J. Chemother. 2020, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Petrelli, F.; Labianca, R.; Zaniboni, A.; Lonardi, S.; Galli, F.; Rulli, E.; Rosati, G.; Corallo, S.; Ronzoni, M.; Cardellino, G.G.; et al. Assessment of duration and effects of 3 vs 6 months of adjuvant chemotherapy in high-risk stage II colorectal cancer: A subgroup analysis of the TOSCA randomized clinical trial. JAMA Oncol. 2020, 6, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Hotta, T.; Takifuji, K.; Arii, K.; Yokoyama, S.; Matsuda, K.; Higashiguchi, T.; Tominaga, T.; Oku, Y.; Yamaue, H. Clinical impact of adjuvant chemotherapy on patients with stage III colorectal cancer: L-LV/5FU chemotherapy as a modified RPMI regimen is an independent prognostic factor for survival. Anticancer Res. 2006, 26, 1425–1432. [Google Scholar] [PubMed]
- Sastre, J.; Navarro, M.; Aranda, E.; Fonseca, E.; Checa, T.; Alonso, M.C.; Gallen, M.; Camps, C.; Anton, A.; Diaz-Rubio, E. Retrospective evaluation of toxicity in three different schedules of adjuvant chemotherapy for patients with resected colorectal cancer. TTD Spanish Cooperative Group. Oncol. Rep. 1999, 6, 1421–1424. [Google Scholar] [CrossRef] [PubMed]
- Mizumoto, Y.; Yokoyama, S.; Matsuda, K.; Iwamoto, H.; Mitani, Y.; Tamura, K.; Nakamura, Y.; Murakami, D.; Oka, M.; Kobayashi, Y.; et al. Modulation of capecitabine administration to improve continuity of adjuvant chemotherapy for patients with colorectal cancer: A phase II study. Mol. Clin. Oncol. 2020, 12, 126–133. [Google Scholar] [CrossRef] [Green Version]
- Iveson, T.; Boyd, K.A.; Kerr, R.S.; Robles-Zurita, J.; Saunders, M.P.; Briggs, A.H.; Cassidy, J.; Hollander, N.H.; Tabernero, J.; Haydon, A.; et al. 3-month versus 6-month adjuvant chemotherapy for patients with high-risk stage II and III colorectal cancer: 3-year follow-up of the SCOT non-inferiority RCT. Health Technol. Assess. 2019, 23, 1–88. [Google Scholar] [CrossRef] [Green Version]
- Sgouros, J.; Aravantinos, G.; Kouvatseas, G.; Rapti, A.; Stamoulis, G.; Bisvikis, A.; Res, H.; Samantas, E. Impact of dose reductions, delays between chemotherapy cycles, and/or shorter courses of adjuvant chemotherapy in stage II and III colorectal cancer patients: A single-center retrospective study. J. Gastrointest. Cancer 2015, 46, 343–349. [Google Scholar] [CrossRef]
- Schuurhuizen, C.S.; Verheul, H.M.; Braamse, A.M.; Buffart, L.M.; Bloemendal, H.J.; Dekker, J.; Konings, I.R. The predictive value of cumulative toxicity for quality of life in patients with metastatic colorectal cancer during first-line palliative chemotherapy. Cancer Manag. Res. 2018, 10, 3015–3021. [Google Scholar] [CrossRef] [Green Version]
- Madi, A.; Fisher, D.; Maughan, T.S.; Colley, J.P.; Meade, A.M.; Maynard, J.; Humphreys, V.; Wasan, H.; Adams, R.A.; Idziaszczyk, S.; et al. Pharmacogenetic analyses of 2183 patients with advanced colorectal cancer; potential role for common dihydropyrimidine dehydrogenase variants in toxicity to chemotherapy. Eur. J. Cancer 2018, 102, 31–39. [Google Scholar] [CrossRef]
- Lund, C.M.; Nielsen, D.; Dehlendorff, C.; Christiansen, A.B.; Ronholt, F.; Johansen, J.S.; Vistisen, K.K. Efficacy and toxicity of adjuvant chemotherapy in elderly patients with colorectal cancer: The ACCORE study. ESMO Open 2016, 1, e000087. [Google Scholar] [CrossRef] [Green Version]
- Sisic, I.; Pojskic, B.; Mekic Abazovic, A.; Kovcin, V. Therapeutic efficacy and toxicity of bolus application of chemotherapy protocol in the treatment of metastatic colorectal cancer. Med. Glas. 2015, 12, 206–211. [Google Scholar] [CrossRef]
- Xiao, B.; Viennois, E.; Chen, Q.; Wang, L.; Han, M.K.; Zhang, Y.; Zhang, Z.; Kang, Y.; Wan, Y.; Merlin, D. Silencing of intestinal glycoprotein CD98 by orally targeted nanoparticles enhances chemosensitization of colon cancer. ACS Nano 2018, 12, 5253–5265. [Google Scholar] [CrossRef]
- Alibolandi, M.; Rezvani, R.; Farzad, S.A.; Taghdisi, S.M.; Abnous, K.; Ramezani, M. Tetrac-conjugated polymersomes for integrin-targeted delivery of camptothecin to colon adenocarcinoma in vitro and in vivo. Int. J. Pharm. 2017, 532, 581–594. [Google Scholar] [CrossRef] [PubMed]
- Handali, S.; Moghimipour, E.; Rezaei, M.; Ramezani, Z.; Kouchak, M.; Amini, M.; Angali, K.A.; Saremy, S.; Dorkoosh, F.A. A novel 5-Fluorouracil targeted delivery to colon cancer using folic acid conjugated liposomes. Biomed. Pharmacother. 2018, 108, 1259–1273. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Rao, Y. Current status of nanoscale drug delivery systems for colorectal cancer liver metastasis. Biomed. Pharmacother. 2019, 114, 108764. [Google Scholar] [CrossRef] [PubMed]
- Rothbarth, J.; van de Velde, C.J. Treatment of liver metastases of colorectal cancer. Ann. Oncol. 2005, 16 (Suppl. 2), ii144–ii149. [Google Scholar] [CrossRef]
- Marquez, J.; Fernandez-Pineiro, I.; Arauzo-Bravo, M.J.; Poschmann, G.; Stuhler, K.; Khatib, A.M.; Sanchez, A.; Unda, F.; Ibarretxe, G.; Bernales, I.; et al. Targeting liver sinusoidal endothelial cells with miR-20a-loaded nanoparticles reduces murine colon cancer metastasis to the liver. Int. J. Cancer 2018, 143, 709–719. [Google Scholar] [CrossRef]
- Teng, Y.; Mu, J.; Hu, X.; Samykutty, A.; Zhuang, X.; Deng, Z.; Zhang, L.; Cao, P.; Yan, J.; Miller, D.; et al. Grapefruit-derived nanovectors deliver miR-18a for treatment of liver metastasis of colon cancer by induction of M1 macrophages. Oncotarget 2016, 7, 25683–25697. [Google Scholar] [CrossRef] [Green Version]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the clinic: An update. Bioeng. Transl. Med. 2019, 4, e10143. [Google Scholar] [CrossRef] [Green Version]
- Bangham, A.D.; Standish, M.M.; Watkins, J.C. Diffusion of univalent ions across the lamellae of swollen phospholipids. J. Mol. Biol. 1965, 13, 238–252. [Google Scholar] [CrossRef]
- Dominska, M.; Blanchard, G.J. Constituent-dependent liposome structure and organization. Langmuir ACS J. Surf. Colloids 2010, 26, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Jaafar-Maalej, C.; Diab, R.; Andrieu, V.; Elaissari, A.; Fessi, H. Ethanol injection method for hydrophilic and lipophilic drug-loaded liposome preparation. J. Liposome Res. 2010, 20, 228–243. [Google Scholar] [CrossRef] [PubMed]
- Petty, H.R.; McConnell, H.M. Cytochemical study of liposome and lipid vesicle phagocytosis. Biochim. Et Biophys. Acta 1983, 735, 77–85. [Google Scholar] [CrossRef]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal formulations in clinical use: An updated review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Huang, L. Liposomal nanostructures for drug delivery in gastrointestinal cancers. J. Pharmacol. Exp. Ther. 2019, 370, 647–656. [Google Scholar] [CrossRef]
- Huang, J.R.; Lee, M.H.; Li, W.S.; Wu, H.C. Liposomal irinotecan for treatment of colorectal cancer in a preclinical model. Cancers 2019, 11, 281. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Feng, Q.; Dou, Z.; Yuan, W.; Sui, C.; Zhang, X.; Xia, G.; Sun, H.; Ma, J. Local targeted therapy of liver metastasis from colon cancer by galactosylated liposome encapsulated with doxorubicin. PLoS ONE 2013, 8, e73860. [Google Scholar] [CrossRef] [Green Version]
- Shariat, S.; Badiee, A.; Amir Jalali, S.; Mansourian, M.; Alireza Mortazavi, S.; Reza Jaafari, M. Preparation and characterization of different liposomal formulations containing P5 HER2/neu-derived peptide and evaluation of their immunological responses and antitumor effects. Iran. J. Basic Med. Sci. 2015, 18, 506–513. [Google Scholar]
- Ostergaard, J.; Moeller, E.H. Ghrelin-liposome interactions: Characterization of liposomal formulations of an acylated 28-amino acid peptide using CE. Electrophoresis 2010, 31, 339–345. [Google Scholar] [CrossRef]
- Wang, T.; Liu, Z.; Zhang, Z.; Tang, S.; Yue, M.; Feng, S.; Hu, M.; Xuan, L.; Chen, Y. Evaluation of antitumor activity of survivin short interfering RNA delivered by lipid nanoparticles in colon cancer in vitro and in vivo. Oncol. Lett. 2017, 14, 2001–2008. [Google Scholar] [CrossRef] [Green Version]
- Guan, J.; Sun, J.; Sun, F.; Lou, B.; Zhang, D.; Mashayekhi, V.; Sadeghi, N.; Storm, G.; Mastrobattista, E.; He, Z. Hypoxia-induced tumor cell resistance is overcome by synergistic GAPDH-siRNA and chemotherapy co-delivered by long-circulating and cationic-interior liposomes. Nanoscale 2017, 9, 9190–9201. [Google Scholar] [CrossRef] [PubMed]
- Kwong, B.; Gai, S.A.; Elkhader, J.; Wittrup, K.D.; Irvine, D.J. Localized immunotherapy via liposome-anchored Anti-CD137 + IL-2 prevents lethal toxicity and elicits local and systemic antitumor immunity. Cancer Res. 2013, 73, 1547–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef] [Green Version]
- Allen, T.M.; Hansen, C. Pharmacokinetics of stealth versus conventional liposomes: Effect of dose. Biochim. Biophys. Acta 1991, 1068, 133–141. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, Y.; Li, X.; Liang, X.; Luo, X. The influence of different long-circulating materials on the pharmacokinetics of liposomal vincristine sulfate. Int. J. Nanomed. 2016, 11, 4187–4197. [Google Scholar] [CrossRef] [Green Version]
- Reddy, K.R. Controlled-release, pegylation, liposomal formulations: New mechanisms in the delivery of injectable drugs. Ann. Pharmacother. 2000, 34, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Kuai, R.; Yuan, W.; Drake, L.; Moon, J.J.; Schwendeman, A. Effect of size and pegylation of liposomes and peptide-based synthetic lipoproteins on tumor targeting. Nanomedicine 2017, 13, 1869–1878. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.L.; Majzoub, R.N.; Shirazi, R.S.; Ewert, K.K.; Chen, Y.J.; Liang, K.S.; Safinya, C.R. Endosomal escape and transfection efficiency of PEGylated cationic liposome-DNA complexes prepared with an acid-labile PEG-lipid. Biomaterials 2012, 33, 4928–4935. [Google Scholar] [CrossRef] [Green Version]
- Gabizon, A.A.; Patil, Y.; La-Beck, N.M. New insights and evolving role of pegylated liposomal doxorubicin in cancer therapy. Drug Resist. Updates 2016, 29, 90–106. [Google Scholar] [CrossRef]
- Smith, S.A.; Selby, L.I.; Johnston, A.P.R.; Such, G.K. The endosomal escape of nanoparticles: Toward more efficient cellular delivery. Bioconjugate Chem. 2019, 30, 263–272. [Google Scholar] [CrossRef]
- Zhang, D.; Xu, H.; Hu, M.N.; Deng, Y.H. “PEG dilemma” for liposomes and its solving approaches. Yao Xue Xue Bao Acta Pharm. Sin. 2015, 50, 252–260. [Google Scholar]
- Akiyoshi, K.; Itaya, A.; Nomura, S.; Ono, N.; Yoshikawa, K. Induction of neuron-like tubes and liposome networks by cooperative effect of gangliosides and phospholipids. FEBS Lett. 2003, 534, 33–38. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Liu, D.; Zhu, L.; Gan, Q.; Le, X. Temperature-dependent structure stability and in vitro release of chitosan-coated curcumin liposome. Food Res. Int. 2015, 74, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Dutta, J.; Dutta, P.K.; Koh, J. A systematic study on chitosan-liposome based systems for biomedical applications. Int. J. Biol. Macromol. 2020, 160, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Alomrani, A.; Badran, M.; Harisa, G.I.; ALshehry, M.; Alhariri, M.; Alshamsan, A.; Alkholief, M. The use of chitosan-coated flexible liposomes as a remarkable carrier to enhance the antitumor efficacy of 5-fluorouracil against colorectal cancer. Saudi Pharm. J. 2019, 27, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Naeem, M.; Awan, U.A.; Subhan, F.; Cao, J.; Hlaing, S.P.; Lee, J.; Im, E.; Jung, Y.; Yoo, J.W. Advances in colon-targeted nano-drug delivery systems: Challenges and solutions. Arch. Pharm. Res. 2020, 43, 153–169. [Google Scholar] [CrossRef]
- Zappavigna, S.; Abate, M.; Cossu, A.M.; Lusa, S.; Campani, V.; Scotti, L.; Luce, A.; Yousif, A.M.; Merlino, F.; Grieco, P.; et al. Urotensin-II-targeted liposomes as a new drug delivery system towards prostate and colon cancer cells. J. Oncol. 2019, 2019, 9293560. [Google Scholar] [CrossRef]
- Liang, X.; Luo, M.; Wei, X.W.; Ma, C.C.; Yang, Y.H.; Shao, B.; Liu, Y.T.; Liu, T.; Ren, J.; Liu, L.; et al. A folate receptor-targeted lipoplex delivering interleukin-15 gene for colon cancer immunotherapy. Oncotarget 2016, 7, 52207–52217. [Google Scholar] [CrossRef] [Green Version]
- Moghimipour, E.; Rezaei, M.; Ramezani, Z.; Kouchak, M.; Amini, M.; Angali, K.A.; Dorkoosh, F.A.; Handali, S. Folic acid-modified liposomal drug delivery strategy for tumor targeting of 5-fluorouracil. Eur. J. Pharm. Sci. 2018, 114, 166–174. [Google Scholar] [CrossRef]
- Juang, V.; Chang, C.H.; Wang, C.S.; Wang, H.E.; Lo, Y.L. pH-Responsive PEG-Shedding and targeting peptide-modified nanoparticles for dual-delivery of irinotecan and microRNA to enhance tumor-specific therapy. Small 2019, 15, e1903296. [Google Scholar] [CrossRef]
- Moghimipour, E.; Rezaei, M.; Ramezani, Z.; Kouchak, M.; Amini, M.; Angali, K.A.; Dorkoosh, F.A.; Handali, S. Transferrin targeted liposomal 5-fluorouracil induced apoptosis via mitochondria signaling pathway in cancer cells. Life Sci. 2018, 194, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Needham, D.; Anyarambhatla, G.; Kong, G.; Dewhirst, M.W. A new temperature-sensitive liposome for use with mild hyperthermia: Characterization and testing in a human tumor xenograft model. Cancer Res. 2000, 60, 1197–1201. [Google Scholar] [PubMed]
- Wang, L.; Yang, C.Q.; Wang, J. Assemble of magnetic nanoparticles into the structure of cisplatin liposome. Yao Xue Xue Bao Acta Pharm. Sin. 2011, 46, 592–598. [Google Scholar]
- Zhang, D.; Shah, P.K.; Culver, H.R.; David, S.N.; Stansbury, J.W.; Yin, X.; Bowman, C.N. Photo-responsive liposomes composed of spiropyran-containing triazole-phosphatidylcholine: Investigation of merocyanine-stacking effects on liposome-fiber assembly-transition. Soft Matter 2019, 15, 3740–3750. [Google Scholar] [CrossRef]
- Mansoori, B.; Mohammadi, A.; Abedi-Gaballu, F.; Abbaspour, S.; Ghasabi, M.; Yekta, R.; Shirjang, S.; Dehghan, G.; Hamblin, M.R.; Baradaran, B. Hyaluronic acid-decorated liposomal nanoparticles for targeted delivery of 5-fluorouracil into HT-29 colorectal cancer cells. J. Cell. Physiol. 2020. [Google Scholar] [CrossRef]
- Banerjee, S.; Sen, K.; Pal, T.K.; Guha, S.K. Poly(styrene-co-maleic acid)-based pH-sensitive liposomes mediate cytosolic delivery of drugs for enhanced cancer chemotherapy. Int. J. Pharm. 2012, 436, 786–797. [Google Scholar] [CrossRef]
- Banerjee, S.; Pal, T.K.; Guha, S.K. Probing molecular interactions of poly (styrene-co-maleic acid) with lipid matrix models to interpret the therapeutic potential of the co-polymer. Biochim. Biophys. Acta 2012, 1818, 537–550. [Google Scholar] [CrossRef] [Green Version]
- May, J.P.; Li, S.D. Hyperthermia-induced drug targeting. Expert Opin. Drug Deliv. 2013, 10, 511–527. [Google Scholar] [CrossRef]
- Bi, H.; Xue, J.; Jiang, H.; Gao, S.; Yang, D.; Fang, Y.; Shi, K. Current developments in drug delivery with thermosensitive liposomes. Asian J. Pharm. Sci. 2019, 14, 365–379. [Google Scholar] [CrossRef]
- Lyon, P.C.; Gray, M.D.; Mannaris, C.; Folkes, L.K.; Stratford, M.; Campo, L.; Chung, D.Y.F.; Scott, S.; Anderson, M.; Goldin, R.; et al. Safety and feasibility of ultrasound-triggered targeted drug delivery of doxorubicin from thermosensitive liposomes in liver tumours (TARDOX): A single-centre, open-label, phase 1 trial. Lancet. Oncol. 2018, 19, 1027–1039. [Google Scholar] [CrossRef] [Green Version]
- Gholami, Y.H.; Engel, A. Theranostic nanoplatforms for treatment and diagnosis of rectal and colon cancer: A brief review. Miniinvasive Surg. 2018, 2018. [Google Scholar] [CrossRef]
- Garcia, B.B.M.; Mertins, O.; Silva, E.R.D.; Mathews, P.D.; Han, S.W. Arginine-modified chitosan complexed with liposome systems for plasmid DNA delivery. Colloids Surf. B Biointerfaces 2020, 193, 111131. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhang, S.; Cui, S.; Yang, B.; Zhao, Y.; Chen, H.; Hao, X.; Shen, Q.; Zhou, J. Chitosan enhanced gene delivery of cationic liposome via non-covalent conjugation. Biotechnol. Lett. 2012, 34, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Gao, X.; Men, K.; Wang, B.; Zhang, S.; Qiu, J.; Huang, M.; Gou, M.; Huang, N.; Qian, Z.; et al. Gene therapy for C-26 colon cancer using heparin-polyethyleneimine nanoparticle-mediated survivin T34A. Int. J. Nanomed. 2011, 6, 2419–2427. [Google Scholar] [CrossRef] [Green Version]
- Hashemi, M.; Omidi, M.; Muralidharan, B.; Tayebi, L.; Herpin, M.J.; Mohagheghi, M.A.; Mohammadi, J.; Smyth, H.D.C.; Milner, T.E. Layer-by-layer assembly of graphene oxide on thermosensitive liposomes for photo-chemotherapy. Acta Biomater. 2018, 65, 376–392. [Google Scholar] [CrossRef]
- Caracciolo, G.; Amenitsch, H. Cationic liposome/DNA complexes: From structure to interactions with cellular membranes. Eur. Biophys. J. EBJ 2012, 41, 815–829. [Google Scholar] [CrossRef]
- Strieth, S.; Eichhorn, M.E.; Werner, A.; Sauer, B.; Teifel, M.; Michaelis, U.; Berghaus, A.; Dellian, M. Paclitaxel encapsulated in cationic liposomes increases tumor microvessel leakiness and improves therapeutic efficacy in combination with Cisplatin. Clin. Cancer Res. 2008, 14, 4603–4611. [Google Scholar] [CrossRef] [Green Version]
- Strieth, S.; Nussbaum, C.F.; Eichhorn, M.E.; Fuhrmann, M.; Teifel, M.; Michaelis, U.; Berghaus, A.; Dellian, M. Tumor-selective vessel occlusions by platelets after vascular targeting chemotherapy using paclitaxel encapsulated in cationic liposomes. Int. J. Cancer 2008, 122, 452–460. [Google Scholar] [CrossRef]
- Fasol, U.; Frost, A.; Buchert, M.; Arends, J.; Fiedler, U.; Scharr, D.; Scheuenpflug, J.; Mross, K. Vascular and pharmacokinetic effects of EndoTAG-1 in patients with advanced cancer and liver metastasis. Ann. Oncol. 2012, 23, 1030–1036. [Google Scholar] [CrossRef]
- Templeton, N.S. Cationic liposome-mediated gene delivery in vivo. Biosci. Rep. 2002, 22, 283–295. [Google Scholar] [CrossRef]
- Lu, Y.; Zhong, L.; Jiang, Z.; Pan, H.; Zhang, Y.; Zhu, G.; Bai, L.; Tong, R.; Shi, J.; Duan, X. Cationic micelle-based siRNA delivery for efficient colon cancer gene therapy. Nanoscale Res. Lett. 2019, 14, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, K.L.; Ou-Yang, F.; Yen, S.H.; Shih, H.L.; Lan, K.H. Cationic liposome coupled endostatin gene for treatment of peritoneal colon cancer. Clin. Exp. Metastasis 2010, 27, 307–318. [Google Scholar] [CrossRef]
- Ando, H.; Abu Lila, A.S.; Fukushima, M.; Matsuoka, R.; Shimizu, T.; Okuhira, K.; Ishima, Y.; Huang, C.L.; Wada, H.; Ishida, T. A simplified method for manufacturing RNAi therapeutics for local administration. Int. J. Pharm. 2019, 564, 256–262. [Google Scholar] [CrossRef]
- Qian, Y.; Liang, X.; Yang, J.; Zhao, C.; Nie, W.; Liu, L.; Yi, T.; Jiang, Y.; Geng, J.; Zhao, X.; et al. Hyaluronan Reduces Cationic Liposome-Induced Toxicity and Enhances the Antitumor Effect of Targeted Gene Delivery in Mice. ACS Appl. Mater. Interfaces 2018, 10, 32006–32016. [Google Scholar] [CrossRef] [PubMed]
- Schreier, H.; Gagne, L.; Bock, T.; Erdos, G.W.; Druzgala, P.; Conary, J.T.; Muller, B.W. Physicochemical properties and in vitro toxicity of cationic liposome cDNA complexes. Pharm. Acta Helv. 1997, 72, 215–223. [Google Scholar] [CrossRef]
- Liu, L.; Ye, Q.; Lu, M.; Chen, S.T.; Tseng, H.W.; Lo, Y.C.; Ho, C. A new approach to deliver anti-cancer nanodrugs with reduced off-target toxicities and improved efficiency by temporarily blunting the reticuloendothelial system with intralipid. Sci. Rep. 2017, 7, 16106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semple, S.C.; Akinc, A.; Chen, J.; Sandhu, A.P.; Mui, B.L.; Cho, C.K.; Sah, D.W.; Stebbing, D.; Crosley, E.J.; Yaworski, E.; et al. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 2010, 28, 172–176. [Google Scholar] [CrossRef]
- Judge, A.D.; Robbins, M.; Tavakoli, I.; Levi, J.; Hu, L.; Fronda, A.; Ambegia, E.; McClintock, K.; MacLachlan, I. Confirming the RNAi-mediated mechanism of action of siRNA-based cancer therapeutics in mice. J. Clin. Investig. 2009, 119, 661–673. [Google Scholar] [CrossRef] [Green Version]
- Xing, J.; Zhang, X.; Wang, Z.; Zhang, H.; Chen, P.; Zhou, G.; Sun, C.; Gu, N.; Ji, M. Novel lipophilic SN38 prodrug forming stable liposomes for colorectal carcinoma therapy. Int. J. Nanomed. 2019, 14, 5201–5213. [Google Scholar] [CrossRef] [Green Version]
- Arouri, A.; Mouritsen, O.G. Phospholipase A(2)-susceptible liposomes of anticancer double lipid-prodrugs. Eur. J. Pharm. Sci. 2012, 45, 408–420. [Google Scholar] [CrossRef]
- Campani, V.; Giarra, S.; De Rosa, G. Lipid-based core-shell nanoparticles: Evolution and potentialities in drug delivery. Open Nano 2018, 3, 5–17. [Google Scholar] [CrossRef]
- Gao, W.; Hu, C.M.; Fang, R.H.; Zhang, L. Liposome-like nanostructures for drug delivery. J. Mater. Chem. B 2013, 1. [Google Scholar] [CrossRef] [PubMed]
- Sharifabad, M.E.; Mercer, T.; Sen, T. Drug-loaded liposome-capped mesoporous core-shell magnetic nanoparticles for cellular toxicity study. Nanomedicine 2016, 11, 2757–2767. [Google Scholar] [CrossRef]
- Wang, H.; Zhao, P.; Su, W.; Wang, S.; Liao, Z.; Niu, R.; Chang, J. PLGA/polymeric liposome for targeted drug and gene co-delivery. Biomaterials 2010, 31, 8741–8748. [Google Scholar] [CrossRef]
- Zhu, D.; Wang, Z.; Zong, S.; Zhang, Y.; Chen, C.; Zhang, R.; Yun, B.; Cui, Y. Investigating the intracellular behaviors of liposomal nanohybrids via SERS: Insights into the influence of metal nanoparticles. Theranostics 2018, 8, 941–954. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Yang, C.; Zhang, L.; Hu, T.; Sun, D.; Cao, H.; Yang, F.; Guo, G.; Gong, C.; Zhang, X.; et al. Killing colon cancer cells through PCD pathways by a novel hyaluronic acid-modified shell-core nanoparticle loaded with RIP3 in combination with chloroquine. Biomaterials 2017, 124, 195–210. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Wang, P.; Men, K.; Gao, X.; Huang, M.; Gou, M.; Chen, L.; Qian, Z.; Wei, Y. Treating colon cancer with a suicide gene delivered by self-assembled cationic MPEG-PCL micelles. Nanoscale 2012, 4, 2400–2407. [Google Scholar] [CrossRef]
- Xu, G.; Shi, H.; Ren, L.; Gou, H.; Gong, D.; Gao, X.; Huang, N. Enhancing the anti-colon cancer activity of quercetin by self-assembled micelles. Int. J. Nanomed. 2015, 10, 2051–2063. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; He, Y.; Ji, J.; Zheng, S.; Cheng, Y. Tumor targeted curcumin delivery by folate-modified MPEG-PCL self-assembly micelles for colorectal cancer therapy. Int. J. Nanomed. 2020, 15, 1239–1252. [Google Scholar] [CrossRef] [Green Version]
- Mishra, V.; Bansal, K.K.; Verma, A.; Yadav, N.; Thakur, S.; Sudhakar, K.; Rosenholm, J.M. Solid lipid nanoparticles: Emerging colloidal nano drug delivery systems. Pharmaceutics 2018, 10, 191. [Google Scholar] [CrossRef] [Green Version]
- Madureira, A.R.; Nunes, S.; Campos, D.A.; Fernandes, J.C.; Marques, C.; Zuzarte, M.; Gullon, B.; Rodriguez-Alcala, L.M.; Calhau, C.; Sarmento, B.; et al. Safety profile of solid lipid nanoparticles loaded with rosmarinic acid for oral use: In vitro and animal approaches. Int. J. Nanomed. 2016, 11, 3621–3640. [Google Scholar] [CrossRef] [Green Version]
- Doktorovova, S.; Kovacevic, A.B.; Garcia, M.L.; Souto, E.B. Preclinical safety of solid lipid nanoparticles and nanostructured lipid carriers: Current evidence from in vitro and in vivo evaluation. Eur. J. Pharm. Biopharm. 2016, 108, 235–252. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.Y.; Liu, T.I.; Yu, T.W.; Kv, R.; Chiang, W.H.; Tsai, Y.C.; Chen, H.H.; Lin, S.C.; Chiu, H.C. Hierarchically targetable polysaccharide-coated solid lipid nanoparticles as an oral chemo/thermotherapy delivery system for local treatment of colon cancer. Biomaterials 2019, 197, 86–100. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, Z.; Yang, Y.; Wang, Z.; Yang, Z.; Li, B.; Xie, X.; Song, J.; Zhang, H.; Li, Y.; et al. Thermo-sensitive liposome co-loaded of vincristine and doxorubicin based on their similar physicochemical properties had synergism on tumor treatment. Pharm. Res. 2016, 33, 1881–1898. [Google Scholar] [CrossRef]
- Kim, K.S.; Youn, Y.S.; Bae, Y.H. Immune-triggered cancer treatment by intestinal lymphatic delivery of docetaxel-loaded nanoparticle. J. Control. Release 2019, 311–312, 85–95. [Google Scholar] [CrossRef]
- Lim, S.J.; Kim, C.K. Formulation parameters determining the physicochemical characteristics of solid lipid nanoparticles loaded with all-trans retinoic acid. Int. J. Pharm. 2002, 243, 135–146. [Google Scholar] [CrossRef]
- Wong, H.L.; Bendayan, R.; Rauth, A.M.; Wu, X.Y. Development of solid lipid nanoparticles containing ionically complexed chemotherapeutic drugs and chemosensitizers. J. Pharm. Sci. 2004, 93, 1993–2008. [Google Scholar] [CrossRef]
- Li, Y.; Taulier, N.; Rauth, A.M.; Wu, X.Y. Screening of lipid carriers and characterization of drug-polymer-lipid interactions for the rational design of polymer-lipid hybrid nanoparticles (PLN). Pharm. Res. 2006, 23, 1877–1887. [Google Scholar] [CrossRef]
- Kuai, R.; Ochyl, L.J.; Bahjat, K.S.; Schwendeman, A.; Moon, J.J. Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat. Mater. 2017, 16, 489–496. [Google Scholar] [CrossRef]
- Kuai, R.; Yuan, W.; Son, S.; Nam, J.; Xu, Y.; Fan, Y.; Schwendeman, A.; Moon, J.J. Elimination of established tumors with nanodisc-based combination chemoimmunotherapy. Sci. Adv. 2017, 4, 13. [Google Scholar] [CrossRef]
- Barauskas, J.; Johnsson, M.; Joabsson, F.; Tiberg, F. Cubic phase nanoparticles (Cubosome): Principles for controlling size, structure, and stability. Langmuir ACS J. Surf. Colloids 2005, 21, 2569–2577. [Google Scholar] [CrossRef] [PubMed]
- Angelov, B.; Angelova, A.; Garamus, V.M.; Drechsler, M.; Willumeit, R.; Mutafchieva, R.; Stepanek, P.; Lesieur, S. Earliest stage of the tetrahedral nanochannel formation in cubosome particles from unilamellar nanovesicles. Langmuir ACS J. Surf. Colloids 2012, 28, 16647–16655. [Google Scholar] [CrossRef] [PubMed]
- Gozdz, W.T. Cubosome topologies at various particle sizes and crystallographic symmetries. Langmuir ACS J. Surf. Colloids 2015, 31, 13321–13326. [Google Scholar] [CrossRef] [PubMed]
- Uyama, M.; Handa, T.; Nakano, M. Novel cubosome system resistant to lipid removal by serum albumin. Chem. Pharm. Bull. 2019, 67, 1099–1103. [Google Scholar] [CrossRef]
- Saber, M.M.; Al-Mahallawi, A.M.; Nassar, N.N.; Stork, B.; Shouman, S.A. Targeting colorectal cancer cell metabolism through development of cisplatin and metformin nano-cubosomes. BMC Cancer 2018, 18, 822. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Zhang, M.; Merlin, D. Advances in plant-derived edible nanoparticle-based lipid nano-drug delivery systems as therapeutic nanomedicines. J. Mater. Chem. B 2018, 6, 1312–1321. [Google Scholar] [CrossRef]
- Mu, J.; Zhuang, X.; Wang, Q.; Jiang, H.; Deng, Z.B.; Wang, B.; Zhang, L.; Kakar, S.; Jun, Y.; Miller, D.; et al. Interspecies communication between plant and mouse gut host cells through edible plant derived exosome-like nanoparticles. Mol. Nutr. Food Res. 2014, 58, 1561–1573. [Google Scholar] [CrossRef]
- Teng, Y.; Ren, Y.; Sayed, M.; Hu, X.; Lei, C.; Kumar, A.; Hutchins, E.; Mu, J.; Deng, Z.; Luo, C.; et al. Plant-Derived Exosomal MicroRNAs Shape the Gut Microbiota. Cell Host Microbe 2018, 24, 637–652.e8. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Xiao, B.; Wang, H.; Han, M.K.; Zhang, Z.; Viennois, E.; Xu, C.; Merlin, D. Edible Ginger-derived Nano-lipids Loaded with Doxorubicin as a Novel Drug-delivery Approach for Colon Cancer Therapy. Mol. Ther. 2016, 24, 1783–1796. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Viennois, E.; Prasad, M.; Zhang, Y.; Wang, L.; Zhang, Z.; Han, M.K.; Xiao, B.; Xu, C.; Srinivasan, S.; et al. Edible ginger-derived nanoparticles: A novel therapeutic approach for the prevention and treatment of inflammatory bowel disease and colitis-associated cancer. Biomaterials 2016, 101, 321–340. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Zhuang, X.; Mu, J.; Deng, Z.B.; Jiang, H.; Zhang, L.; Xiang, X.; Wang, B.; Yan, J.; Miller, D.; et al. Delivery of therapeutic agents by nanoparticles made of grapefruit-derived lipids. Nat. Commun. 2013, 4, 1867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, X.; Deng, Z.B.; Mu, J.; Zhang, L.; Yan, J.; Miller, D.; Feng, W.; McClain, C.J.; Zhang, H.G. Ginger-derived nanoparticles protect against alcohol-induced liver damage. J. Extracell. Vesicles 2015, 4, 28713. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.; Yang, C.; Viennois, E.; Zhang, M.; Merlin, D. Isolation, purification, and characterization of Ginger-derived Nanoparticles (GDNPs) from ginger, rhizome of zingiber officinale. Bioprotoc 2019, 9. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, M.; Lama, S.; Wang, L.; Merlin, D. Natural-lipid nanoparticle-based therapeutic approach to deliver 6-shogaol and its metabolites M2 and M13 to the colon to treat ulcerative colitis. J. Control. Release 2020, 323, 293–310. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhang, M.; Sung, J.; Wang, L.; Jung, Y.; Merlin, D. Isolation and characterization of exosomes from mouse feces. Bioprotoc 2020, 10. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, M.; Sung, J.; Wang, L.; Jung, Y.; Merlin, D. Autologous exosome transfer: A new personalized treatment concept to prevent colitis in a murine model. J. Crohns Colitis 2019. [Google Scholar] [CrossRef]
- Liang, G.; Zhu, Y.; Ali, D.J.; Tian, T.; Xu, H.; Si, K.; Sun, B.; Chen, B.; Xiao, Z. Engineered exosomes for targeted co-delivery of miR-21 inhibitor and chemotherapeutics to reverse drug resistance in colon cancer. J. Nanobiotechnology 2020, 18, 10. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Amin, M.; Mansourian, M.; Koning, G.A.; Badiee, A.; Jaafari, M.R.; Ten Hagen, T.L.M. Development of a novel cyclic RGD peptide for multiple targeting approaches of liposomes to tumor region. J. Control. Release 2015, 220, 308–315. [Google Scholar] [CrossRef]
- Liu, K.C.; Arivajiagane, A.; Wu, S.J.; Tzou, S.C.; Chen, C.Y.; Wang, Y.M. Development of a novel thermal-sensitive multifunctional liposome with antibody conjugation to target EGFR-expressing tumors. Nanomedicine 2019, 15, 285–294. [Google Scholar] [CrossRef]
- Farran, B.; Montenegro, R.C.; Kasa, P.; Pavitra, E.; Huh, Y.S.; Han, Y.K.; Kamal, M.A.; Nagaraju, G.P.; Rama Raju, G.S. Folate-conjugated nanovehicles: Strategies for cancer therapy. Mater. Sci. Eng. C Mater. Biol. Appl. 2020, 107, 110341. [Google Scholar] [CrossRef]
- Handali, S.; Moghimipour, E.; Kouchak, M.; Ramezani, Z.; Amini, M.; Angali, K.A.; Saremy, S.; Dorkoosh, F.A.; Rezaei, M. New folate receptor targeted nano liposomes for delivery of 5-fluorouracil to cancer cells: Strong implication for enhanced potency and safety. Life Sci. 2019, 227, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Merlin, D. Nanoparticle-mediated drug delivery systems for the treatment of IBD: Current perspectives. Int. J. Nanomed. 2019, 14, 8875–8889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, P.K.; Islam, F.; Lam, A.K. The roles of cancer stem cells and therapy resistance in colorectal carcinoma. Cells 2020, 9, 1392. [Google Scholar] [CrossRef] [PubMed]
- Holen, K.D. Target practice: Figuring out which, when, and why to use systemic therapies for metastatic colon cancer. Cancer Investig. 2006, 24, 98–105. [Google Scholar] [CrossRef]
- Wang, K.; Song, K.; Ma, Z.; Yao, Y.; Liu, C.; Yang, J.; Xiao, H.; Zhang, J.; Zhang, Y.; Zhao, W. Identification of EMT-related high-risk stage II colorectal cancer and characterisation of metastasis-related genes. Br. J. Cancer 2020. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez Gonzalez, M.; Gutierrez, M.L.; Sayagues, J.M.; Munoz-Bellvis, L.; Orfao, A. Genomic profiling of sporadic liver metastatic colorectal cancer. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Gener, P.; Gouveia, L.P.; Sabat, G.R.; de Sousa Rafael, D.F.; Fort, N.B.; Arranja, A.; Fernandez, Y.; Prieto, R.M.; Ortega, J.S.; Arango, D.; et al. Fluorescent CSC models evidence that targeted nanomedicines improve treatment sensitivity of breast and colon cancer stem cells. Nanomedicine 2015, 11, 1883–1892. [Google Scholar] [CrossRef]
- Chaitra, L.P.; Prashant, A.; Gowthami, C.S.; Hajira, B.; Suma, M.N.; Mahesh, S.S.; Manjunath, G.V.; Sheeladevi, C.S. Detection of cancer stem cell-related markers in different stages of colorectal carcinoma patients of Indian origin by immunohistochemistry. J. Cancer Res. Ther. 2019, 15, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.B.; Yu, Y.; Du, J.; Levi, E.; Phillip, P.A.; Majumdar, A.P. Age-related increase in colorectal cancer stem cells in macroscopically normal mucosa of patients with adenomas: A risk factor for colon cancer. Biochem. Biophys. Res. Commun. 2009, 378, 344–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morral, C.; Stanisavljevic, J.; Hernando-Momblona, X.; Mereu, E.; Alvarez-Varela, A.; Cortina, C.; Stork, D.; Slebe, F.; Turon, G.; Whissell, G.; et al. Zonation of ribosomal DNA transcription defines a stem cell hierarchy in colorectal cancer. Cell Stem Cell 2020, 26, 845–861. [Google Scholar] [CrossRef]
- Watanabe, T.; Okumura, T.; Hirano, K.; Yamaguchi, T.; Sekine, S.; Nagata, T.; Tsukada, K. Circulating tumor cells expressing cancer stem cell marker CD44 as a diagnostic biomarker in patients with gastric cancer. Oncol. Lett. 2017, 13, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Debele, T.A.; Yu, L.Y.; Yang, C.S.; Shen, Y.A.; Lo, C.L. pH- and GSH-sensitive hyaluronic Acid-MP conjugate micelles for intracellular delivery of doxorubicin to colon cancer cells and cancer stem cells. Biomacromolecules 2018, 19, 3725–3737. [Google Scholar] [CrossRef] [PubMed]
- Mansoori, B.; Mohammadi, A.; Shajari, N.; Davudian, S.; Salehi, S.; Baradaran, B. Nano-liposome-based target toxicity machine: An alternative/complementary approach in atopic diseases. Artif. Cells Nanomed. Biotechnol. 2017, 45, 1292–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.L.; Wang, Z.J.; Wei, G.H.; Yang, Y.; Wang, X.W. Changes in extracellular matrix in different stages of colorectal cancer and their effects on proliferation of cancer cells. World J. Gastrointest. Oncol. 2020, 12, 267–275. [Google Scholar] [CrossRef]
- Gil, J.; Ramsey, D.; Pawlowski, P.; Szmida, E.; Leszczynski, P.; Bebenek, M.; Sasiadek, M.M. The influence of tumor microenvironment on ATG4D gene expression in colorectal cancer patients. Med. Oncol. 2018, 35, 159. [Google Scholar] [CrossRef] [Green Version]
- Pascussi, J.M.; Giraud, J.; Samalin, E.; Grillet, F.; Pannequin, J. Fundamentals of the metastatic process. Bull. Cancer 2016, 103, S39–S47. [Google Scholar] [CrossRef]
- Wu, P.H.; Opadele, A.E.; Onodera, Y.; Nam, J.M. Targeting integrins in cancer nanomedicine: Applications in cancer diagnosis and therapy. Cancers 2019, 11, 1783. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, C.; Suares, D.; Yergeri, M.C. Tumor microenvironment targeted nanotherapy. Front. Pharmacol. 2018, 9, 1230. [Google Scholar] [CrossRef]
- Duan, X.; Chan, C.; Han, W.; Guo, N.; Weichselbaum, R.R.; Lin, W. Immunostimulatory nanomedicines synergize with checkpoint blockade immunotherapy to eradicate colorectal tumors. Nat. Commun. 2019, 10, 1899. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Duan, X.; Guo, N.; Chan, C.; Poon, C.; Weichselbaum, R.R.; Lin, W. Core-shell nanoscale coordination polymers combine chemotherapy and photodynamic therapy to potentiate checkpoint blockade cancer immunotherapy. Nat. Commun. 2016, 7, 12499. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Wu, C.; Zhu, C.; He, Y.; Yang, D.; Cheng, Y.; Gao, X. Oral administration of liposome-apatinib and locally delivery of docetaxel/MPEG-PCL by fibrin glue synergistically improve therapeutic effect in colorectal cancer. J. Biomed. Nanotechnol. 2018, 14, 2077–2091. [Google Scholar] [CrossRef] [PubMed]
- Bae, K.B.; Kim, S.H.; Kang, M.S.; Kim, D.H. An animal model of colorectal cancer liver metastasis with a high metastasis rate and clonal dynamics. Anticancer Res. 2020, 40, 3297–3306. [Google Scholar] [CrossRef]
- Samuelsson, E.; Shen, H.; Blanco, E.; Ferrari, M.; Wolfram, J. Contribution of Kupffer cells to liposome accumulation in the liver. Colloids and surf. B Biointerfaces 2017, 158, 356–362. [Google Scholar] [CrossRef]
- Pohlen, U.; Buhr, H.J.; Berger, G.; Ritz, J.P.; Holmer, C. Hepatic arterial infusion (HAI) with PEGylated liposomes containing 5-FU improves tumor control of liver metastases in a rat model. Investig. New Drugs 2012, 30, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Ichihara, H.; Hino, M.; Umebayashi, M.; Matsumoto, Y.; Ueoka, R. Intravenous injection of hybrid liposomes suppresses the liver metastases in xenograft mouse models of colorectal cancer in vivo. Eur. J. Med. Chem. 2012, 57, 143–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.; Zhang, Z.; Zhang, Y.; Lv, H.; Zhou, J.; Li, C.; Hou, L.; Zhang, Q. Dual-functional liposomes based on pH-responsive cell-penetrating peptide and hyaluronic acid for tumor-targeted anticancer drug delivery. Biomaterials 2012, 33, 9246–9258. [Google Scholar] [CrossRef] [PubMed]
- Duzgunes, N.; de Ilarduya, C.T. Genetic nanomedicine: Gene delivery by targeted lipoplexes. Methods Enzymol. 2012, 509, 355–367. [Google Scholar] [CrossRef]
- Zhdanov, R.; Bogdanenko, E.; Moskovtsev, A.; Podobed, O.; Duzgunes, N. Liposome-mediated gene delivery: Dependence on lipid structure, glycolipid-mediated targeting, and immunological properties. Methods Enzymol. 2003, 373, 433–465. [Google Scholar] [CrossRef]
- Belfiore, L.; Saunders, D.N.; Ranson, M.; Thurecht, K.J.; Storm, G.; Vine, K.L. Towards clinical translation of ligand-functionalized liposomes in targeted cancer therapy: Challenges and opportunities. J. Control. Release 2018, 277, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Hatakeyama, H. Development of a novel liposomal DDS by manipulating pharmacokinetics and intracellular trafficking for drug therapy and nucleic acid medicine. Yakugaku Zasshi 2018, 138, 591–598. [Google Scholar] [CrossRef] [Green Version]
- Davidson, B.L.; Breakefield, X.O. Viral vectors for gene delivery to the nervous system. Nat. Rev. Neurosci. 2003, 4, 353–364. [Google Scholar] [CrossRef]
- Stone, D.; David, A.; Bolognani, F.; Lowenstein, P.R.; Castro, M.G. Viral vectors for gene delivery and gene therapy within the endocrine system. J. Endocrinol. 2000, 164, 103–118. [Google Scholar] [CrossRef] [Green Version]
- Kaneda, Y. Development of non-viral gene delivery system and its applications. Nihon Rinsho 1996, 54, 2829–2838. [Google Scholar] [PubMed]
- Zhang, X.; Men, K.; Zhang, Y.; Zhang, R.; Yang, L.; Duan, X. Local and systemic delivery of mRNA encoding survivin-T34A by lipoplex for efficient colon cancer gene therapy. Int. J. Nanomed. 2019, 14, 2733–2751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, H.H.; Budzynski, W.; Koganty, R.R.; Krantz, M.J.; Reddish, M.A.; Rogers, J.A.; Longenecker, B.M.; Samuel, J. Liposomal formulations of synthetic MUC1 peptides: Effects of encapsulation versus surface display of peptides on immune responses. Bioconjugate Chem. 1998, 9, 451–458. [Google Scholar] [CrossRef]
- Fabrizi, E.; di Martino, S.; Pelacchi, F.; Ricci-Vitiani, L. Therapeutic implications of colon cancer stem cells. World J. Gastroenterol. 2010, 16, 3871–3877. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, E.; Bester, A.C.; Shifman, S.; Kerem, B. Transcriptional dynamics in colorectal carcinogenesis: New insights into the role of c-Myc and miR17 in benign to cancer transformation. Cancer Res. 2014, 74, 5532–5540. [Google Scholar] [CrossRef] [Green Version]
- Angitapalli, R.; Litwin, A.M.; Kumar, P.R.; Nasser, E.; Lombardo, J.; Mashtare, T.; Wilding, G.E.; Fakih, M.G. Adjuvant FOLFOX chemotherapy and splenomegaly in patients with stages II-III colorectal cancer. Oncology 2009, 76, 363–368. [Google Scholar] [CrossRef]
- Guo, J.; Yu, Z.; Das, M.; Huang, L. Nano codelivery of oxaliplatin and folinic acid achieves synergistic chemo-immunotherapy with 5-fluorouracil for colorectal cancer and liver metastasis. ACS Nano 2020, 14, 5075–5089. [Google Scholar] [CrossRef]
- Arshad, U.; Sutton, P.A.; Ashford, M.B.; Treacher, K.E.; Liptrott, N.J.; Rannard, S.P.; Goldring, C.E.; Owen, A. Critical considerations for targeting colorectal liver metastases with nanotechnology. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnology 2020, 12, e1588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulbake, A.; Jain, A.; Jain, A.; Jain, A.; Jain, S.K. Insight to drug delivery aspects for colorectal cancer. World J. Gastroenterol. 2016, 22, 582–599. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.M. Getting into the colon: Approaches to target colorectal cancer. Expert Opin. Drug Deliv. 2014, 11, 1343–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fountzilas, E.; Krishnan, E.; Janku, F.; Fu, S.; Karp, D.D.; Naing, A.; Subbiah, V.; Hong, D.S.; Piha-Paul, S.A.; Vining, D.J.; et al. A phase I clinical trial of hepatic arterial infusion of oxaliplatin and oral capecitabine, with or without intravenous bevacizumab, in patients with advanced cancer and predominant liver involvement. Cancer Chemother. Pharmacol. 2018, 82, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Tsimberidou, A.M.; Moulder, S.; Fu, S.; Wen, S.; Naing, A.; Bedikian, A.Y.; Daring, S.; Uehara, C.; Ng, C.; Wallace, M.; et al. Phase I clinical trial of hepatic arterial infusion of cisplatin in combination with intravenous liposomal doxorubicin in patients with advanced cancer and dominant liver involvement. Cancer Chemother. Pharmacol. 2010, 66, 1087–1093. [Google Scholar] [CrossRef] [Green Version]
- Tashiro, J.; Yamaguchi, S.; Ishii, T.; Suzuki, A.; Kondo, H.; Morita, Y.; Hara, K.; Koyama, I. Inferior oncological prognosis of surgery without oral chemotherapy for stage III colon cancer in clinical settings. World J. Surg. Oncol. 2014, 12, 145. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Bajracharya, R.; Min, J.Y.; Han, J.W.; Park, B.J.; Han, H.K. Strategic approaches for colon targeted drug delivery: An overview of recent advancements. Pharmaceutics 2020, 12, 68. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Zhu, G.; Lu, B.; Peng, Q. Oral nano-delivery systems for colon targeting therapy. Pharm. Nanotechnol. 2017, 5, 83–94. [Google Scholar] [CrossRef]
- Charalambides, D.; Segal, I. Colonic pH: A comparison between patients with colostomies due to trauma and colorectal cancer. Am. J. Gastroenterol. 1992, 87, 74–78. [Google Scholar]
- Thornton, J.R. High colonic pH promotes colorectal cancer. Lancet 1981, 1, 1081–1083. [Google Scholar] [CrossRef]
- Rajpoot, K.; Jain, S.K. Oral delivery of pH-responsive alginate microbeads incorporating folic acid-grafted solid lipid nanoparticles exhibits enhanced targeting effect against colorectal cancer: A dual-targeted approach. Int. J. Biol. Macromol. 2020, 151, 830–844. [Google Scholar] [CrossRef] [PubMed]
- Rajpoot, K.; Jain, S.K. Irinotecan hydrochloride trihydrate loaded folic acid-tailored solid lipid nanoparticles for targeting colorectal cancer: Development, characterization, and in vitro cytotoxicity study using HT-29 cells. J. Microencapsul. 2019, 36, 659–676. [Google Scholar] [CrossRef] [PubMed]
- Rajpoot, K.; Jain, S.K. Colorectal cancer-targeted delivery of oxaliplatin via folic acid-grafted solid lipid nanoparticles: Preparation, optimization, and in vitro evaluation. Artif. Cells Nanomed. Biotechnol. 2018, 46, 1236–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vetrano, S.; Danese, S. Colitis, microbiota, and colon cancer: An infernal triangle. Gastroenterology 2013, 144, 461–463. [Google Scholar] [CrossRef]
- Oussoren, C.; Eling, W.M.; Crommelin, D.J.; Storm, G.; Zuidema, J. The influence of the route of administration and liposome composition on the potential of liposomes to protect tissue against local toxicity of two antitumor drugs. Biochim. Biophys. Acta 1998, 1369, 159–172. [Google Scholar] [CrossRef] [Green Version]
- Naciute, M.; Niemi, V.; Kemp, R.A.; Hook, S. Lipid-encapsulated oral therapeutic peptide vaccines reduce tumour growth in an orthotopic mouse model of colorectal cancer. Eur. J. Pharm. Biopharm. 2020, 152, 183–192. [Google Scholar] [CrossRef]
- Yu, Q.; Qiu, Y.; Wang, X.; Tang, J.; Liu, Y.; Mei, L.; Li, M.; Yang, M.; Tang, L.; Gao, H.; et al. Efficient siRNA transfer to knockdown a placenta specific lncRNA using RGD-modified nano-liposome: A new preeclampsia-like mouse model. Int. J. Pharm. 2018, 546, 115–124. [Google Scholar] [CrossRef]
- Fathi, S.; Oyelere, A.K. Liposomal drug delivery systems for targeted cancer therapy: Is active targeting the best choice? Future Med. Chem. 2016, 8, 2091–2112. [Google Scholar] [CrossRef]
- Tummala, S.; Gowthamarajan, K.; Satish Kumar, M.N.; Wadhwani, A. Oxaliplatin immuno hybrid nanoparticles for active targeting: An approach for enhanced apoptotic activity and drug delivery to colorectal tumors. Drug Deliv. 2016, 23, 1773–1787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vedovatto, S.; Facchini, J.C.; Batista, R.K.; Paim, T.C.; Lionzo, M.I.Z.; Wink, M.R. Development of chitosan, gelatin and liposome film and analysis of its biocompatibility in vitro. Int. J. Biol. Macromol. 2020, 160, 750–757. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.H.; Tian, D.; Yang, Z.C.; Li, J.L. Exosomal miR-21 promotes proliferation, invasion and therapy resistance of colon adenocarcinoma cells through its target PDCD4. Sci. Rep. 2020, 10, 8271. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.H.L.; Wang, T.; Yin, W.; Tran, T.T.D.; Nguyen, T.N.G.; Lee, B.J.; Duan, W. Aspirin-loaded nanoexosomes as cancer therapeutics. Int. J. Pharm. 2019, 572, 118786. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, X.; Du, L.; Wang, Y.; Liu, X.; Tian, H.; Wang, L.; Li, P.; Zhao, Y.; Duan, W.; et al. Exosome-transmitted miR-128-3p increase chemosensitivity of oxaliplatin-resistant colorectal cancer. Mol. Cancer 2019, 18, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Tam, M.; Samaei, S.; Lerouge, S.; Barralet, J.; Stevenson, M.M.; Cerruti, M. Mucoadhesive chitosan hydrogels as rectal drug delivery vessels to treat ulcerative colitis. Acta Biomater. 2017, 48, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.L.; Feng, C.L.; Zheng, W.S.; Huang, S.; Zhang, W.X.; Wu, H.N.; Zhan, Y.; Han, Y.X.; Wu, S.; Jiang, J.D. Tumor-selective lipopolyplex encapsulated small active RNA hampers colorectal cancer growth in vitro and in orthotopic murine. Biomaterials 2017, 141, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Wang, L.; Xu, H.Q.; Huang, X.E.; Qian, Y.D.; Xiang, J. Clinical comparison between paclitaxel liposome (Lipusu (R)) and paclitaxel for treatment of patients with metastatic gastric cancer. Asian Pac. J. Cancer Prev. 2013, 14, 2591–2594. [Google Scholar] [CrossRef] [Green Version]
- Hagemeister, F.; Rodriguez, M.A.; Deitcher, S.R.; Younes, A.; Fayad, L.; Goy, A.; Dang, N.H.; Forman, A.; McLaughlin, P.; Medeiros, L.J.; et al. Long term results of a phase 2 study of vincristine sulfate liposome injection (Marqibo (®)) substituted for non-liposomal vincristine in cyclophosphamide, doxorubicin, vincristine, prednisone with or without rituximab for patients with untreated aggressive non-Hodgkin lymphomas. Br. J. Haematol. 2013, 162, 631–638. [Google Scholar] [CrossRef]
- FDA approves liposomal vincristine (Marqibo) for rare leukemia. Oncology 2012, 26, 841.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cargo | Formulation | Target | Category | Route | Stage | Ref. |
|---|---|---|---|---|---|---|
| 5-fluorouracil | Chitosan-coated liposomes | HT29 cells | LCLs | N/A | Characterization | [65] |
| Doxorubicin | Urotensin-II-conjugated liposomes | WiDr or LoVo cells | Active targeting | N/A | Characterization | [67] |
| 5-fluorouracil | FA-coated liposomes | HT29, CT26, and Caco2 cells | Active targeting | Subcutaneous injection | Preclinical | [34,69,142] |
| 5-fluorouracil | SMA-liposomes | HT29 cells | pH-sensitive | N/A | Characterization | [76,77] |
| Doxorubicin | ThermoDox® | CRC metastatic liver cancer | Thermo-sensitive | IV infusion | Clinical trial | [80] |
| Paclitaxel | Cationic liposomes (EndoTAG-1) | Advanced CRC, liver metastasis | Cationic liposomes | IV injection | Clinical trial | [89] |
| Endostatin gene | Cationic liposomes | CT26 and HCT116 cells | Cationic liposomes | Intraperitoneal injection | Preclinical | [92] |
| PLK1 siRNA | SNALPs | PLK1 overexpressed CRC | LCL (PEG-modified) cationic liposomes | IV injection | Preclinical | [98] |
| Retinoids | DPPC or DPPC/DPPE-PEG | HT29 and Colon205 cells | Prodrug | N/A | Characterization | [100] |
| Moeixitecan | DPPC/HSPC/DSPE-PEG | HT29 cells | Prodrug | Subcutaneous injection | Preclinical | [99] |
| mRIP3-pDNA | HA-PLGA | CT26 cells | Core-shell, active targeting | IV injection | Preclinical | [106] |
| siRNA siMcl1/siBcl-xl | DOTAP/PEG-PCL | C26 CRC cells | Cationic micelle, gene therapy | Intratumorally injection | Preclinical | [91] |
| Curcumin | FA-conjugated mPEG-PCL | CT26 cells | Self-Assembly Micelles | IV injection | Preclinical | [109] |
| Docetaxel | Cationic SLNs | Distal ileum, CT26 cells | SLNs | Oral | Preclinical | [115] |
| Doxorubicin | HDL mimics | CT26, MC38 cells | Nanodisc | IV injection | Preclinical | [120] |
| Cisplatin, metformin | Pluronic-F127, glyceryl monooleate, polyvinyl alcohol | HCT116 cells | Nano-cubosome | N/A | Characterization | [125] |
| Doxorubicin | FA-coated Ginger LNPs | Colon26 cells | PDLNP, Active targeting | IV injection | Preclinical | [129] |
| 6-shogaol | Ginger LNPs | Colon | PDLNP | Oral | Preclinical | [133,134] |
| 5-fluorouracil, miR-21i | Engineered exosome | 5-fluorouracil resistant HCT116 cells | Exosome, Multiple-targeting | IV injection | Preclinical | [137] |
| 5-fluorouracil | HA-modified liposomes | HT29 cells | Active targeting (CD44) | N/A | Characterization | [75] |
| 5-fluorouracil | PEGylated liposomes | CRC metastatic liver cancer | LCLs | Hepatic arterial infusion | Preclinical | [165] |
| Fluorescent dye | Mixture of vesicular and micellar molecules | HCT116, metastatic liver cancer | Hybrid liposomes | IV injection | Characterization | [166] |
| Paclitaxel | HA, cell-penetrating peptide | CRC metastatic liver cancer | pH-sensitive, cell-penetrating | IV injection | Preclinical | [167] |
| Survivin siRNA | Lipoplex | LoVo cells | Cationic LCLs, Gene therapy | Intraperitoneal injection | Preclinical | [50] |
| Mucin-1 peptide (BP-25) | Cationic liposome | T-cell | Immune therapy, Cationic liposomes | Subcutaneous | Preclinical | [176] |
| Docetaxel | Cationic SLN | Apical sodium bile acid transporter, T-cell | Immune therapy, SLN | Lymphatic transport | Preclinical | [115] |
| Folinic acid, 5-fluorouracil, oxaliplatin, anti-PD-L1 antibody | PEGylated lipid nanoparticle | CT26-FL3, metastatic liver cancer | Multiple-targeting, Immune therapy | IV injection | Preclinical | [180] |
| Irinotecan, miR-200 | Peptide-modified liposome, SLN | HCT116, CT26 cells | Multiple-targeting (pH-sensitive, cell-penetrating, mitochondria-targeting) | IV injection | Preclinical | [70] |
| Oxaliplatin, anti-TRAIL | Immunohybrid liposomes | HT29 cells | Multiple-targeting | IV injection | Preclinical | [199] |
| Aspirin | CRC cell derived exosomes | HT29, CRC stem cells | Active targeting | IV injection | Preclinical | [202] |
| miR-128-3p | Exosomes | Oxaliplatin-resistant HCT116OxR cells | Gene therapy | Intratumorally injection | Preclinical | [203] |
| p21-saRNA-322 | HA-lipid shell nanoparticles | HT29 | Gene therapy, core-shell | Rectal delivery | Preclinical | [205] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, C.; Merlin, D. Lipid-Based Drug Delivery Nanoplatforms for Colorectal Cancer Therapy. Nanomaterials 2020, 10, 1424. https://doi.org/10.3390/nano10071424
Yang C, Merlin D. Lipid-Based Drug Delivery Nanoplatforms for Colorectal Cancer Therapy. Nanomaterials. 2020; 10(7):1424. https://doi.org/10.3390/nano10071424
Chicago/Turabian StyleYang, Chunhua, and Didier Merlin. 2020. "Lipid-Based Drug Delivery Nanoplatforms for Colorectal Cancer Therapy" Nanomaterials 10, no. 7: 1424. https://doi.org/10.3390/nano10071424