Comparative Study of Different Acidic Surface Structures in Solid Catalysts Applied for the Isobutene Dimerization Reaction

 and
and
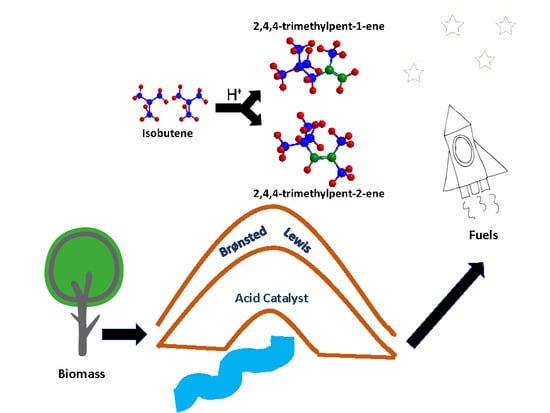
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Catalysts Preparation
2.2. Supports and Catalysts Characterization
2.3. Catalytic Evaluation in Gas Phase Isobutene Dimerization
3. Results
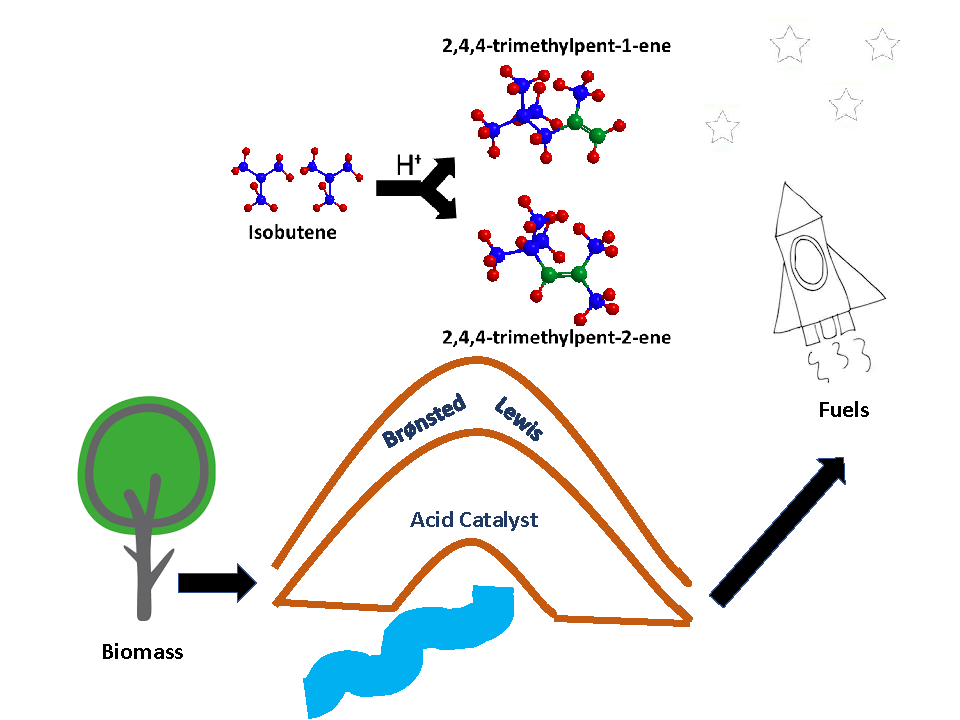
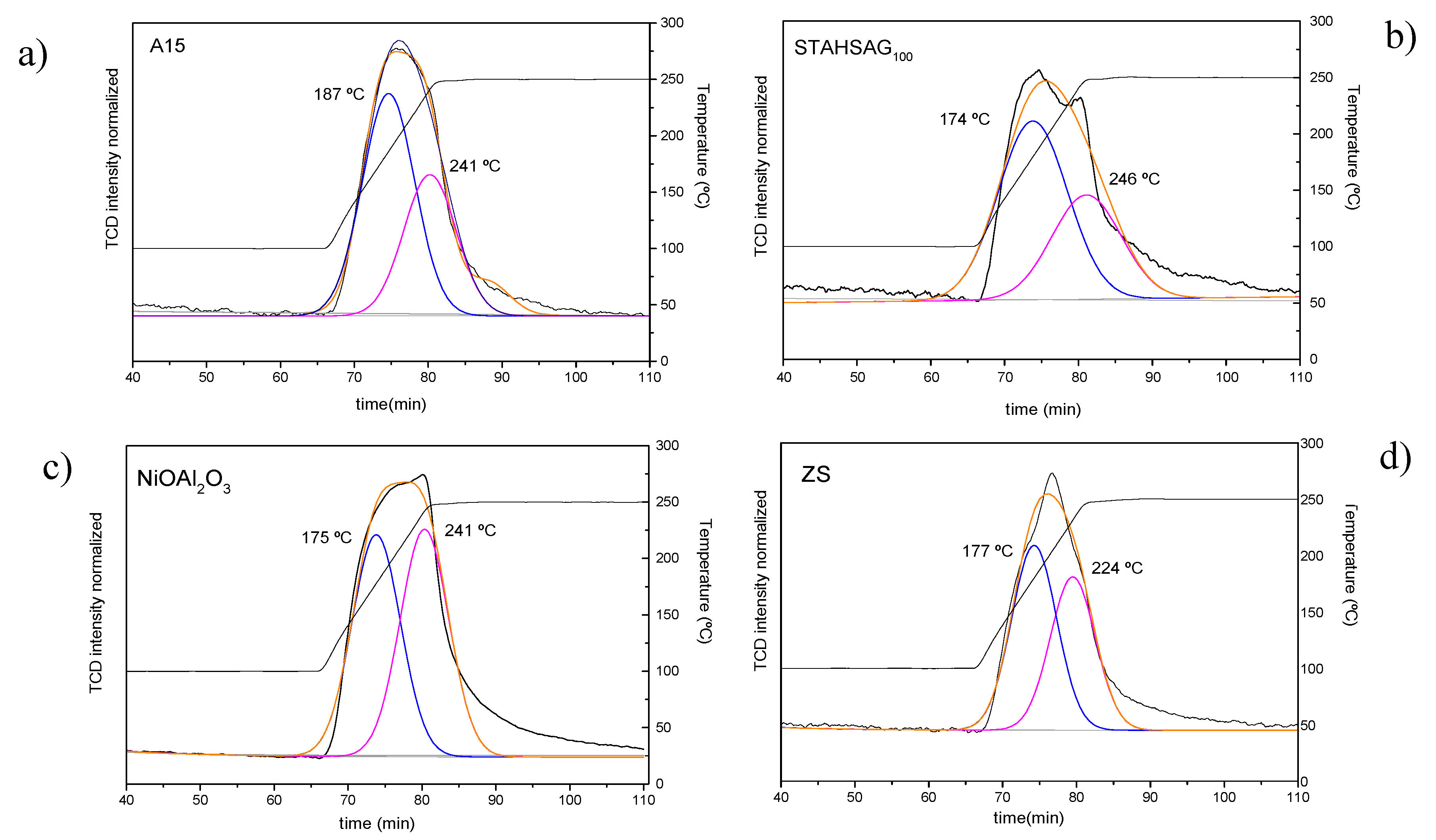
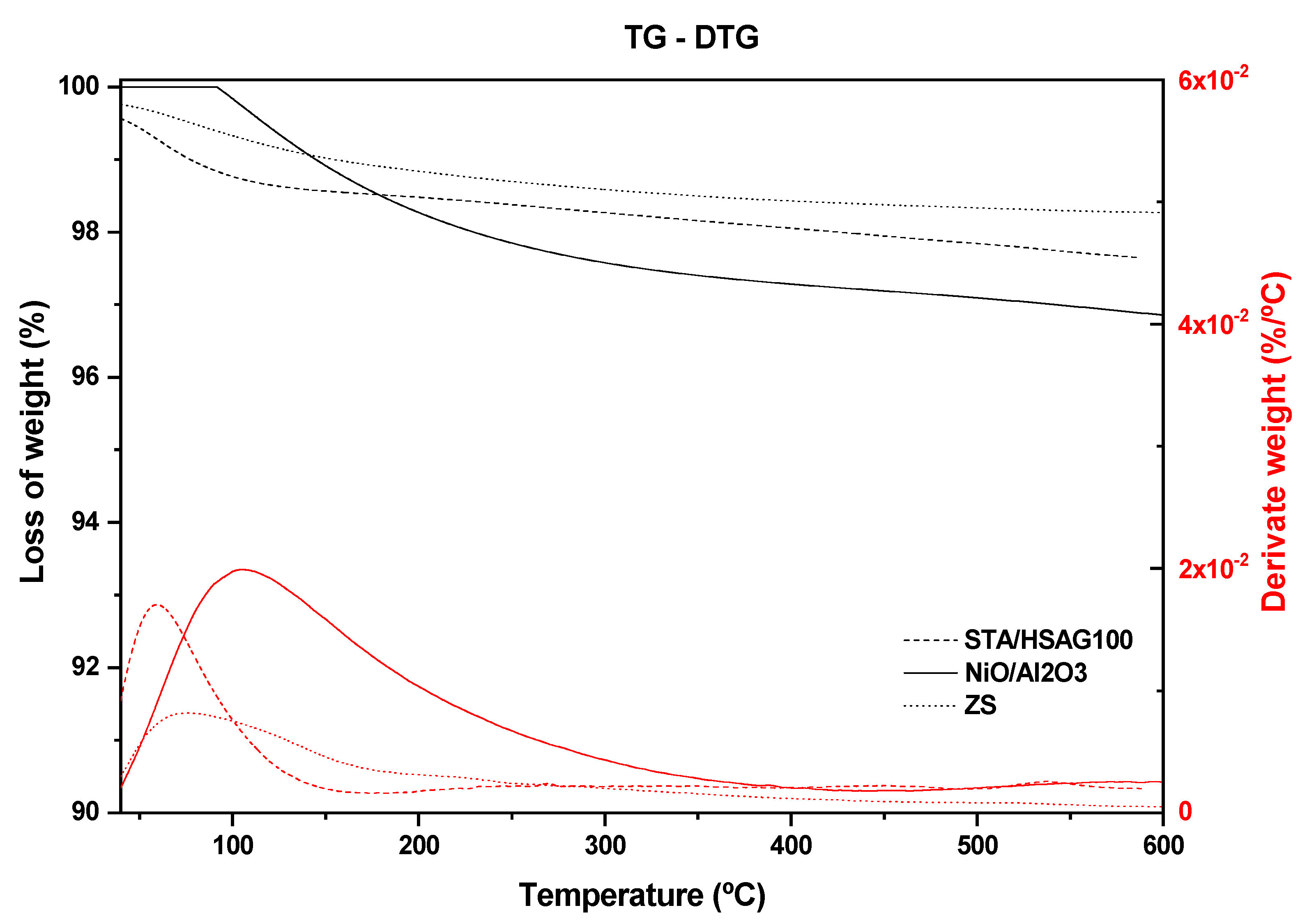
3.1. Catalysts Characterization
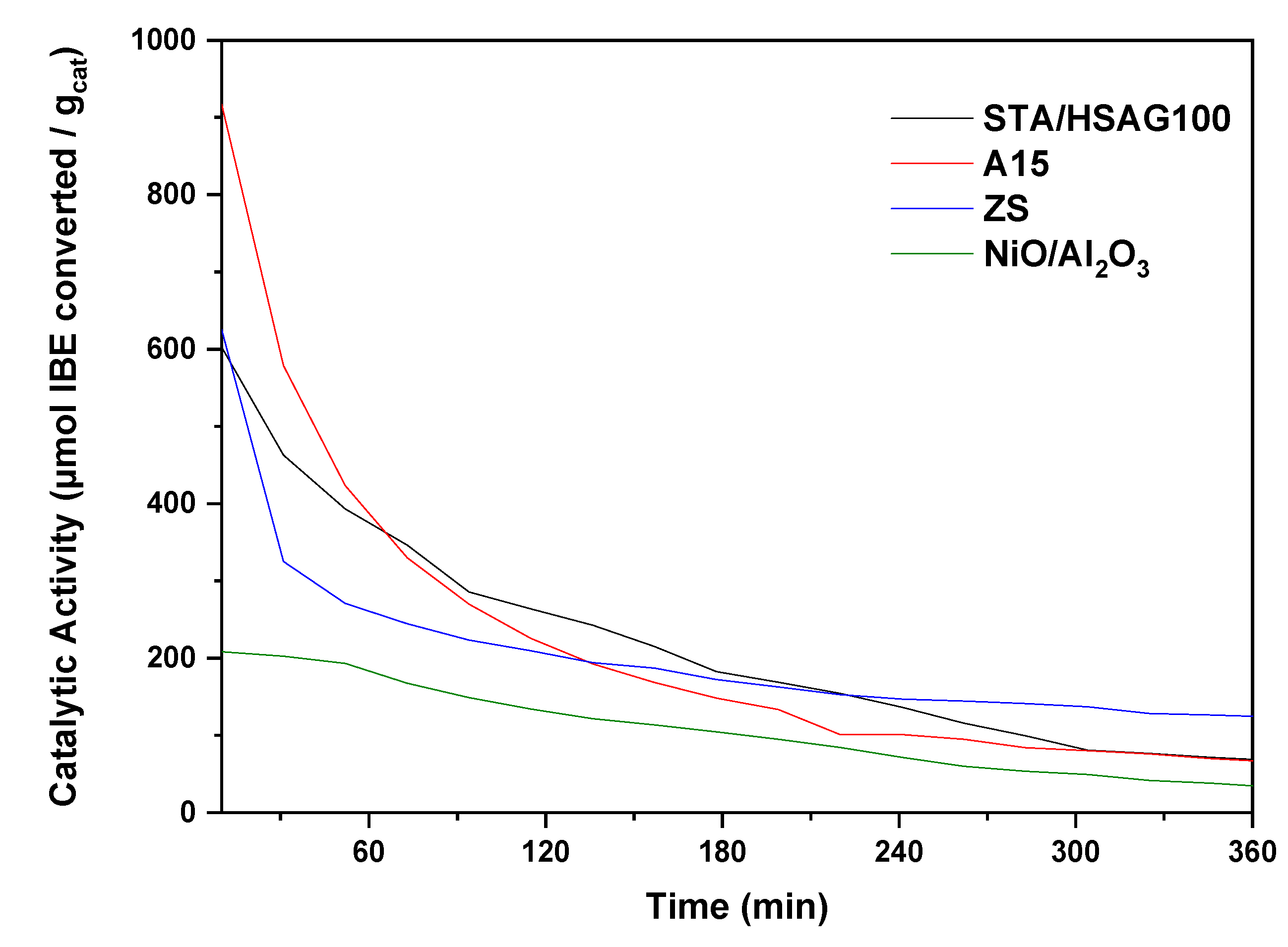
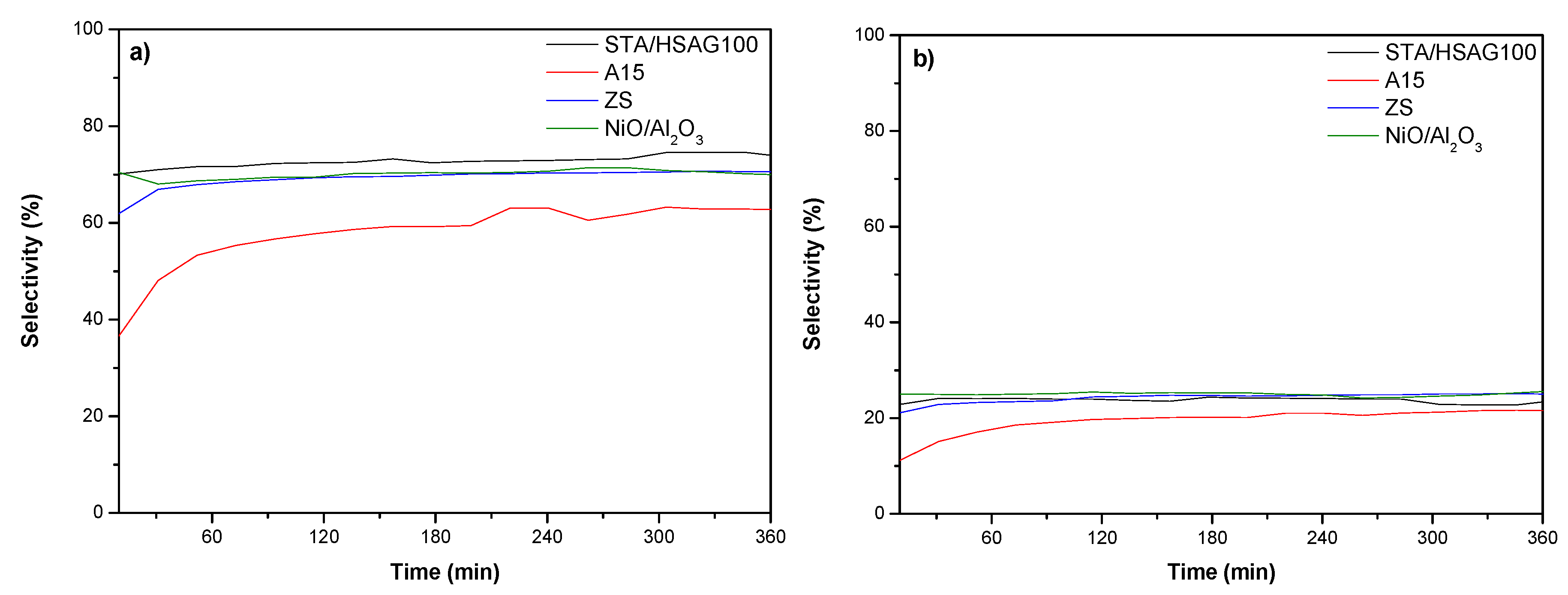
3.2. Catalytic Results
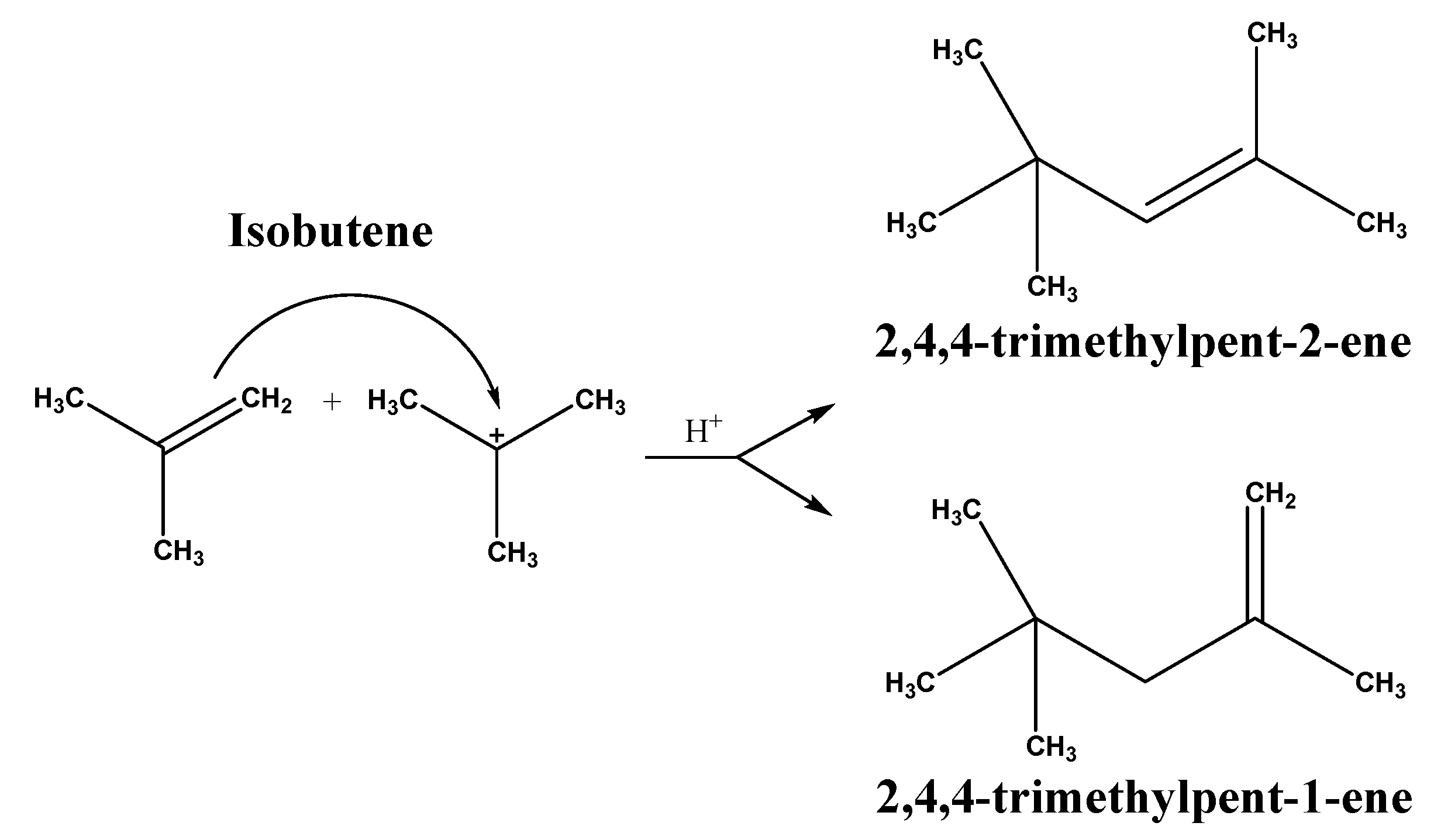
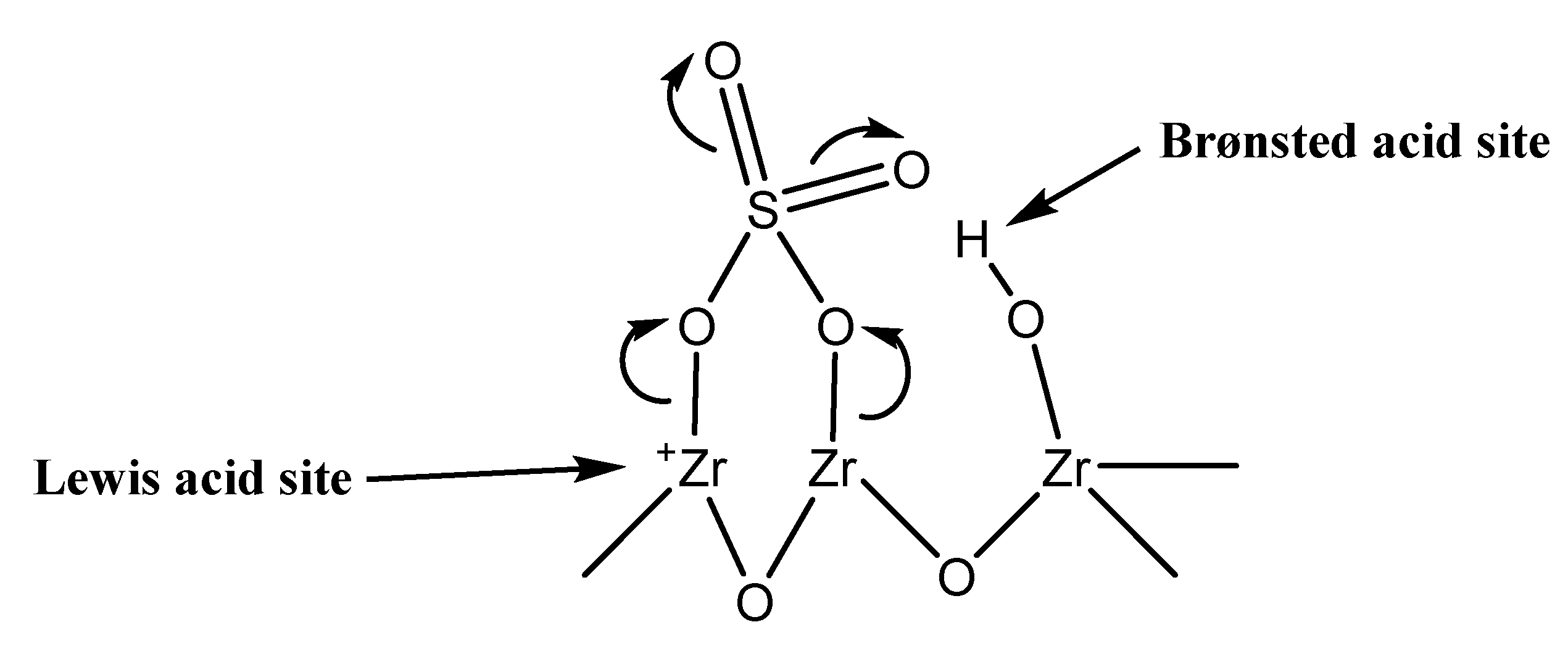
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Oil and Petroleum Products—A Statistical Overview—Statistics Explained. Available online: https://ec.europa.eu/eurostat/statistics-explained/index.php?title=Oil_and_petroleum_products_-_a_statistical_overview (accessed on 2 February 2020).
- Archive:Consumption of Energy—Statistics Explained. Available online: https://ec.europa.eu/eurostat/statistics-explained/index.php?title=Archive:Consumption_of_energy (accessed on 2 February 2020).
- Sarathy, S.M.; Farooq, A.; Kalghatgi, G.T. Recent progress in gasoline surrogate fuels. Prog. Energy Combust. Sci. 2018, 65, 67–108. [Google Scholar] [CrossRef]
- Mortensen, P.M.; Grunwaldt, J.D.; Jensen, P.A.; Knudsen, K.G.; Jensen, A.D. A review of catalytic upgrading of bio-oil to engine fuels. Appl. Catal. A Gen. 2011, 407, 1–19. [Google Scholar] [CrossRef]
- Dagle, V.L.; Smith, C.; Flake, M.; Albrecht, K.O.; Gray, M.J.; Ramasamy, K.K.; Dagle, R.A. Integrated process for the catalytic conversion of biomass-derived syngas into transportation fuels. Green Chem. 2016, 18, 1880–1891. [Google Scholar] [CrossRef]
- Antunes, B.M.; Rodrigues, A.E.; Lin, Z.; Portugal, I.; Silva, C.M. Alkenes oligomerization with resin catalysts. Fuel Process. Technol. 2015, 138, 86–99. [Google Scholar] [CrossRef]
- Marchionna, M.; Di Girolamo, M.; Patrini, R. Light olefins dimerization to high quality gasoline components. Catal. Today 2001, 65, 397–403. [Google Scholar] [CrossRef]
- Rorrer, J.E.; Toste, F.D.; Bell, A.T. Mechanism and Kinetics of Isobutene Formation from Ethanol and Acetone over Zn xZr yO z. ACS Catal. 2019, 9, 10588–10604. [Google Scholar] [CrossRef]
- Hidalgo, J.M.; Zbuzek, M.; Černý, R.; Jíša, P. Current uses and trends in catalytic isomerization, alkylation and etherification processes to improve gasoline quality. Cent. Eur. J. Chem. 2014, 12, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Scurto, A.M.; Subramaniam, B. Improved 1-butene/isobutane alkylation with acidic ionic liquids and tunable acid/ionic liquid mixtures. J. Catal. 2009, 268, 243–250. [Google Scholar] [CrossRef]
- Di Girolamo, M.; Marchionna, M. Acidic and basic ion exchange resins for industrial applications. J. Mol. Catal. 2001, 177, 33–40. [Google Scholar] [CrossRef]
- Liu, J.; Ge, Y.; Song, Y.; Du, M.; Zhou, X.; Wang, J.A. Dimerization of iso-butene on sodium exchanged Amberlyst-15 resins. Catal. Commun. 2019, 119, 57–61. [Google Scholar] [CrossRef]
- Alcántara, R.; Alcántara, E.; Canoira, L.; Franco, M.J.; Herrera, M.; Navarro, A. Trimerization of isobutene over Amberlyst-15 catalyst. React. Funct. Polym. 2000, 45, 19–27. [Google Scholar] [CrossRef]
- Mantilla, A.; Tzompantzi, F.; Ferrat, G.; López-Ortega, A.; Alfaro, S.; Gómez, R.; Torres, M. Oligomerization of isobutene on sulfated titania: Effect of reaction conditions on selectivity. Catal. Today 2005, 107–108, 707–712. [Google Scholar] [CrossRef]
- Padilla, J.M.; Del Angel, G.; Tzompantzi, F.J.; Manrı, M.E. One pot preparation of NiO/ZrO2 sulfated catalysts and its evaluation for the isobutene oligomerization. Catal. Today 2008, 135, 154–159. [Google Scholar] [CrossRef]
- Mantilla, A.; Ferrat, G.; López-Ortega, A.; Romero, E.; Tzompantzi, F.; Torres, M.; Ortíz-Islas, E.; Gómez, R. Catalytic behavior of sulfated TiO2in light olefins oligomerization. J. Mol. Catal. Chem. 2005, 228, 333–338. [Google Scholar] [CrossRef]
- Davis, R.J. The Role of Transition Metal Promoters on Sulfated Zirconia Catalysts for Low-Temperature Butane Isomerization. J. Catal. 1996, 133, 125–133. [Google Scholar]
- Querini, C.A.; Roa, E. Deactivation of solid acid catalysts during isobutane alkylation with C4 olefins. Appl. Catal. A Gen. 1997, 163, 199–215. [Google Scholar] [CrossRef]
- Chen, G.; Li, J.; Yang, X.; Wu, Y. Surface-appropriate lipophobicity-Application in isobutene oligomerization over Teflon-modified silica-supported 12-silicotungstic acid. Appl. Catal. A Gen. 2006, 310, 16–23. [Google Scholar] [CrossRef]
- Corma, A. Solid acid catalysts. Curr. Opin. Solid State Mater. Sci. 1997, 2, 63–75. [Google Scholar] [CrossRef]
- Lefebvre, F. Acid Catalysis by Heteropolyacids: Transformations of Alkanes. Curr. Catal. 2017, 6, 77–89. [Google Scholar] [CrossRef]
- Yoon, J.W.; Jhung, S.H.; Choo, D.H.; Lee, S.J.; Lee, K.Y.; Chang, J.S. Oligomerization of isobutene over dealuminated Y zeolite catalysts. Appl. Catal. A Gen. 2008, 337, 73–77. [Google Scholar] [CrossRef]
- Yoon, J.W.; Chang, J.S.; Lee, H.D.; Kim, T.J.; Jhung, S.H. Trimerization of isobutene over a zeolite beta catalyst. J. Catal. 2007, 245, 253–256. [Google Scholar] [CrossRef]
- Dalla Costa, B.O.; Querini, C.A. Isobutane alkylation with solid catalysts based on beta zeolite. Appl. Catal. A Gen. 2010, 385, 144–152. [Google Scholar] [CrossRef]
- Corma, A.; Martinez, A.; Martinez, C. Isobutane/2-butene alkylation on MCM-22 catalyst. Influence of zeolite structure and acidity on activity and selectivity. Catal. Lett. 1994, 28, 187–201. [Google Scholar] [CrossRef]
- Corma, A.; Martínez, A. Chemistry, catalysts, and processes for isoparaffin-olefin alkylation: Actual situation and future trends. Catal. Rev. 1993, 35, 483–570. [Google Scholar] [CrossRef]
- Fernández-Morales, J.M.; Lozano, L.A.; Castillejos-López, E.; Rodríguez-Ramos, I.; Guerrero-Ruiz, A.; Zamaro, J.M. Direct sulfation of a Zr-based metal-organic framework to attain strong acid catalysts. Microporous Mesoporous Mater. 2019, 290, 109686. [Google Scholar] [CrossRef]
- Trickett, C.A.; Osborn Popp, T.M.; Su, J.; Yan, C.; Weisberg, J.; Huq, A.; Urban, P.; Jiang, J.; Kalmutzki, M.J.; Liu, Q.; et al. Identification of the strong Brønsted acid site in a metal–organic framework solid acid catalyst. Nat. Chem. 2019, 11, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Ehrmaier, A.; Liu, Y.; Peitz, S.; Jentys, A.; Chin, Y.H.C.; Sanchez-Sanchez, M.; Bermejo-Deval, R.; Lercher, J. Dimerization of Linear Butenes on Zeolite-Supported Ni 2+. ACS Catal. 2019, 9, 315–324. [Google Scholar] [CrossRef]
- Ehrmaier, A.; Peitz, S.; Sanchez-Sanchez, M.; Bermejo-Deval, R.; Lercher, J. On the role of co-cations in nickel exchanged LTA zeolite for butene dimerization. Microporous Mesoporous Mater. 2019, 284, 241–246. [Google Scholar] [CrossRef]
- Comito, R.J.; Metzger, E.D.; Wu, Z.; Zhang, G.; Hendon, C.H.; Miller, J.T.; Dincă, M. Selective Dimerization of Propylene with Ni-MFU-4l. Organometallics 2017, 36, 1681–1683. [Google Scholar] [CrossRef] [Green Version]
- Almohalla, M.; Rodríguez-Ramos, I.; Guerrero-Ruiz, A. Comparative study of three heteropolyacids supported on carbon materials as catalysts for ethylene production from bioethanol. Catal. Sci. Technol. 2017, 7, 1892–1901. [Google Scholar] [CrossRef]
- Álvarez-Rodríguez, J.; Rodríguez-Ramos, I.; Guerrero-Ruiz, A.; Gallegos-Suarez, E.; Arcoya, A. Influence of the nature of support on Ru-supported catalysts for selective hydrogenation of citral. Chem. Eng. J. 2012, 204–205, 169–178. [Google Scholar] [CrossRef]
- Lónyi, F.; Valyon, J. On the interpretation of the NH 3 -TPD patterns of. Microporous Mesoporous Mater. 2001, 47, 293–301. [Google Scholar] [CrossRef]
- Wijaya, Y.P.; Winoto, H.P.; Park, Y.K.; Suh, D.J.; Lee, H.; Ha, J.M.; Jae, J. Heteropolyacid catalysts for Diels-Alder cycloaddition of 2,5-dimethylfuran and ethylene to renewable p-xylene. Catal. Today 2017, 293–294, 167–175. [Google Scholar] [CrossRef]
- Li, N.; Wang, A.; Zheng, M.; Wang, X.; Cheng, R.; Zhang, T. Probing into the catalytic nature of Co/sulfated zirconia for selective reduction of NO with methane. J. Catal. 2004, 225, 307–315. [Google Scholar] [CrossRef]
- Charisiou, N.D.; Iordanidis, A.; Polychronopoulou, K.; Yentekakis, I.V.; Goula, M.A. Studying the stability of Ni supported on modified with CeO2 alumina catalysts for the biogas dry reforming reaction. Mater. Today Proc. 2018, 5, 27607–27616. [Google Scholar] [CrossRef]
- Wang, A.; Arora, P.; Bernin, D.; Kumar, A.; Kamasamudram, K.; Olsson, L. Investigation of the robust hydrothermal stability of Cu/LTA for NH3-SCR reaction. Appl. Catal. B Environ. 2019, 246, 242–253. [Google Scholar] [CrossRef]
- Katada, N.; Tsubaki, T.; Niwa, M. Measurements of number and strength distribution of Brønsted and Lewis acid sites on sulfated zirconia by ammonia IRMS-TPD method. Appl. Catal. A Gen. 2008, 340, 76–86. [Google Scholar] [CrossRef]
- Saboya, R.M.A.; Cecilia, J.A.; García-Sancho, C.; Sales, A.V.; de Luna, F.M.T.; Rodríguez-Castellón, E.; Cavalcante, C.L. Assessment of commercial resins in the biolubricants production from free fatty acids of castor oil. Catal. Today 2017, 279, 274–285. [Google Scholar] [CrossRef]
- Fan, G.; Liao, C.; Fang, T.; Luo, S.; Song, G. Amberlyst 15 as a new and reusable catalyst for the conversion of cellulose into cellulose acetate. Carbohydr. Polym. 2014, 112, 203–209. [Google Scholar] [CrossRef]
- Ouni, T.; Honkela, M.; Kolah, A.; Aittamaa, J. Isobutene dimerisation in a miniplant-scale reactor. Chem. Eng. Process. Process Intensif. 2006, 45, 329–339. [Google Scholar] [CrossRef]
- Goortani, B.M.; Gaurav, A.; Deshpande, A.; Ng, F.T.T.; Rempel, G.L. Production of isooctane from isobutene: Energy integration and carbon dioxide abatement via catalytic distillation. Ind. Eng. Chem. Res. 2015, 54, 3570–3581. [Google Scholar] [CrossRef]
- Saus, A.; Schmidl, E. Benzyl sulfonic acid siloxane as a catalyst: Oligomerization of isobutene. J. Catal. 1985, 94, 187–194. [Google Scholar] [CrossRef]
- Corma, A.; Ortega, F.J. Influence of adsorption parameters on catalytic cracking and catalyst decay. J. Catal. 2005, 233, 257–265. [Google Scholar] [CrossRef]
- García Bosch, N.; Bachiller-Baeza, B.; Rodriguez-Ramos, I.; Guerrero-Ruiz, A. Fructose transformations in ethanol using carbon supported polyoxometalate acidic solids for 5-Etoxymethylfurfural production. ChemCatChem 2018. [Google Scholar] [CrossRef]
- Bringué, R.; Ramírez, E.; Iborra, M.; Tejero, J.; Cunill, F. Influence of acid ion-exchange resins morphology in a swollen state on the synthesis of ethyl octyl ether from ethanol and 1-octanol. J. Catal. 2013, 304, 7–21. [Google Scholar] [CrossRef] [Green Version]
- Yan, G.X.; Wang, A.; Wachs, I.E.; Baltrusaitis, J. Critical review on the active site structure of sulfated zirconia catalysts and prospects in fuel production. Appl. Catal. A Gen. 2019, 572, 210–225. [Google Scholar] [CrossRef]
- El-Dafrawy, S.M.; Hassan, S.M.; Farag, M. Kinetics and mechanism of Pechmann condensation reaction over sulphated zirconia-supported zinc oxide. J. Mater. Res. Technol. 2020, 9, 13–21. [Google Scholar] [CrossRef]
- Liu, N.; Ma, Z.; Wang, S.; Shi, L.; Hu, X.; Meng, X. Palladium-doped sulfated zirconia: Deactivation behavior in isomerization of n-hexane. Fuel 2020, 262, 116566. [Google Scholar] [CrossRef]
- Ecormier, M.A.; Wilson, K.; Lee, A.F. Structure-reactivity correlations in sulphated-zirconia catalysts for the isomerisation of α-pinene. J. Catal. 2003, 215, 57–65. [Google Scholar] [CrossRef]
- Knözinger, H.; Ratnasamy, P. Catalysis Reviews: Science and Engineering Catalytic Aluminas: Surface Models and Characterization of surface sites. Catal. Rev. 1978, 17, 31–70. [Google Scholar] [CrossRef]
- Sarkar, A.; Seth, D. Active Sites of a NiSO4/γ-Al2O3 Catalyst for the Oligomerization of Active Sites of a NiSO 4 / c -Al 2 O 3 Catalyst for the Oligomerization of Isobutene. Top. Catal. 2014. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | Weight Loss |
|---|---|
| A15 | 41.3% |
| STA/HSAG100 | 2.4% |
| ZS | 1.7% |
| NiO/Al2O3 | 3.2% |
| Catalysts | Conv0 | Conv100 | Conv300 |
|---|---|---|---|
| A15 | 49 | 17 | 5 |
| STA/HSAG100 | 36 | 14 | 5 |
| ZS | 17 | 6 | 4 |
| NiO/Al2O3 | 7 | 5 | 2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Morales, J.M.; Castillejos, E.; Asedegbega-Nieto, E.; Dongil, A.B.; Rodríguez-Ramos, I.; Guerrero-Ruiz, A. Comparative Study of Different Acidic Surface Structures in Solid Catalysts Applied for the Isobutene Dimerization Reaction. Nanomaterials 2020, 10, 1235. https://doi.org/10.3390/nano10061235
Fernández-Morales JM, Castillejos E, Asedegbega-Nieto E, Dongil AB, Rodríguez-Ramos I, Guerrero-Ruiz A. Comparative Study of Different Acidic Surface Structures in Solid Catalysts Applied for the Isobutene Dimerization Reaction. Nanomaterials. 2020; 10(6):1235. https://doi.org/10.3390/nano10061235
Chicago/Turabian StyleFernández-Morales, José M., Eva Castillejos, Esther Asedegbega-Nieto, Ana Belén Dongil, Inmaculada Rodríguez-Ramos, and Antonio Guerrero-Ruiz. 2020. "Comparative Study of Different Acidic Surface Structures in Solid Catalysts Applied for the Isobutene Dimerization Reaction" Nanomaterials 10, no. 6: 1235. https://doi.org/10.3390/nano10061235