Exploration of the Interaction Strength at the Interface of Anionic Chalcogen Anchors and Gold (111)-Based Nanomaterials


Abstract
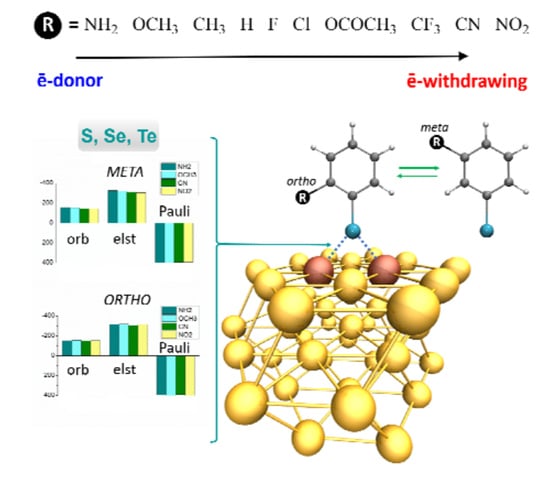
:
1. Introduction
2. Models, Computation, and Methodology
3. Results and Discussion
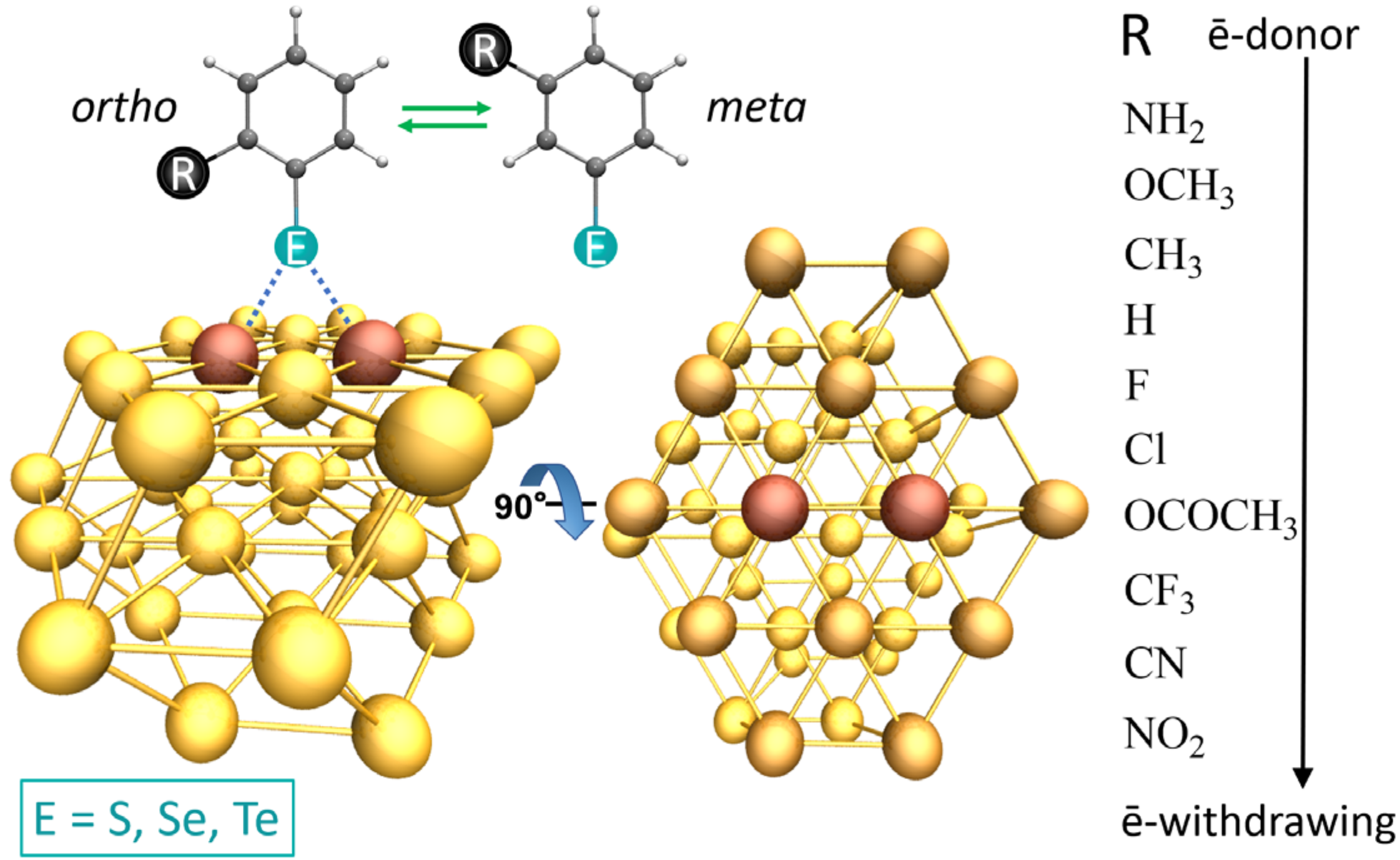
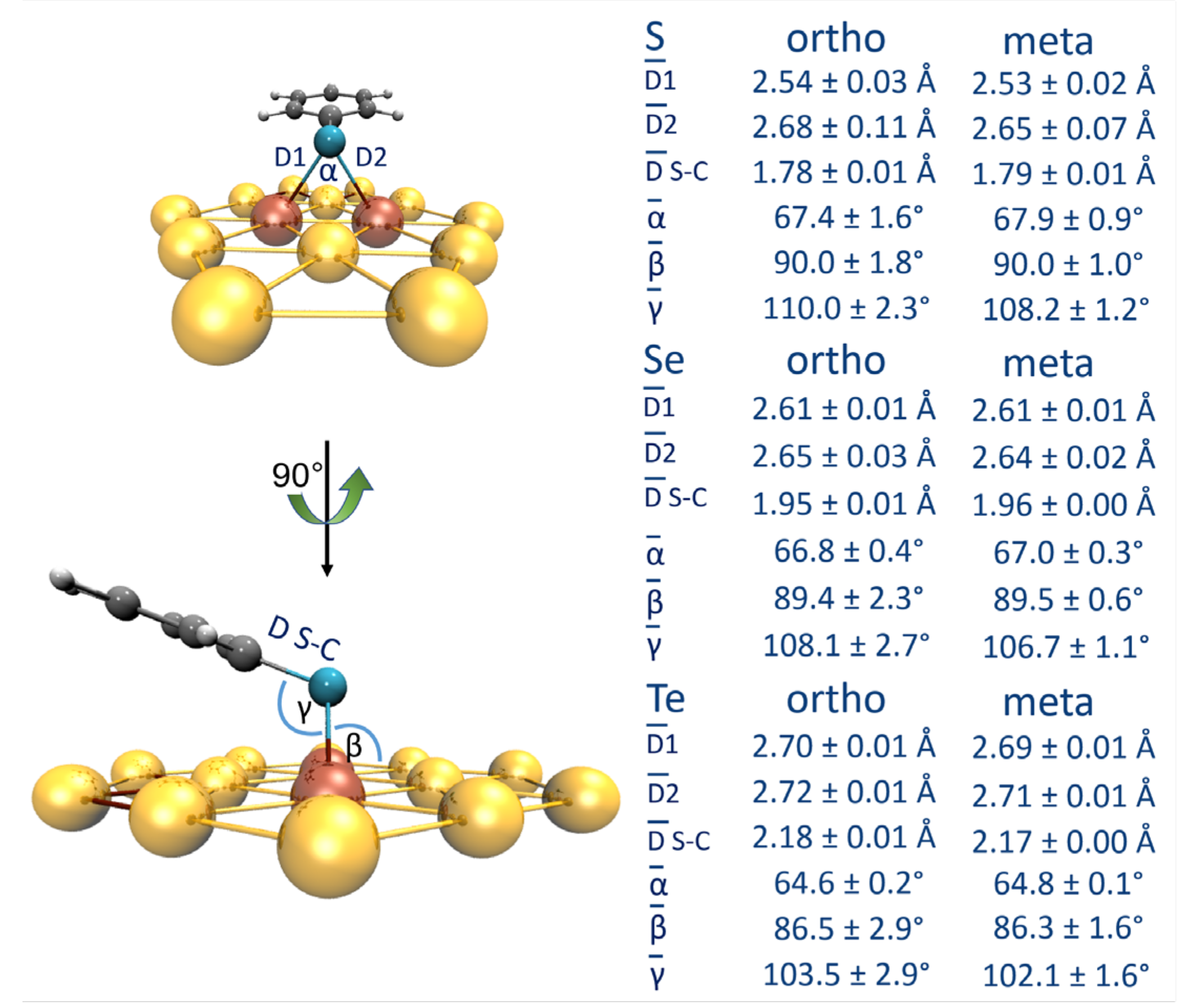
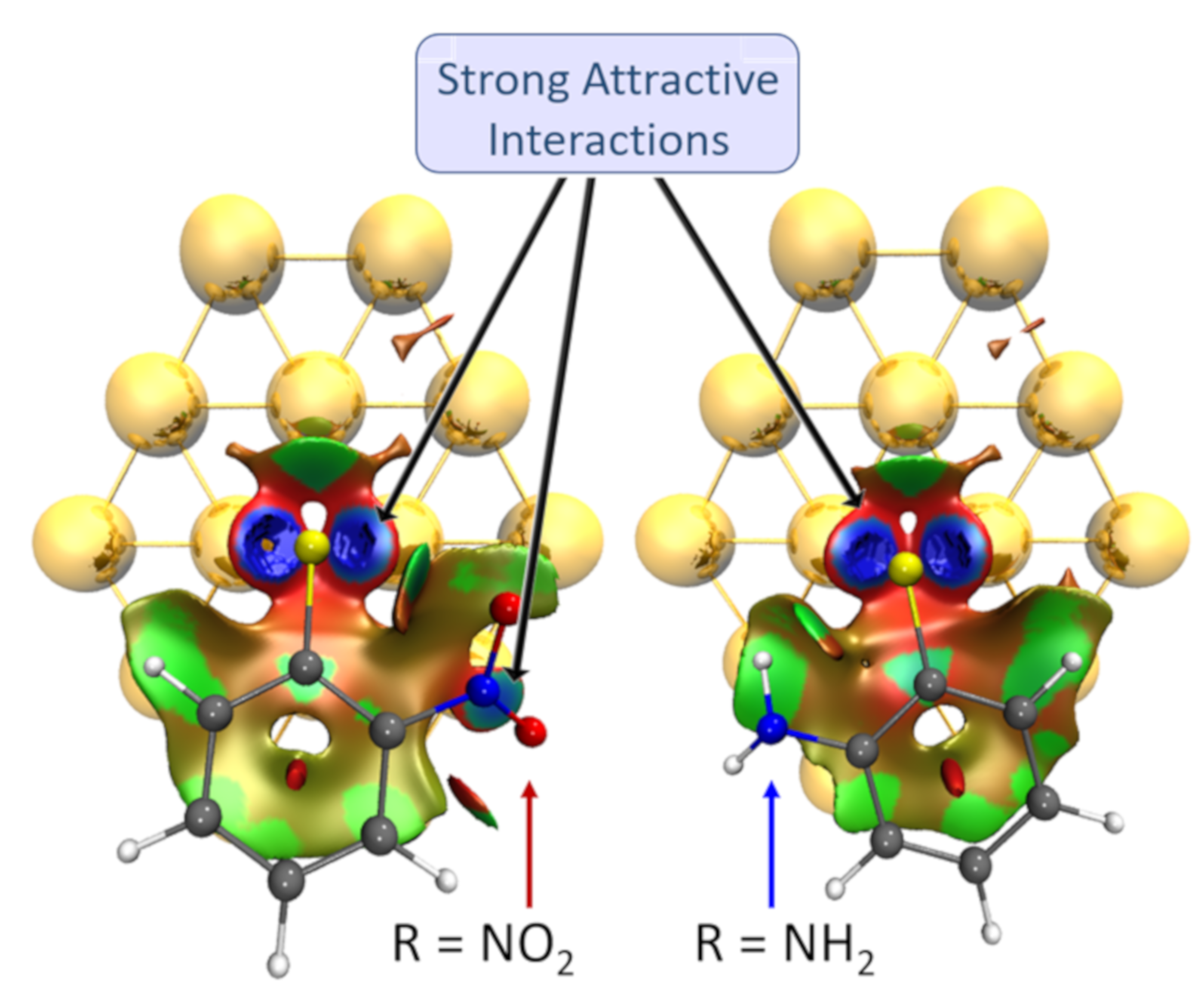
3.1. Binding Mode Conformation
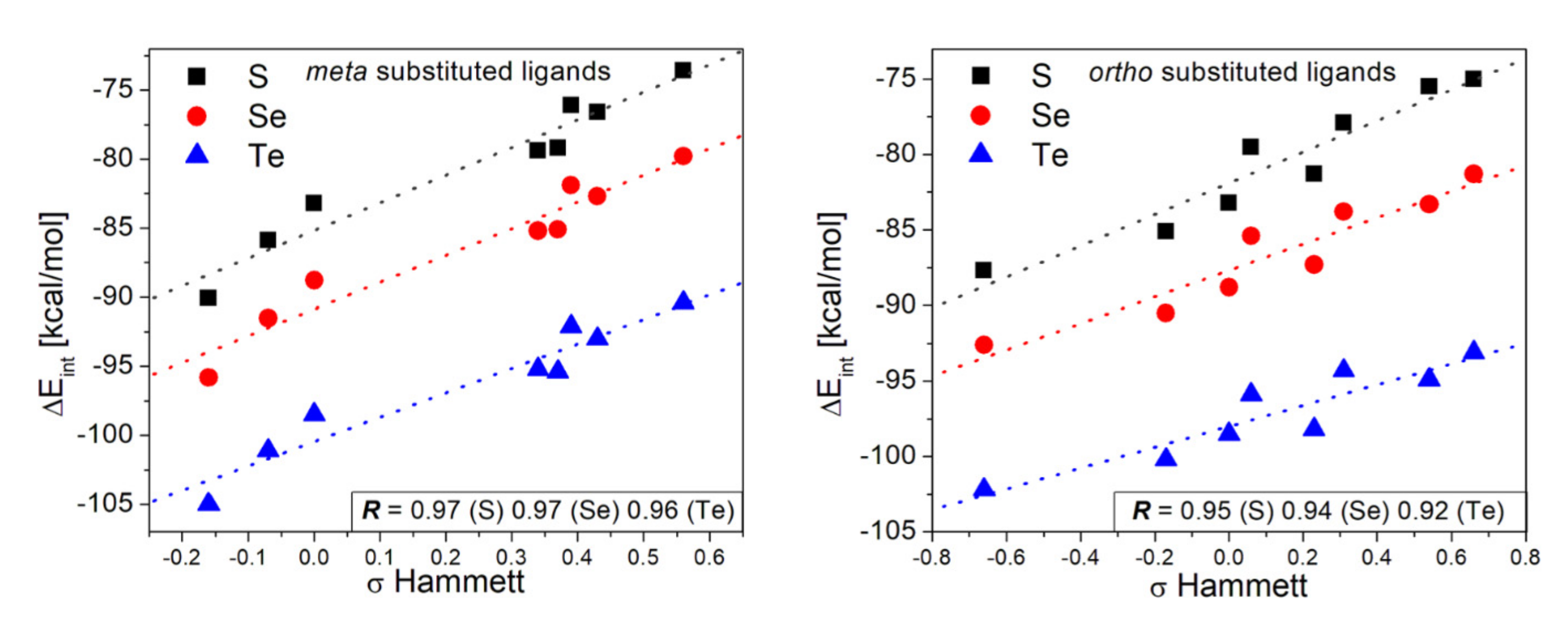
3.2. Interaction Strength Analysis
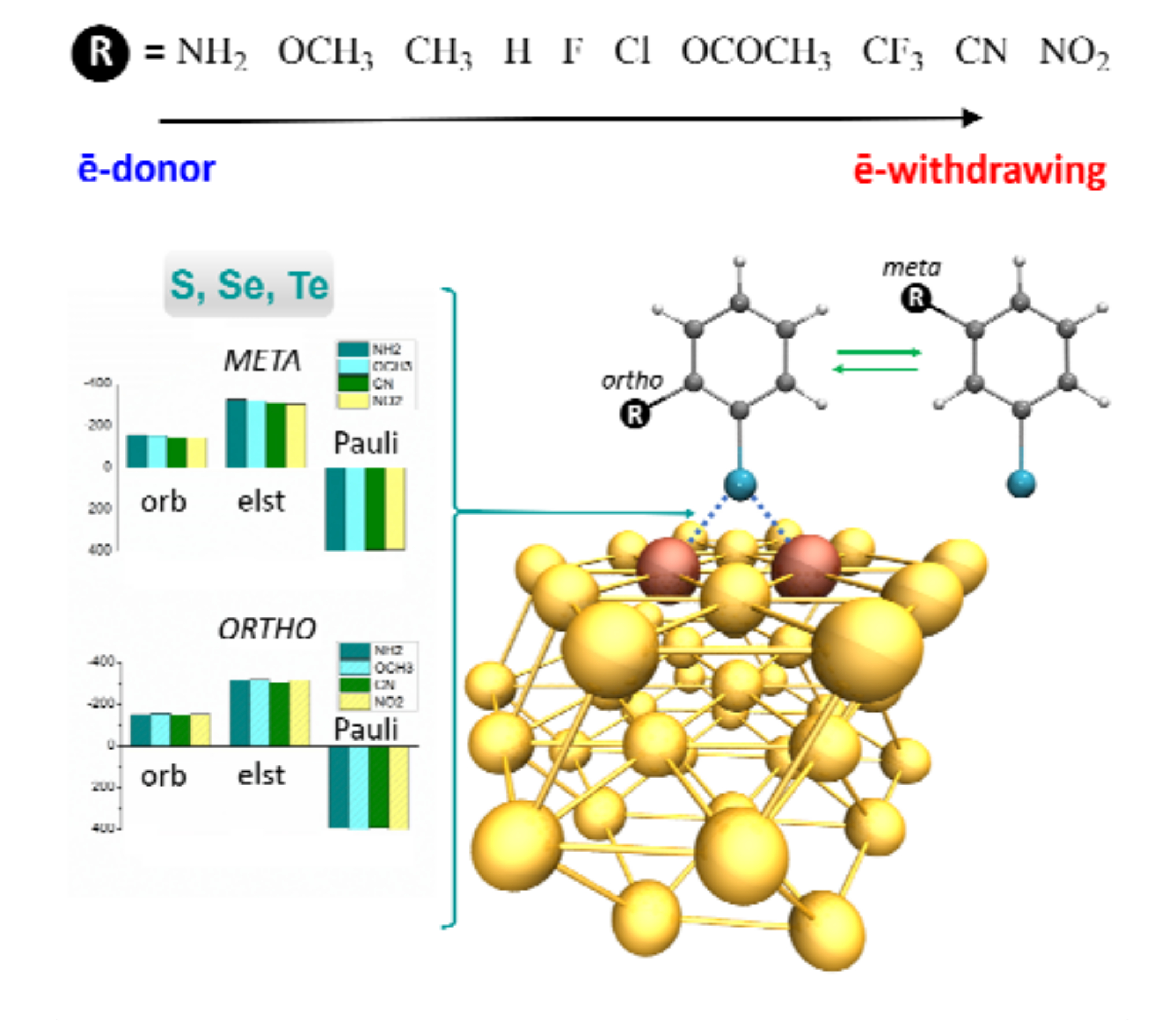
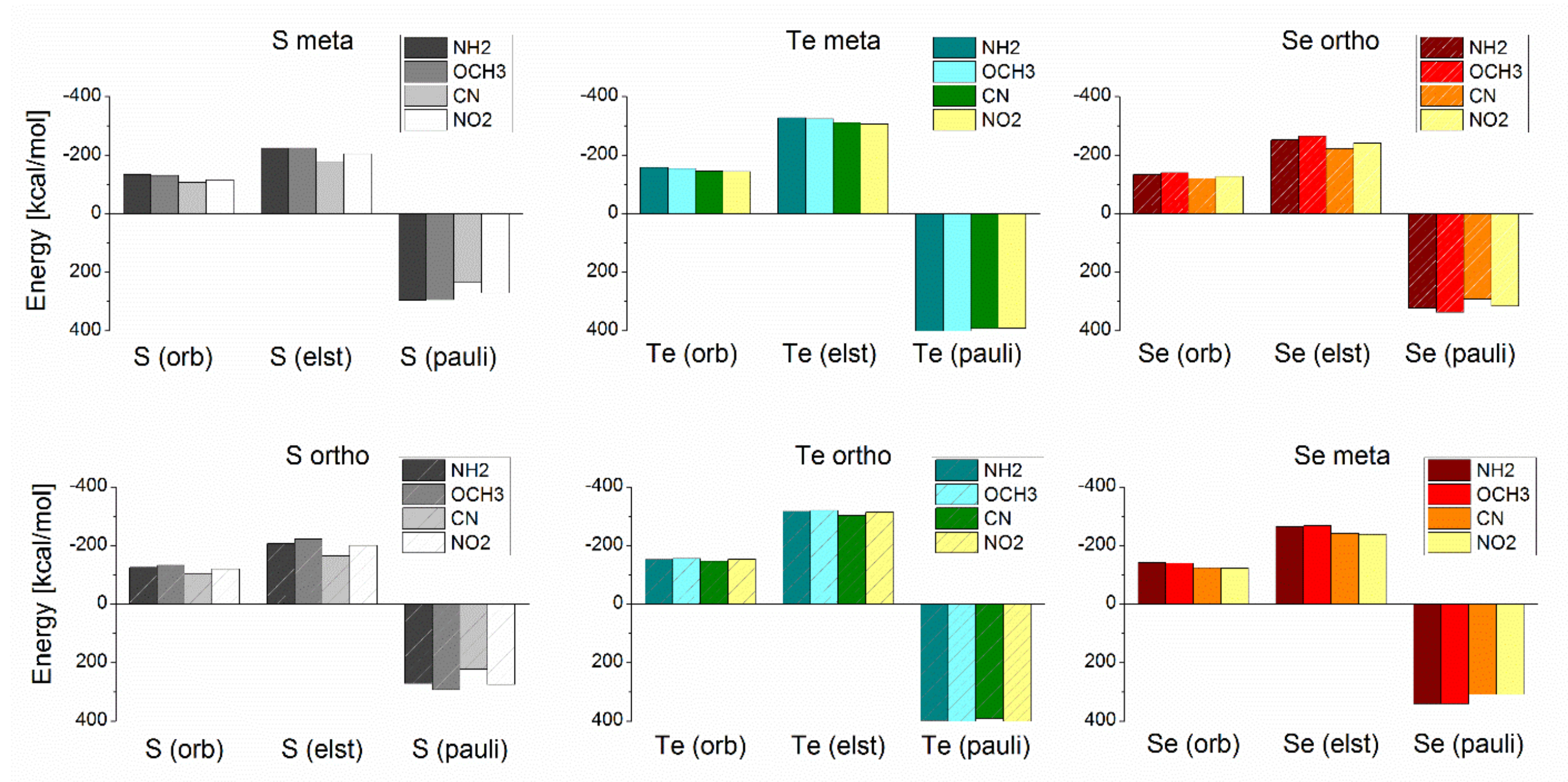
3.3. Energy Decomposition Analysis
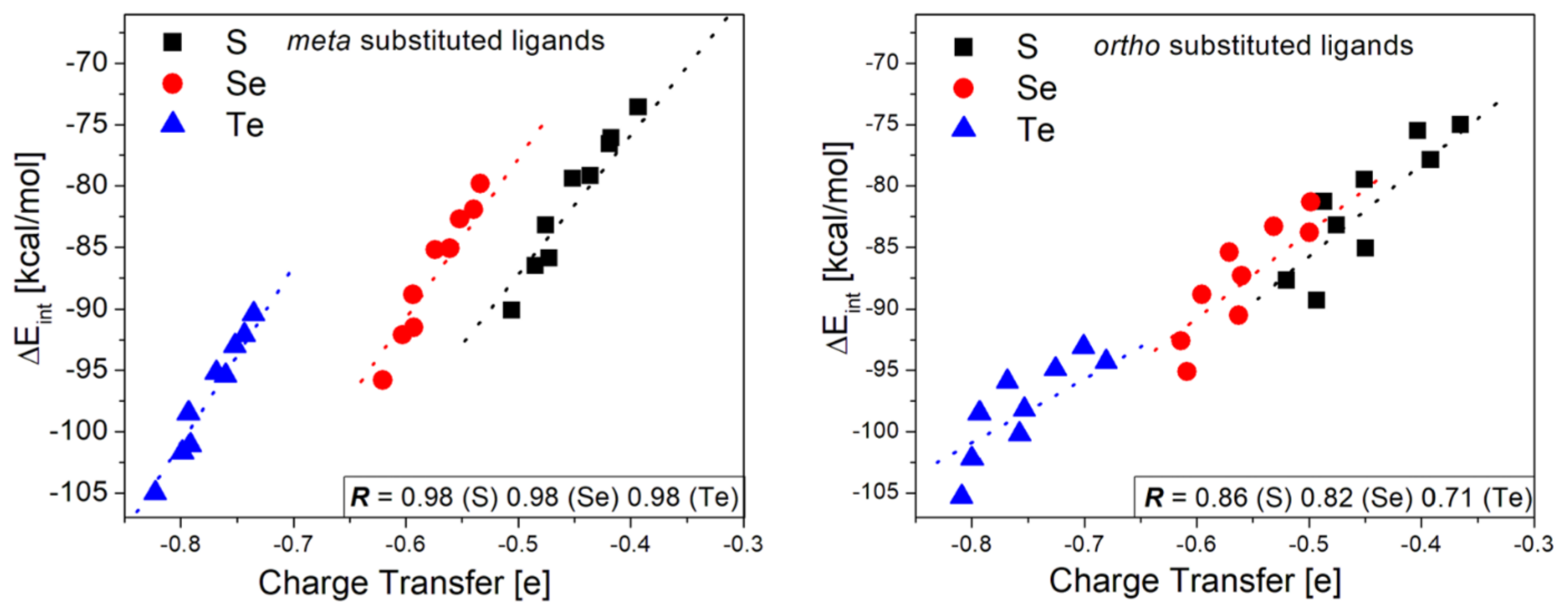
3.4. Charge Transfer Analysis
3.5. Multi-Substituted Selenophenolate Ligands
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lee, S.; Puck, A.; Graupe, M.; Colorado, R.; Shon, Y.-S.; Lee, T.R.; Perry, S.S. Structure, Wettability, and Frictional Properties of Phenyl-Terminated Self-Assembled Monolayers on Gold. Langmuir 2001, 17, 7364–7370. [Google Scholar] [CrossRef]
- Houston, J.E.; Kim, H.I. Adhesion, Friction, and Mechanical Properties of Functionalized Alkanethiol Self-Assembled Monolayers. Acc. Chem. Res. 2002, 35, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Kara, A.; Pasquali, L.; Bendounan, A.; Sirotti, F.; Esaulov, V.A. On sulfur core level binding energies in thiol self-assembly and alternative adsorption sites: An experimental and theoretical study. J. Chem. Phys. 2015, 143, 104702. [Google Scholar] [CrossRef] [PubMed]
- Haruta, M. When Gold Is Not Noble: Catalysis by Nanoparticles. Chem. Rec. 2003, 3, 75–87. [Google Scholar] [CrossRef]
- Daniel, M.-C.; Astruc, D. Gold Nanoparticles: Assembly, Supramolecular Chemistry, Quantum-Size-Related Properties, and Applications toward Biology, Catalysis, and Nanotechnology. Chem. Rev. 2004, 104, 293–346. [Google Scholar] [CrossRef]
- Hashmi, A.S.K.; Hutchings, G.J. Gold Catalysis. Angew. Chem. Int. Ed. 2006, 45, 7896–7936. [Google Scholar] [CrossRef]
- Thompson, D.T. Using gold nanoparticles for catalysis. Nano Today 2007, 2, 40–43. [Google Scholar] [CrossRef]
- Kang, B.; Mackey, M.A.; El-Sayed, M.A. Nuclear Targeting of Gold Nanoparticles in Cancer Cells Induces DNA Damage, Causing Cytokinesis Arrest and Apoptosis. J. Am. Chem. Soc. 2010, 132, 1517–1519. [Google Scholar] [CrossRef]
- Pasquato, L.; Pengo, P.; Scrimin, P. Functional gold nanoparticles for recognition and catalysis. J. Mater. Chem. 2004, 14, 3481–3487. [Google Scholar] [CrossRef]
- Love, J.C.; Estroff, L.A.; Kriebel, J.K.; Nuzzo, R.G.; Whitesides, G.M. Self-Assembled Monolayers of Thiolates on Metals as a Form of Nanotechnology. Chem. Rev. 2005, 105, 1103–1170. [Google Scholar] [CrossRef]
- Newton, L.; Slater, T.; Clark, N.; Vijayaraghavan, A. Self assembled monolayers (SAMs) on metallic surfaces (gold and graphene) for electronic applications. J. Mater. Chem. C 2013, 1, 376–393. [Google Scholar] [CrossRef]
- Inkpen, M.S.; Liu, Z.; Li, H.; Campos, L.M.; Neaton, J.B.; Venkataraman, L. Non-chemisorbed gold–sulfur binding prevails in self-assembled monolayers. Nat. Chem. 2019, 11, 351–358. [Google Scholar] [CrossRef] [PubMed]
- McGuiness, C.L.; Diehl, G.A.; Blasini, D.; Smilgies, D.-M.; Zhu, M.; Samarth, N.; Weidner, T.; Ballav, N.; Zharnikov, M.; Allara, D.L. Molecular Self-Assembly at Bare Semiconductor Surfaces: Cooperative Substrate−Molecule Effects in Octadecanethiolate Monolayer Assemblies on GaAs(111), (110), and (100). ACS Nano 2010, 4, 3447–3465. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Jamison, A.C.; Lee, T.R. Surface Dipoles: A Growing Body of Evidence Supports Their Impact and Importance. Acc. Chem. Res. 2015, 48, 3007–3015. [Google Scholar] [CrossRef] [PubMed]
- Sardar, R.; Funston, A.M.; Mulvaney, P.; Murray, R.W. Gold Nanoparticles: Past, Present, and Future. Langmuir 2009, 25, 13840–13851. [Google Scholar] [CrossRef]
- Mameka, N.; Lührs, L.; Heissler, S.; Gliemann, H.; Wöll, C. Tailoring the Strength of Nanoporous Gold by Self-Assembled Monolayers of Alkanethiols. ACS Appl. Nano Mater. 2018, 1, 6613–6621. [Google Scholar] [CrossRef]
- Vericat, C.; Vela, M.E.; Benitez, G.; Carro, P.; Salvarezza, R.C. Self-assembled monolayers of thiols and dithiols on gold: New challenges for a well-known system. Chem. Soc. Rev. 2010, 39, 1805–1834. [Google Scholar] [CrossRef]
- Pensa, E.; Cortés, E.; Corthey, G.; Carro, P.; Vericat, C.; Fonticelli, M.H.; Benítez, G.; Rubert, A.A.; Salvarezza, R.C. The Chemistry of the Sulfur–Gold Interface: In Search of a Unified Model. Acc. Chem. Res. 2012, 45, 1183–1192. [Google Scholar] [CrossRef]
- Häkkinen, H. The gold–sulfur interface at the nanoscale. Nat. Chem. 2012, 4, 443–455. [Google Scholar] [CrossRef]
- Zenasni, O.; Marquez, M.D.; Jamison, A.C.; Lee, H.J.; Czader, A.; Lee, T.R. Inverted Surface Dipoles in Fluorinated Self-Assembled Monolayers. Chem. Mater. 2015, 27, 7433–7446. [Google Scholar] [CrossRef]
- Chen, H.; Heng, C.K.; Puiu, P.D.; Zhou, X.D.; Lee, A.C.; Lim, T.M.; Tan, S.N. Detection of Saccharomyces cerevisiae immobilized on self-assembled monolayer (SAM) of alkanethiolate using electrochemical impedance spectroscopy. Anal. Chim. Acta 2005, 554, 52–59. [Google Scholar] [CrossRef]
- Li, S.; Luo, Q.; Zhang, Z.; Shen, G.; Wu, H.; Chen, A.; Liu, X.; Li, M.; Zhang, A. Electrochemistry Study of Permselectivity and Interfacial Electron Transfers of a Branch-Tailed Fluorosurfactant Self-Assembled Monolayer on Gold. Molecules 2018, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weidner, T.; Shaporenko, A.; Müller, J.; Schmid, M.; Cyganik, P.; Terfort, A.; Zharnikov, M. Effect of the Bending Potential on Molecular Arrangement in Alkaneselenolate Self-Assembled Monolayers. J. Phys. Chem. C 2008, 112, 12495–12506. [Google Scholar] [CrossRef]
- Wang, M.; Ren, K.; Wang, L. Iron-Catalyzed Ligand-Free Carbon-Selenium (or Tellurium) Coupling of Arylboronic Acids with Diselenides and Ditellurides. Adv. Synth. Catal. 2009, 351, 1586–1594. [Google Scholar] [CrossRef]
- Liotta, D.; Sunay, U.; Santiesteban, H.; Markiewicz, W. Phenyl selenide anion, a superior reagent for the SN2 cleavage of esters and lactones. J. Org. Chem. 1981, 46, 2605–2610. [Google Scholar] [CrossRef]
- Trenner, J.; Depken, C.; Weber, T.; Breder, A. Direct Oxidative Allylic and Vinylic Amination of Alkenes through Selenium Catalysis. Angew. Chem. Int. Ed. 2013, 52, 8952–8956. [Google Scholar] [CrossRef]
- Singh, F.V.; Wirth, T. Selenium-Catalyzed Regioselective Cyclization of Unsaturated Carboxylic Acids Using Hypervalent Iodine Oxidants. Org. Lett. 2011, 13, 6504–6507. [Google Scholar] [CrossRef]
- Yu, L.; Li, H.; Zhang, X.; Ye, J.; Liu, J.; Xu, Q.; Lautens, M. Organoselenium-Catalyzed Mild Dehydration of Aldoximes: An Unexpected Practical Method for Organonitrile Synthesis. Org. Lett. 2014, 16, 1346–1349. [Google Scholar] [CrossRef]
- Prasad, C.D.; Balkrishna, S.J.; Kumar, A.; Bhakuni, B.S.; Shrimali, K.; Biswas, S.; Kumar, S. Transition-Metal-Free Synthesis of Unsymmetrical Diaryl Chalcogenides from Arenes and Diaryl Dichalcogenides. J. Org. Chem. 2013, 78, 1434–1443. [Google Scholar] [CrossRef]
- Shaporenko, A.; Cyganik, P.; Buck, M.; Terfort, A.; Zharnikov, M. Self-Assembled Monolayers of Aromatic Selenolates on Noble Metal Substrates. J. Phys. Chem. B 2005, 109, 13630–13638. [Google Scholar] [CrossRef]
- Nakayama, Y.; Watanabe, K.; Ueyama, N.; Nakamura, A.; Harada, A.; Okuda, J. Titanium Complexes Having Chelating Diaryloxo Ligands Bridged by Tellurium and Their Catalytic Behavior in the Polymerization of Ethylene. Organometallics 2000, 19, 2498–2503. [Google Scholar] [CrossRef]
- Takashima, Y.; Nakayama, Y.; Watanabe, K.; Itono, T.; Ueyama, N.; Nakamura, A.; Yasuda, H.; Harada, A.; Okuda, J. Polymerizations of Cyclic Esters Catalyzed by Titanium Complexes Having Chalcogen-Bridged Chelating Diaryloxo Ligands. Macromolecules 2002, 35, 7538–7544. [Google Scholar] [CrossRef]
- Weidner, T.; Shaporenko, A.; Müller, J.; Höltig, M.; Terfort, A.; Zharnikov, M. Self-Assembled Monolayers of Aromatic Tellurides on (111)-Oriented Gold and Silver Substrates. J. Phys. Chem. C 2007, 111, 11627–11635. [Google Scholar] [CrossRef]
- Ossowski, J.; Wächter, T.; Silies, L.; Kind, M.; Noworolska, A.; Blobner, F.; Gnatek, D.; Rysz, J.; Bolte, M.; Feulner, P.; et al. Thiolate versus Selenolate: Structure, Stability, and Charge Transfer Properties. ACS Nano 2015, 9, 4508–4526. [Google Scholar] [CrossRef]
- Miranda-Rojas, S.; Muñoz-Castro, A.; Arratia-Pérez, R.; Mendizábal, F. Theoretical insights into the adsorption of neutral{,} radical and anionic thiophenols on gold(111). Phys. Chem. Chem. Phys. 2013, 15, 20363–20370. [Google Scholar] [CrossRef]
- Miranda-Rojas, S.; Salazar-Molina, R.; Kästner, J.; Arratia-Pérez, R.; Mendizábal, F. Theoretical exploration of seleno and tellurophenols as promising alternatives to sulfur ligands for anchoring to gold (111) materials. RSC Adv. 2016, 6, 4458–4468. [Google Scholar] [CrossRef]
- Ahlrichs, R.; Bär, M.; Häser, M.; Horn, H.; Kölmel, C. Electronic structure calculations on workstation computers: The program system turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the density functional ladder: Nonempirical meta–generalized gradient approximation designed for molecules and solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [Green Version]
- Johansson, M.P.; Lechtken, A.; Schooss, D.; Kappes, M.M.; Furche, F. 2D-3D transition of gold cluster anions resolved. Phys. Rev. A 2008, 77, 53202. [Google Scholar] [CrossRef]
- Goel, S.; Velizhanin, K.A.; Piryatinski, A.; Tretiak, S.; Ivanov, S.A. DFT Study of Ligand Binding to Small Gold Clusters. J. Phys. Chem. Lett. 2010, 1, 927–931. [Google Scholar] [CrossRef]
- Rappoport, D.; Furche, F. Property-optimized Gaussian basis sets for molecular response calculations. J. Chem. Phys. 2010, 133, 134105. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, A.; Horn, H.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Andrae, D.; Häußermann, U.; Dolg, M.; Stoll, H.; Preuß, H. Energy-adjustedab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Igel-Mann, G.; Stoll, H.; Preuss, H. Pseudopotentials for main group elements (IIIa through VIIa). Mol. Phys. 1988, 65, 1321–1328. [Google Scholar] [CrossRef]
- Bergner, A.; Dolg, M.; Küchle, W.; Stoll, H.; Preuß, H. Ab initio energy-adjusted pseudopotentials for elements of groups 13–17. Mol. Phys. 1993, 80, 1431–1441. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Hujo, W.; Grimme, S. Performance of the van der Waals Density Functional VV10 and (hybrid)GGA Variants for Thermochemistry and Noncovalent Interactions. J. Chem. Theory Comput. 2011, 7, 3866–3871. [Google Scholar] [CrossRef]
- Grimme, S. Comment on: “On the Accuracy of DFT Methods in Reproducing Ligand Substitution Energies for Transition Metal Complexes in Solution: The Role of Dispersive Interactions” by H. Jacobsen and L. Cavallo. ChemPhysChem 2012, 13, 1407–1409. [Google Scholar] [CrossRef]
- Eichkorn, K.; Treutler, O.; Öhm, H.; Häser, M.; Ahlrichs, R. Auxiliary basis sets to approximate Coulomb potentials. Chem. Phys. Lett. 1995, 240, 283–290. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Ziegler, T.; Rauk, A. On the calculation of bonding energies by the Hartree Fock Slater method. Theor. Chim. Acta 1977, 46, 1–10. [Google Scholar] [CrossRef]
- Te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic total energy using regular approximations. J. Chem. Phys. 1994, 101, 9783–9792. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [Green Version]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef]
- Sánchez-Sanz, G.; Trujillo, C.; Alkorta, I.; Elguero, J. Understanding Regium Bonds and their Competition with Hydrogen Bonds in Au2:HX Complexes. ChemPhysChem. 2019, 20, 1572–1580. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| [Au42-EPhRo]− | ΔEint(TPSS-D3) | ΔEint(TPSS) | % Dispersion | ||||||
|---|---|---|---|---|---|---|---|---|---|
| S | Se | Te | S | Se | Te | S | Se | Te | |
| Au42-EC6H4(NH2)o | −87.7 | −92.6 | −102.2 | −55.4 | −59.2 | −66.6 | 36.8 | 36.0 | 34.8 |
| Au42-EC6H4(OCH3)o | −89.3 | −95.1 | −105.3 | −57.1 | −61.2 | −69.3 | 36.1 | 35.7 | 34.1 |
| Au42-EC6H4(CH3)o | −85.1 | −90.5 | −100.2 | −54.4 | −58.0 | −65.3 | 36.1 | 35.9 | 34.8 |
| Au42-EC6H5 | −83.2 | −88.8 | −98.5 | −55.4 | −59.1 | −66.7 | 33.4 | 33.5 | 32.3 |
| Au42-EC6H4(F)o | −79.5 | −85.4 | −95.9 | −51.4 | −55.5 | −64.1 | 35.4 | 35.0 | 33.2 |
| Au42-EC6H4(Cl)o | −81.3 | −87.3 | −98.2 | −50.0 | −54.0 | −62.7 | 38.5 | 38.1 | 36.1 |
| Au42-EC6H4(OCOCH3)o | −77.9 | −83.8 | −94.3 | −43.8 | −48.1 | −57.2 | 43.7 | 42.6 | 39.4 |
| Au42-EC6H4(CF3)o | −75.5 | −83.3 | −94.9 | −45.3 | −51.7 | −61.3 | 40.0 | 37.9 | 35.4 |
| Au42-EC6H4(CN)o | −75.0 | −81.3 | −93.1 | −43.6 | −48.1 | −57.3 | 41.8 | 40.9 | 38.5 |
| Au42-EC6H4(NO2)o | −76.7 | −84.3 | −97.8 | −42.9 | −48.5 | −59.5 | 44.0 | 42.5 | 39.1 |
| [Au42–EPhRo]− | ΔEint(TPSS-D3) | ΔEint(TPSS) | % Dispersion | ||||||
|---|---|---|---|---|---|---|---|---|---|
| S | Se | Te | S | Se | Te | S | Se | Te | |
| Au42-EC6H4(NH2)m | −90.1 | −95.8 | −105.0 | −58.7 | −62.5 | −69.4 | 34.9 | 34.7 | 33.9 |
| Au42-EC6H4(OCH3)m | −86.5 | −92.1 | −101.7 | −55.7 | −59.5 | −67.0 | 35.6 | 35.4 | 34.1 |
| Au42-EC6H4(CH3)m | −85.9 | −91.5 | −101.1 | −55.6 | −59.6 | −67.3 | 35.3 | 34.9 | 33.5 |
| Au42-EC6H5 | −83.2 | −88.8 | −98.5 | −55.4 | −59.0 | −66.7 | 33.4 | 33.6 | 32.3 |
| Au42-EC6H4(F)m | −79.4 | −85.2 | −95.2 | −51.4 | −55.4 | −63.1 | 35.3 | 35.0 | 33.7 |
| Au42-EC6H4(Cl)m | −79.2 | −85.1 | −95.4 | −48.9 | −53.2 | −61.5 | 38.3 | 37.5 | 35.5 |
| Au42-EC6H4(OCOCH3)m | −76.1 | −81.9 | −92.1 | −47.0 | −51.3 | −59.2 | 38.2 | 37.4 | 35.7 |
| Au42-EC6H4(CF3)m | −76.6 | −82.7 | −93.0 | −46.9 | −51.2 | −59.3 | 38.8 | 38.1 | 36.2 |
| Au42-EC6H4(CN)m | −73.6 | −79.8 | −90.4 | −44.4 | −49.0 | −57.4 | 39.7 | 38.6 | 36.5 |
| Au42-EC6H4(NO2)m | −74.7 | −80.6 | −91.0 | −43.3 | −47.7 | −56.0 | 42.1 | 40.8 | 38.4 |
| Model System | % Orb (meta) | % Orb (ortho) | % Elect (meta) | % Elect (ortho) |
|---|---|---|---|---|
| Au42-SC6H4(NH2) | 37.4 | 37.6 | 62.6 | 62.4 |
| Au42-SeC6H4(NH2) | 34.8 | 34.7 | 65.2 | 65.3 |
| Au42-TeC6H4(NH2) | 32.6 | 32.4 | 67.4 | 67.6 |
| Au42-SC6H4(OCH3) | 37.0 | 37.2 | 63.0 | 62.8 |
| Au42-SeC6H4(OCH3) | 34.3 | 34.5 | 65.7 | 65.5 |
| Au42-TeC6H4(OCH3) | 32.3 | 32.7 | 67.7 | 67.3 |
| Au42-SC6H4(CN) | 37.6 | 38.7 | 62.4 | 61.3 |
| Au42-SeC6H4(CN) | 33.8 | 34.7 | 66.2 | 65.3 |
| Au42-TeC6H4(CN) | 31.6 | 32.5 | 68.4 | 67.5 |
| Au42-SC6H4(NO2) | 36.0 | 37.4 | 64.0 | 62.6 |
| Au42-SeC6H4(NO2) | 33.9 | 35.1 | 66.1 | 64.9 |
| Au42-TeC6H4(NO2) | 31.9 | 32.7 | 68.1 | 67.3 |
| S | Se | Te | ||||
|---|---|---|---|---|---|---|
| Substituent | Au42 | ∆S a | Au42 | ∆Se a | Au42 | ∆Te a |
| -NH2 | −0.52 | 0.28 | −0.61 | 0.41 | −0.80 | 0.65 |
| -OCH3 | −0.49 | 0.26 | −0.61 | 0.41 | −0.81 | 0.66 |
| -CH3 | −0.45 | 0.25 | −0.56 | 0.39 | −0.76 | 0.63 |
| -H | −0.48 | 0.27 | −0.60 | 0.42 | −0.79 | 0.66 |
| -F | −0.45 | 0.24 | −0.57 | 0.39 | −0.77 | 0.63 |
| -Cl | −0.49 | 0.23 | −0.56 | 0.36 | −0.75 | 0.61 |
| -OCOCH3 | −0.39 | 0.18 | −0.50 | 0.32 | −0.68 | 0.54 |
| -CF3 | −0.40 | 0.20 | −0.53 | 0.34 | −0.73 | 0.58 |
| -CN | −0.37 | 0.16 | −0.50 | 0.30 | −0.70 | 0.55 |
| -NO2 | −0.35 | 0.14 | −0.46 | 0.27 | −0.66 | 0.50 |
| S | Se | Te | ||||
|---|---|---|---|---|---|---|
| Substituent | Au42 | ∆Sa | Au42 | ∆Se a | Au42 | ∆Te a |
| -NH2 | −0.51 | 0.27 | −0.62 | 0.42 | −0.82 | 0.67 |
| -OCH3 | −0.48 | 0.27 | −0.60 | 0.42 | −0.80 | 0.66 |
| -CH3 | −0.47 | 0.27 | −0.59 | 0.42 | −0.79 | 0.66 |
| -H | −0.48 | 0.27 | −0.59 | 0.42 | −0.79 | 0.66 |
| -F | −0.45 | 0.25 | −0.57 | 0.40 | −0.77 | 0.64 |
| -Cl | −0.44 | 0.23 | −0.56 | 0.38 | −0.76 | 0.63 |
| -OCOCH3 | −0.42 | 0.22 | −0.54 | 0.36 | −0.74 | 0.62 |
| -CF3 | −0.42 | 0.22 | −0.55 | 0.38 | −0.75 | 0.63 |
| -CN | −0.39 | 0.21 | −0.53 | 0.36 | −0.74 | 0.61 |
| -NO2 | −0.36 | 0.20 | −0.49 | 0.34 | −0.71 | 0.58 |
| ΔEint(TPSS-D3) | ΔEint(TPSS) | % Dispersion | |
|---|---|---|---|
| (NH2)m-m-p | −97.3 | −71.0 | 37.2 |
| (NH2)o-o-p | −96.3 | −68.7 | 40.2 |
| (NH2)m-o-o | −95.3 | −67.0 | 42.4 |
| (NH2)m-m-o | −94.5 | −67.3 | 40.5 |
| (NH2)m-m | −93.0 | −68.7 | 35.3 |
| (NH2)o-p | −92.9 | −68.6 | 35.4 |
| (NH2)m-p | −91.8 | −68.1 | 34.8 |
| (NH2)o-o | −90.7 | −64.7 | 40.2 |
| (NH2)m-o | −87.9 | −62.2 | 41.3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miranda-Rojas, S.; Mendizabal, F. Exploration of the Interaction Strength at the Interface of Anionic Chalcogen Anchors and Gold (111)-Based Nanomaterials. Nanomaterials 2020, 10, 1237. https://doi.org/10.3390/nano10061237
Miranda-Rojas S, Mendizabal F. Exploration of the Interaction Strength at the Interface of Anionic Chalcogen Anchors and Gold (111)-Based Nanomaterials. Nanomaterials. 2020; 10(6):1237. https://doi.org/10.3390/nano10061237
Chicago/Turabian StyleMiranda-Rojas, Sebastián, and Fernando Mendizabal. 2020. "Exploration of the Interaction Strength at the Interface of Anionic Chalcogen Anchors and Gold (111)-Based Nanomaterials" Nanomaterials 10, no. 6: 1237. https://doi.org/10.3390/nano10061237