Effect of Domain Size, Boundary, and Loading Conditions on Mechanical Properties of Amorphous Silica: A Reactive Molecular Dynamics Study


Abstract
:1. Introduction
2. Numerical Approach
2.1. Reactive Molecular Dynamics Simulation
2.2. Development of a-SiO
2.3. Cases Studied and Computations
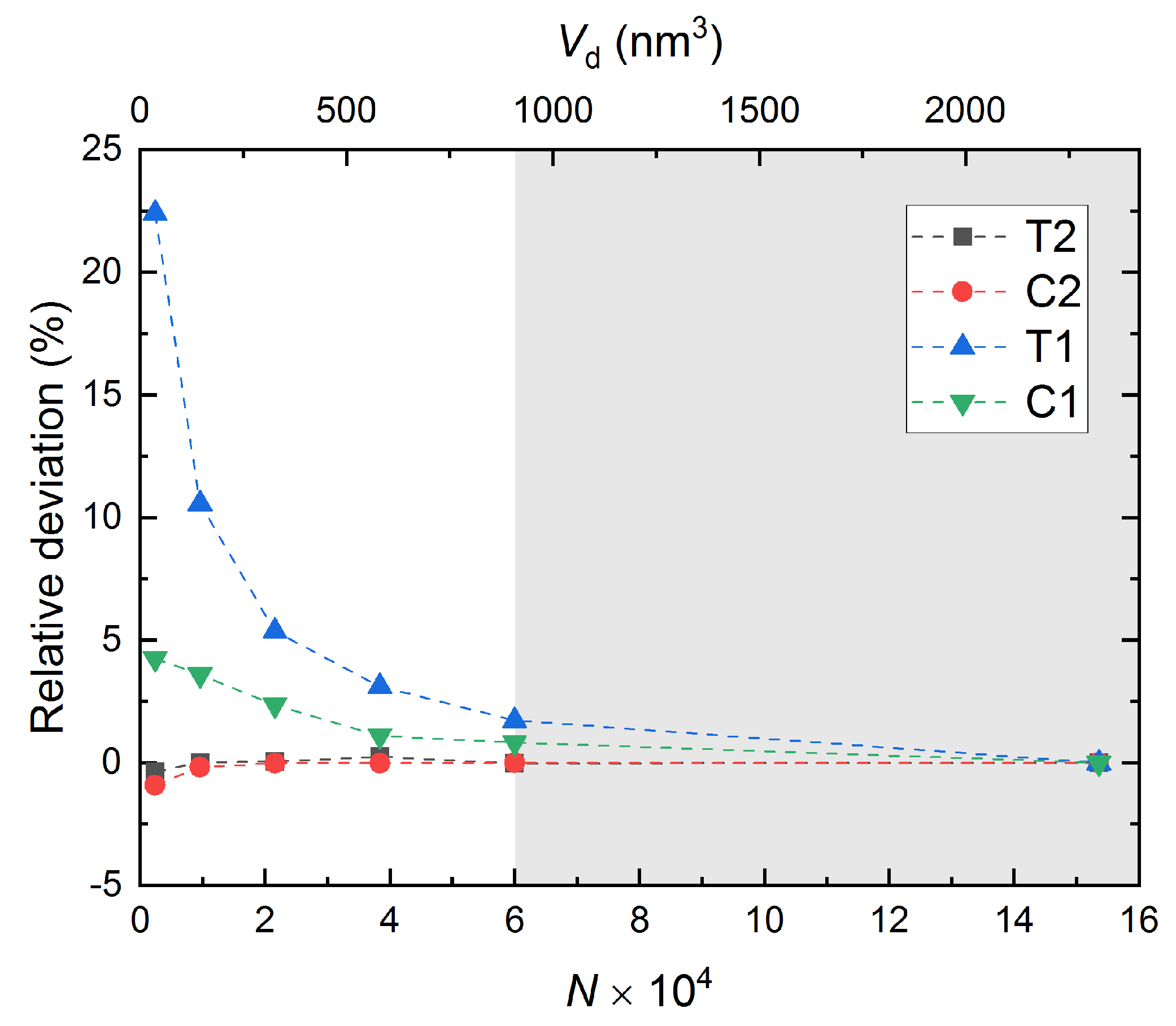
- T2—simulation domain is fully periodic and subjected to tensile loading on both sides in the x-direction, cf. Figure 2b.
- C2—simulation domain is fully periodic and subjected to compressive loading on both sides in the x-direction, cf. Figure 2c.
- T1—simulation domain is periodic in the y- and z-directions, while in the x-direction, the tensile loading is applied to the top surface as the bottom surface is fixed, cf. Figure 2d.
- C1—simulation domain is periodic in the y- and z-directions, while in the x-direction, the compressive loading is applied to the top surface as the bottom surface is fixed, cf. Figure 2e.
3. Results and Discussion
3.1. Global Stress-Strain Curve
3.2. Young’s Modulus and Bimodularity
3.3. Poisson’s Ratio and Isotropy
3.4. Distribution of Si-O Bond
3.5. Computational Cost and Accuracy
4. Conclusions
- Mechanical properties converge with increasing domain size.
- With the presence of free surfaces in semi-periodic cases, the impact of domain size is much more significant than full-periodic cases.
- Amorphous silica exhibits strong bimodular behavior and slight anisotropy at the atomic level. Young’s modulus in tension is higher than in compression, while Poisson’s ratio in x-y plane and x-z plane are slightly different from each other.
- A “safe zone” defined as a zone where accuracy and computational cost are balanced. Defining such a zone is necessary for multiscale models, as well as defining RVE at nanoscale. In this zone, bulk properties can be reproduced with good accuracy.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MD | Molecular dynamics |
| RMD | Reactive molecular dynamics |
| BCs | Boundary conditions |
| LCs | Loading conditions |
| RDF | Radial distribution function |
Appendix A. Simulation Data

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Simulation Set | N | (GPa) | ||
|---|---|---|---|---|
| T2 | 0.24 | 73.052 | 0.377 | 0.395 |
| 0.96 | 73.319 | 0.376 | 0.396 | |
| 2.16 | 73.359 | 0.377 | 0.394 | |
| 3.84 | 73.500 | 0.377 | 0.395 | |
| 6.00 | 73.300 | 0.377 | 0.395 | |
| 15.36 | 73.318 | 0.377 | 0.395 | |
| C2 | 0.24 | 61.231 | 0.350 | 0.347 |
| 0.96 | 61.677 | 0.355 | 0.348 | |
| 2.16 | 61.782 | 0.355 | 0.350 | |
| 3.84 | 61.779 | 0.355 | 0.350 | |
| 6.00 | 61.786 | 0.354 | 0.349 | |
| 15.36 | 61.791 | 0.355 | 0.349 | |
| T1 | 0.24 | 92.412 | 0.324 | 0.346 |
| 0.96 | 83.484 | 0.350 | 0.366 | |
| 2.16 | 79.549 | 0.362 | 0.375 | |
| 3.84 | 77.850 | 0.364 | 0.382 | |
| 6.00 | 76.802 | 0.368 | 0.384 | |
| 15.36 | 75.500 | 0.370 | 0.389 | |
| C1 | 0.24 | 65.246 | 0.370 | 0.368 |
| 0.96 | 64.832 | 0.323 | 0.339 | |
| 2.16 | 64.056 | 0.315 | 0.332 | |
| 3.84 | 63.270 | 0.317 | 0.327 | |
| 6.00 | 63.092 | 0.314 | 0.324 | |
| 15.36 | 62.581 | 0.311 | 0.322 |
References
- Taloni, A.; Vodret, M.; Costantini, G.; Zapperi, S. Size effects on the fracture of microscale and nanoscale materials. Nat. Rev. Mater. 2018, 3, 211–224. [Google Scholar] [CrossRef]
- Abazari, A.M.; Safavi, S.M.; Rezazadeh, G.; Villanueva, L.G. Size Effects on Mechanical Properties of Micro/Nano Structures. arXiv 2015, arXiv:1508.01322. [Google Scholar] [CrossRef] [PubMed]
- Bitzek, E.; Kermode, J.R.; Gumbsch, P. Atomistic aspects of fracture. Int. J. Fract. 2015, 191, 13–30. [Google Scholar] [CrossRef]
- Patil, S.P.; Heider, Y. A Review on Brittle Fracture Nanomechanics by All-Atom Simulations. Nanomaterials 2019, 9, 1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasan, M.R.; Vo, T.Q.; Kim, B. Manipulating thermal resistance at the solid–fluid interface through monolayer deposition. RSC Adv. 2019, 9, 4948–4956. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, C.T.; Beskok, A. Charged nanoporous graphene membranes for water desalination. Phys. Chem. Chem. Phys. 2019, 21, 9483–9494. [Google Scholar] [CrossRef] [PubMed]
- Nasim, M.; Vo, T.Q.; Mustafi, L.; Kim, B.; Lee, C.S.; Chu, W.S.; Chun, D.M. Deposition mechanism of graphene flakes directly from graphite particles in the kinetic spray process studied using molecular dynamics simulation. Comput. Mater. Sci. 2019, 169, 109091. [Google Scholar] [CrossRef]
- Segall, D.; Ismail-Beigi, S.; Arias, T. Elasticity of nanometer-sized objects. Phys. Rev. B 2002, 65, 214109. [Google Scholar] [CrossRef] [Green Version]
- Liang, H.; Upmanyu, M.; Huang, H. Size-dependent elasticity of nanowires: nonlinear effects. Phys. Rev. B 2005, 71, 241403. [Google Scholar] [CrossRef] [Green Version]
- Wu, H. Molecular dynamics study on mechanics of metal nanowire. Mech. Res. Commun. 2006, 33, 9–16. [Google Scholar] [CrossRef]
- Luo, J.; Wang, J.; Bitzek, E.; Huang, J.Y.; Zheng, H.; Tong, L.; Yang, Q.; Li, J.; Mao, S.X. Size-dependent brittle-to-ductile transition in silica glass nanofibers. Nano Lett. 2015, 16, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, V.B. Atomistic calculations of elastic properties of metallic fcc crystal surfaces. Phys. Rev. B 2005, 71, 094104. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Huang, H. Are surfaces elastically softer or stiffer? Appl. Phys. Lett. 2004, 84, 1940–1942. [Google Scholar] [CrossRef]
- Heino, P.; Häkkinen, H.; Kaski, K. Molecular-dynamics study of mechanical properties of copper. EPL Europhys. Lett. 1998, 41, 273. [Google Scholar] [CrossRef] [Green Version]
- Hao, T.; Hossain, Z.M. Atomistic mechanisms of crack nucleation and propagation in amorphous silica. Phys. Rev. B 2019, 100, 014204. [Google Scholar] [CrossRef]
- Sun, J.Y.; Zhu, H.Q.; Qin, S.H.; Yang, D.L.; He, X.T. A review on the research of mechanical problems with different moduli in tension and compression. J. Mech. Sci. Technol. 2010, 24, 1845–1854. [Google Scholar] [CrossRef]
- Li, X.; Sun, J.y.; Dong, J.; He, X.t. One-dimensional and two-dimensional analytical solutions for functionally graded beams with different moduli in tension and compression. Materials 2018, 11, 830. [Google Scholar] [CrossRef] [Green Version]
- Van Duin, A.C.; Dasgupta, S.; Lorant, F.; Goddard, W.A. ReaxFF: A reactive force field for hydrocarbons. J. Phys. Chem. A 2001, 105, 9396–9409. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekhar, S.; Satyanarayana, K.; Pramada, P.; Raghavan, P.; Gupta, T. Review processing, properties and applications of reactive silica from rice husk—An overview. J. Mater. Sci. 2003, 38, 3159–3168. [Google Scholar] [CrossRef]
- Aggarwal, P.; Singh, R.P.; Aggarwal, Y. Use of nano-silica in cement based materials—A review. Cogent Eng. 2015, 2, 1078018. [Google Scholar] [CrossRef]
- Pyrak-Nolte, L.J.; DePaolo, D.J.; Pietraß, T. Controlling Subsurface Fractures and Fluid Flow: A Basic Research Agenda; Technical Report; USDOE Office of Science: Washington, DC, USA, 2015. [Google Scholar]
- Chowdhury, S.C.; Haque, B.Z.G.; Gillespie, J.W. Molecular dynamics simulations of the structure and mechanical properties of silica glass using ReaxFF. J. Mater. Sci. 2016, 51, 10139–10159. [Google Scholar] [CrossRef]
- Pedone, A.; Malavasi, G.; Menziani, M.C.; Segre, U.; Cormack, A.N. Molecular dynamics studies of stress-strain behavior of silica glass under a tensile load. Chem. Mater. 2008, 20, 4356–4366. [Google Scholar] [CrossRef]
- Bauchy, M.; Laubie, H.; Qomi, M.A.; Hoover, C.; Ulm, F.J.; Pellenq, R.M. Fracture toughness of calcium–silicate–hydrate from molecular dynamics simulations. J. Non-Cryst. Solids 2015, 419, 58–64. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, P.; Zhang, Y.; Tonks, M.R. Multi-scale modeling of microstructure dependent intergranular brittle fracture using a quantitative phase-field based method. Comput. Mater. Sci. 2016, 113, 38–52. [Google Scholar] [CrossRef] [Green Version]
- Patil, S.P.; Heider, Y.; Padilla, C.A.H.; Cruz-Chú, E.R.; Markert, B. A comparative molecular dynamics-phase-field modeling approach to brittle fracture. Comput. Methods Appl. Mech. Eng. 2016, 312, 117–129. [Google Scholar] [CrossRef]
- Hansen-Dörr, A.C.; Wilkens, L.; Croy, A.; Dianat, A.; Cuniberti, G.; Kästner, M. Combined molecular dynamics and phase-field modelling of crack propagation in defective graphene. Comput. Mater. Sci. 2019, 163, 117–126. [Google Scholar] [CrossRef] [Green Version]
- Rimsza, J.M.; Jones, R.E.; Criscenti, L.J. Crack propagation in silica from reactive classical molecular dynamics simulations. J. Am. Ceram. Soc. 2018, 101, 1488–1499. [Google Scholar] [CrossRef]
- Van Beest, B.; Kramer, G.J.; Van Santen, R. Force fields for silicas and aluminophosphates based on ab initio calculations. Phys. Rev. Lett. 1990, 64, 1955. [Google Scholar] [CrossRef] [Green Version]
- Munetoh, S.; Motooka, T.; Moriguchi, K.; Shintani, A. Interatomic potential for Si–O systems using Tersoff parameterization. Comput. Mater. Sci. 2007, 39, 334–339. [Google Scholar] [CrossRef]
- Stillinger, F.H.; Weber, T.A. Computer simulation of local order in condensed phases of silicon. Phys. Rev. B 1985, 31, 5262. [Google Scholar] [CrossRef] [Green Version]
- Pedone, A.; Malavasi, G.; Menziani, M.C.; Cormack, A.N.; Segre, U. A new self-consistent empirical interatomic potential model for oxides, silicates, and silica-based glasses. J. Phys. Chem. B 2006, 110, 11780–11795. [Google Scholar] [CrossRef] [PubMed]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Fogarty, J.C.; Aktulga, H.M.; Grama, A.Y.; Van Duin, A.C.; Pandit, S.A. A reactive molecular dynamics simulation of the silica-water interface. J. Chem. Phys. 2010, 132, 174704. [Google Scholar] [CrossRef] [PubMed]
- Mei, H.; Yang, Y.; van Duin, A.C.; Sinnott, S.B.; Mauro, J.C.; Liu, L.; Fu, Z. Effects of water on the mechanical properties of silica glass using molecular dynamics. Acta Mater. 2019, 178, 36–44. [Google Scholar] [CrossRef]
- Mozzi, R.; Warren, B. The structure of vitreous silica. J. Appl. Crystallogr. 1969, 2, 164–172. [Google Scholar] [CrossRef]
- Rimsza, J.M.; Jones, R.E.; Criscenti, L.J. Chemical effects on subcritical fracture in silica from molecular dynamics simulations. J. Geophys. Res. Solid Earth 2018, 123, 9341–9354. [Google Scholar] [CrossRef]
- Tsai, D. The virial theorem and stress calculation in molecular dynamics. J. Chem. Phys. 1979, 70, 1375–1382. [Google Scholar] [CrossRef]
- Freund, L.B.; Suresh, S. Thin Film Materials: Stress, Defect Formation and Surface Evolution; Cambridge University Press: Cambridge, UK, 2004. [Google Scholar]
- Deschamps, T.; Margueritat, J.; Martinet, C.; Mermet, A.; Champagnon, B. Elastic moduli of permanently densified silica glasses. Sci. Rep. 2014, 4, 7193. [Google Scholar] [CrossRef] [Green Version]
- Wiederhorn, S.M. Fracture surface energy of glass. J. Am. Ceram. Soc. 1969, 52, 99–105. [Google Scholar] [CrossRef]
- Wallenberger, F.T.; Watson, J.C.; Li, H. Glass Fibers; ASM International: Materials Park, OH, USA, 2001; pp. 27–34. [Google Scholar]
- Bansal, N.P.; Doremus, R.H. Handbook of Glass Properties; Elsevier: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Chowdhury, S.C.; Wise, E.A.; Ganesh, R.; Gillespie, J.W., Jr. Effects of surface crack on the mechanical properties of Silica: A molecular dynamics simulation study. Eng. Fract. Mech. 2019, 207, 99–108. [Google Scholar] [CrossRef]
- Yu, Y.; Wang, B.; Wang, M.; Sant, G.; Bauchy, M. Revisiting silica with ReaxFF: Towards improved predictions of glass structure and properties via reactive molecular dynamics. J. Non-Cryst. Solids 2016, 443, 148–154. [Google Scholar] [CrossRef] [Green Version]
- Kondo, Y.; Ru, Q.; Takayanagi, K. Thickness induced structural phase transition of gold nanofilm. Phys. Rev. Lett. 1999, 82, 751. [Google Scholar] [CrossRef]
- Hasmy, A.; Medina, E. Thickness induced structural transition in suspended fcc metal nanofilms. Phys. Rev. Lett. 2002, 88, 096103. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, X. Predicting Young’s modulus of nanowires from first-principles calculations on their surface and bulk materials. J. Appl. Phys. 2008, 104, 113517. [Google Scholar] [CrossRef] [Green Version]
- An, H.; Fleming, S.; Cox, G. Visualization of second-order nonlinear layer in thermally poled fused silica glass. Appl. Phys. Lett. 2004, 85, 5819–5821. [Google Scholar] [CrossRef]
- Khor, F.; Cronin, D.S.; Watson, B.; Gierczycka, D.; Malcolm, S. Importance of asymmetry and anisotropy in predicting cortical bone response and fracture using human body model femur in three-point bending and axial rotation. J. Mech. Behav. Biomed. Mater. 2018, 87, 213–229. [Google Scholar] [CrossRef]
- Teichtmeister, S.; Miehe, C. Phase-Field Modeling of Fracture in Anisotropic Media. PAMM 2015, 15, 159–160. [Google Scholar] [CrossRef]
- Rountree, C.L.; Vandembroucq, D.; Talamali, M.; Bouchaud, E.; Roux, S. Plasticity-induced structural anisotropy of silica glass. Phys. Rev. Lett. 2009, 102, 195501. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Funamori, N.; Yagi, T. Differential strain and residual anisotropy in silica glass. J. Appl. Phys. 2013, 114, 103509. [Google Scholar] [CrossRef]
- Koslowski, M.; Strachan, A. Uncertainty propagation in a multiscale model of nanocrystalline plasticity. Reliab. Eng. Syst. Saf. 2011, 96, 1161–1170. [Google Scholar] [CrossRef] [Green Version]
- Ostoja-Starzewski, M. Microstructural randomness versus representative volume element in thermomechanics. J. Appl. Mech. 2001, 69, 25–35. [Google Scholar] [CrossRef]
- Sun, C.; Vaidya, R. Prediction of composite properties from a representative volume element. Compos. Sci. Technol. 1996, 56, 171–179. [Google Scholar] [CrossRef]
- Valavala, P.; Odegard, G.; Aifantis, E. Influence of representative volume element size on predicted elastic properties of polymer materials. Model. Simul. Mater. Sci. Eng. 2009, 17, 045004. [Google Scholar] [CrossRef] [Green Version]








| Structural Parameters | Our Simulation Results | Experimental Results [36] |
|---|---|---|
| Si-Si RDF 1st peak position (nm) | 0.3071 | 0.3077 |
| Si-Si RDF 2nd peak position (nm) | 0.5193 | 0.5182 |
| O-O RDF 1st peak position (nm) | 0.2538 | 0.2626 |
| O-O RDF 2nd peak position (nm) | 0.4896 | 0.5097 |
| Si-O RDF 1st peak position (nm) | 0.1633 | 0.1608 |
| Si-O RDF 2nd peak position (nm) | 0.3969 | 0.4061 |
| (nm) | (nm) | |
|---|---|---|
| 4.902 | 36.285 | 0.24 |
| 9.804 | 145.139 | 0.96 |
| 14.706 | 326.562 | 2.16 |
| 19.608 | 580.555 | 3.84 |
| 24.510 | 907.118 | 6.00 |
| 39.216 | 2322.221 | 15.36 |
| Reference Study | E (GPa) | ||
|---|---|---|---|
| Experiment | Freund and Suresh [39] | 80 | 0.22 |
| Deschamps et al. [40] | 71.5 | 0.176 | |
| Wiederhorn [41] | 72.1 | ... | |
| Wallenberger et al. [42] | 69 | ... | |
| Bansal and Doremus [43] | 72.9 | ... | |
| Bound | 69–80 | 0.176–0.22 | |
| ReaxFF simulation | Hao and Hossain [15] | 69.1 | 0.25–0.32 |
| Chowdhury et al. [22] | 75.4–76.68 | ... | |
| Chowdhury et al. [44] | 74 | 0.39 | |
| Rimsza et al. [28] | ... | 0.31 | |
| Yu et al. [45] | 80.4 ± 1.9 | ... | |
| Mei et al. [35] | 60 | 0.296 | |
| Bound | 60–82.3 | 0.25–0.39 |
| Simulation Set | N | UU | Simulation Set | N | UU |
|---|---|---|---|---|---|
| T2 | 0.24 | 0.164 | T1 | 0.24 | 0.411 |
| 0.96 | 0.323 | 0.96 | 0.420 | ||
| 2.16 | 0.575 | 2.16 | 0.804 | ||
| 3.84 | 1.089 | 3.84 | 1.105 | ||
| 6.00 | 1.553 | 6.00 | 1.704 | ||
| 15.36 | 3.107 | 15.36 | 3.129 | ||
| C2 | 0.24 | 0.140 | C1 | 0.24 | 0.140 |
| 0.96 | 0.241 | 0.96 | 0.241 | ||
| 2.16 | 0.368 | 2.16 | 0.365 | ||
| 3.84 | 0.539 | 3.84 | 0.532 | ||
| 6.00 | 0.745 | 6.00 | 0.740 | ||
| 15.36 | 1.561 | 15.36 | 1.565 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vo, T.; Reeder, B.; Damone, A.; Newell, P. Effect of Domain Size, Boundary, and Loading Conditions on Mechanical Properties of Amorphous Silica: A Reactive Molecular Dynamics Study. Nanomaterials 2020, 10, 54. https://doi.org/10.3390/nano10010054
Vo T, Reeder B, Damone A, Newell P. Effect of Domain Size, Boundary, and Loading Conditions on Mechanical Properties of Amorphous Silica: A Reactive Molecular Dynamics Study. Nanomaterials. 2020; 10(1):54. https://doi.org/10.3390/nano10010054
Chicago/Turabian StyleVo, Truong, Brett Reeder, Angelo Damone, and Pania Newell. 2020. "Effect of Domain Size, Boundary, and Loading Conditions on Mechanical Properties of Amorphous Silica: A Reactive Molecular Dynamics Study" Nanomaterials 10, no. 1: 54. https://doi.org/10.3390/nano10010054