3.1. Cisplatin Adsorption
Recently, we have found [
19] that ovalene is a very suitable PAH in terms of binding energy convergence and related computational cost in order to study the noncovalent interactions between CP and a graphic plane and that the PBE-D3 (BJ) level of theory is capable of providing reliable estimations of the related binding energies.
The first step in the present work has been to validate the most adequate DFT level for the adsorption of CP on the GO prototypes containing the epoxy and hydroxy groups. To do that we have performed DFT optimizations at the PBE-D3 (BJ) level of the CP–C
H
O and CP–C
H
OH complexes whose most stable structures are reported in
Figure 2 together with that previously obtained [
19] for the CP–C
H
cluster. The corresponding interaction energies are reported in
Table 1 and they have been computed also at the B3LYP-D3 (BJ) and M062X levels, which are generally considered [
34,
57] as recommended DFT approaches to study noncovalently bonded intermolecular systems. From the comparison with the MP2C/CBS results, which can be considered here as the reference ones, it can be seen that for all cases the PBE-D3 (BJ) and M062X approaches globally provide an underestimation of the interaction energy, while the opposite is observed for the B3LYP-D3 (BJ) predictions, which lead indeed to larger discrepancies of about 150 meV. More in details, the PBE-D3 (BJ) results provide a better agreement (within 25 meV) for the interaction with the aromatic region GO prototype, which is consistent with our previous findings reported in Ref. [
19], while those carried out at the M062X level better describe (within 20–25 meV) that with the other GO supports. In any case, the differences found for the PBE-D3 (BJ) estimations are globally below 50 meV (and around 4%) for all prototypes and, considering its lower computational cost over the M062X approach, we have decided to use it to perform the geoemetry optimization for the complexes involving larger GO prototypes, both in the gas phase and including an implicit solvent model. Moreover, this choice is motivated by the fact that the PBE-D3 (BJ) functional is capable to predict accurate thermodynamic properties for the cisplatin first hydrolysis reaction, as will be discussed in the next section.
As a result, in
Table 2 we, therefore, present the interaction energy obtained for the adsorption of CP on GO molecular prototypes of increasing size. For circumcoronene and its epoxy(hydroxy) derivatives the reported energies correspond to the full optimization of the complex geometries. However, mostly motivated by the need to make the DFT computations affordable, in the case of circumcircumcoronene-based supports we have just retained the same intermolecular configurations found for the smaller (circumcoronene-based) GO platforms, considering also that significant changes in the CP adsorption geometry should not be expected. As a matter of fact, the relative geometries of the CP–C
H
and CP–C
H
O(C
H
OH) complexes are very similar to those reported in
Figure 2 and are not reported here for the sake of simplicity. CP–C
H
O and CP–C
H
OH geometries can be found in
Figure S2 of the Supporting Information.
It can be seen that the interaction energy increases as the size of the GO prototypes become larger: in the case of functionalized sites, a more attractive binding of about 9 and 7% for the epoxy and hydroxy supports, respectively, is observed when going from C
H
to C
H
, while for the largest prototypes (last row of
Table 2) the corresponding improvement is of just about 2.4 and 1%. As for the aromatic region substrate, the gain in the interaction energy is similar and additional contributions of about 7 and 3 % are found.
These results suggest that the binding energy is well converged for the circumcircumcoronene-based adsorbing platforms and that it is likely that larger prototypes would lead to a slightly larger interaction of less than 1%, as already shown in Ref. [
19] (see
Table 2 therein) for graphene prototypes of increasing size.
In order to further assess the reliability of the considered GO molecular models we have considered four additional prototypes based on CH: Two of them are characterized by the functional group attached on the most central carbon ring; the other two also consider two different oxygen groups attached to the carbon basal plane and close to each other.
The optimized structures of the complexes between CP and these additional prototypes, which are also meant to take into account adsorption sites on GO with a high density of oxidized groups, are shown in
Figure S1 of the Supporting Information. The corresponding total interaction energies are reported in
Table S1 of the Supporting Information and they are close to those reported in the second row of
Table 2 and related to the mono-(and “decentred”-)functionalized prototypes based on C
H
. These findings demonstrate that the physisorption of cisplatin does not significantly depend on the location of the oxidized group (as long as it is far from the plane edges). Moreover, they also show that the drug interaction with two contiguous oxygen groups leads to a negligible improvement of its adsorption energy with respect to that arising with a single oxygen group.
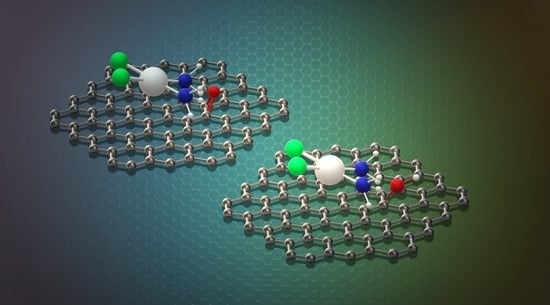
A careful look at
Figure 2 suggests that the nature of the binding for the CP adsorption on the considered GO prototypes must be different: even if in all cases the -NH
ends of CP lean toward the carbon plane, with the Pt atom separated from it of about 3.4 Å, in the case of the the aromatic region the hydrogen atoms tend to interact with the
cloud, whereas for functionalized sites they are oriented in the direction of the epoxy and hydroxy oxygens.
In order to get insight into the nature of binding, an analysis of the energy contributions to the chemical interaction has been performed using the EDA approach [
51,
52], which is an alternative [
58] to the canonical DFT-SAPT [
59] method, when the use of the latter is not computationally convenient. The agreement between corresponding energy contributions as predicted by the EDA and SAPT-based methods are in general qualitatively good since both approaches characterize the noncovalent character of the chemical binding in a very similar way [
58]. In order to validate this assumption, an EDA analysis has been first carried out for the smaller CP-Pyrene (CP-P) complex and the results have been compared with those previously reported at DFT-SAPT level [
19], finding that the partial contributions are qualitatively similar around equilibrium distances, with the dispersion plus induction (or orbital) energies being the main contribution to the total interaction energy (see
Table S2 in the Supporting Information for more details).
These results allowed us to safely apply the EDA approach to the total interaction energy found for CP adsorbed on larger prototypes. In particular, in
Table 3 we report the partial contributions related to the optimized geometries of the CP–C
H
complexes, which can be considered sufficiently large and representative of the overall interaction acting between CP and GO. Significant differences can be indeed seen between the aromatic and the oxidized prototypes: the total interaction energy of CP adsorbed on the aromatic prototype is about −900 meV while for that for the epoxy and hydroxy functionalized supports become larger of about 50% in both cases.
These differences can be related to the specific role of the different energy contributions. More in details, it can be appreciated that the main attractive contribution to the total interaction energy has changed in the case of the functionalized prototypes: while the dispersion energy term remains almost unperturbed, the electrostatic contribution becomes dominant since it displays an increase of 119% and 155% for the epoxy and hydroxy supports, respectively. Regarding the Pauli repulsive contribution, it rises of about 50% and 73% for epoxy and hydroxy prototypes, respectively, but this increase is mostly compensated in both cases by the enlargment of the orbital attractive term.
Therefore, while the dominant attractive contribution for the CP-aromatic prototype complex is the dispersion interaction, which only shows a marginal increase in epoxy and hydroxy species, for the oxidized prototypes the electrostatic contribution is the most important one and sums up leading to significantly larger total binding energies. This enhanced electrostatic contribution can be related to the hydrogen bond-like interaction arising between the -NH
ends of CP and the oxygen groups on the carbon supports. In order to confirm such hypothesis calculations on intermolecular prototypes, such as NH
-H
O, NH
-OHCH
and NH
-O( CH
)
(see also
Figure S2 in the Supporting Information), representing archetypal hydrogen bonds have been carried out. Similar EDA results as for the CP-oxidized prototypes interaction have been obtained and the greatest contribution to the total interaction energy corresponds to the electrostatic term (see
Table S3 in the Supporting Information). In addition, for these archetypal systems, the O–H distance (between the hydrogen of the ammonia and oxygen of the hydroxy and epoxy groups) is around 2 Å while the N–H–O angle is about 160°. Such values are very similar to those obtained for the CP-GO prototypes complexes for which we find distances of about 2.0 and 1.95 Å and corresponding angles of about 161° and 159° for the epoxy and hydroxy prototypes, respectively (see
Figure S2 in the Supporting Information). Therefore, it can be concluded that the enhancement of CP physisorption on GO oxidized prototypes is mainly due to the formation of hydrogen bonds in which hydrogens of the CP ammonia groups interact with the oxygen of the hydroxy and epoxy groups.
To better assess the viability of the CP adsorption in a more realistic medium such as an aqueous solution we have performed geometry optimizations by means of the SMD continuum solvation model, whose results are shown in
Figure 3 together with the corresponding gas-phase estimations: it can be appreciated that both models provide quite similar configurations although a small increase in the angle between the CP and the GO prototype can be appreciated for the SMD results, which leads to a slightly larger distance (of about 0.15 Å) between the Pt and the aromatic C plane. This suggests that the nature of the bonding should not significantly change in the gas phase and water solution. In
Table 4 we finally report present estimations for the adsorption enthalpy (
H
) and free energy (
G
), both in gas phase and aqueous solution, computed at 298.15 K and 1 bar for CP physically sorbed on GO and on rGO. For the adsorption on GO we have chosen the results obtained for the circumcoronene-based supports. The predicted values for rGO have been obtained from the circumcoronene results by scaling the adsorption enthalpies using the interaction energies computed for the related C
H
and C
H
supports, which are reported in
Table 2. It can be seen that for all considered sites the CP adsorption on GO and rGO is a significantly exothermic and exoergonic process in both the gas phase and aqueous solution. As expected, the most favorable adsorption occurs in correspondence of the hydroxy group, followed by the expoxy and aromatic region sites. In particular,
H
and
G
values related to the aqueous solution CP adsorption in correspondence of the oxidized sites are significantly larger (between 10 and 30% and between 15 and 45%, respectively) than those for the aromatic region, confirming the fundamental role of hydrogen bonding contributions also in the condensed phase. An indication of such favorable enthalpy and free energy adsorption variations, though calculated at a much lower and only qualitative level of theory (B3LYP/3-21G (d)), also emerged in the case of GO-coated chitosan, due to hydrogen bonding between the cisplatin ammonia and GO epoxy and hydroxy groups [
22].
In order to take into account the possibility of the competition of the CP self-clusterization process, enthalpy and free energy values obtained for the CP dimerization are also reported in
Table 4. It can be seen that in water solution the free energy variation for CP dimerization is lower or comparable with respect to those corresponding to the CP adsorption on the hydroxy and epoxy sites, whereas it is slightly larger (around 40–60 meV) than that for the aromatic region case.
In conclusion, the above reported results suggest that, on the whole, the CP physical adsorption on GO and rGO is competitive with the drug self-aggregation and it can help to increase its solubility in water solutions, exploiting the high hydrophilicity of the peripheral carboxylic groups located on the edge of the GO and rGO platelets. Moreover, the higher affinity of CP adsorption in correspondence of the oxidized sites, due to active hydrogen bonding contributions, suggests the possible dependence of drug loading and release on pH. This would be a particularly valuable feature, because it can highly enhance the specificity of CP loaded nanocarriers for tumor cells. The hypoxic, acidic environment of cancerous tissue should indeed be able to induce drug release by substituting the weaker hydrogen bonds between -NH
groups and oxidized functions of (r)GO with stronger hydrogen bonds involving more acidic protonated groups. At physiological pH, on the other hand, CP molecules would remain adsorbed onto the surface, decreasing the possibility of undesired collateral reactions. Such a pH-dependent behavior was in fact found [
20] in the case of doxorubicin noncovalently bound to GO supports, which show maximum loading capacity in basic solutions and minimum in acidic environments.
3.2. Cisplatin Reactivity
In order to address the reactivity of the physically adsorbed CP we have studied its first hydrolysis reaction which is generally considered [
60] as the limiting step requiring the largest activation energy and leading to the monoaqua complex. The latter is, together with the diaqua analog, the reactive species responsible of the Pt covalent binding to DNA nucleobases and eventually of CP cytotoxicity.
First, we have tested the capacity of the present DFT approach (adopted in the previous section to study the cisplatin physical adsorption) to predict the drug activation and reaction energies. Cisplatin first hydrolysis proceeds through a S
2 substitution in which the nucleophile water molecule displaces the chloride ion leaving group (see
Figure 4) and involves a distorted trigonal-bipyramidal transition state (TS) [
61,
62,
63].
The enthalpy and free energy variations corresponding to the hydrolysis reaction and its activation for a free (unadsorbed) CP molecule are reported in
Table 5. It can be seen that the estimations obtained with the present PBE-D3 (BJ)/SMD model are close to the experimental findings in aqueous solution and they can be considered of comparable or even better accuracy with respect to previous theoretical predictions [
62,
64]: in fact, present predictions provide a better accord for the
H
and
G
values since they are able to reproduce both the slight endothermicity and endergonicity of the hydrolysis reaction.
Therefore, we believe that the present DFT approach is adequate to study the CP hydrolysis thermochemistry and we have also employed it to investigate the drug reactivity when it is physically adsorbed on GO and rGO platelets. To this aim, in order to make the optimization of the TS and products structures computationally affordable we have just considered the adsorption on the prototypes based on C
H
(see the first column of
Figure 1). The structures of the stationary points corresponding to the CP hydrolysis reactants, transition state and products, when adsorbed on the hydroxy site of a GO (or rGO) platelet are shown in
Figure 5 while the related enthalpy and free energy variations in aqueous solution are reported in
Table 6 together with those for the aromatic region and epoxy site.
It can be appreciated that negligible changes in the activation enthalpy and free energy are found with respect to the case of the free (unadsorbed) CP. In the case of the global reaction a slight lower endothermic and endergonic (of about 100 meV) process is found for the adsorbed drug. Therefore present findings indicate that the hydrolysis of CP is little affected by the condition of its physical adsorption on GO and rGO platelets. However, it must be pointed out that an effect of the reaction kinetics should be expected due to steric assumptions: in fact, when the CP is adsorbed, one of the two sides of the planar molecule is not available for the nucleophile approach which should, therefore, affect the reaction rate.

 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}