Effects of Antibiotics on the Bacterial Community, Metabolic Functions and Antibiotic Resistance Genes in Mariculture Sediments during Enrichment Culturing

Abstract
:1. Introduction
2. Materials and Methods
2.1. Target Antibiotics
2.2. Sample Collection and Experimental Design
2.3. Bacterial Communities Based on Culture-Dependent Analysis
2.4. Culture-Independent Bacterial Community Analysis Based on ILLUMINA HIGH-THROUGHPUT 16S rRNA Gene Sequencing
2.5. Prediction of Bacterial Functions and Analysis of Antibiotic Resistance Genes
3. Results and Discussion
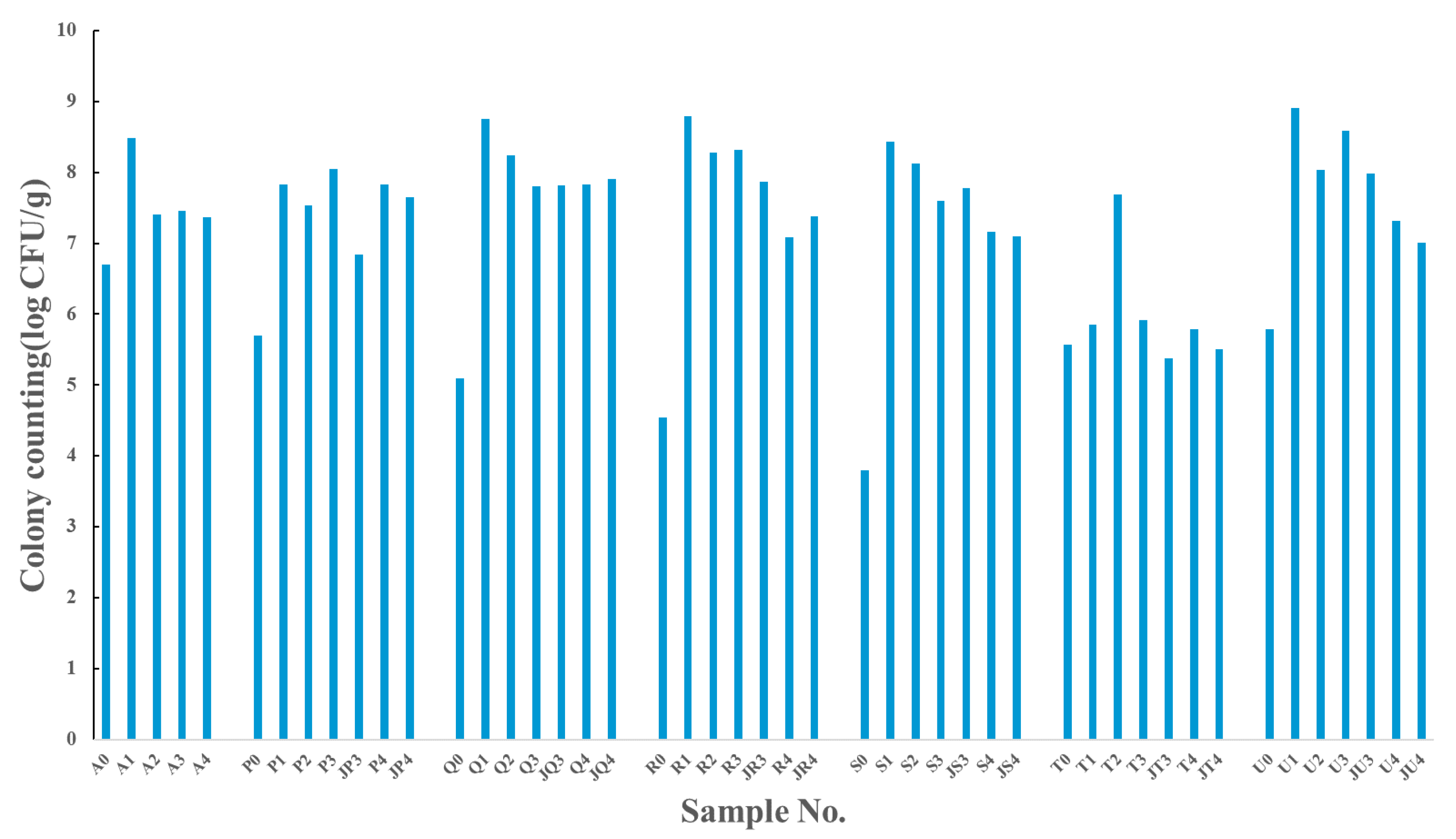
3.1. The Changes in the Abundances and Diversity of Cultivable Bacteria under Different Antibiotic Pressure
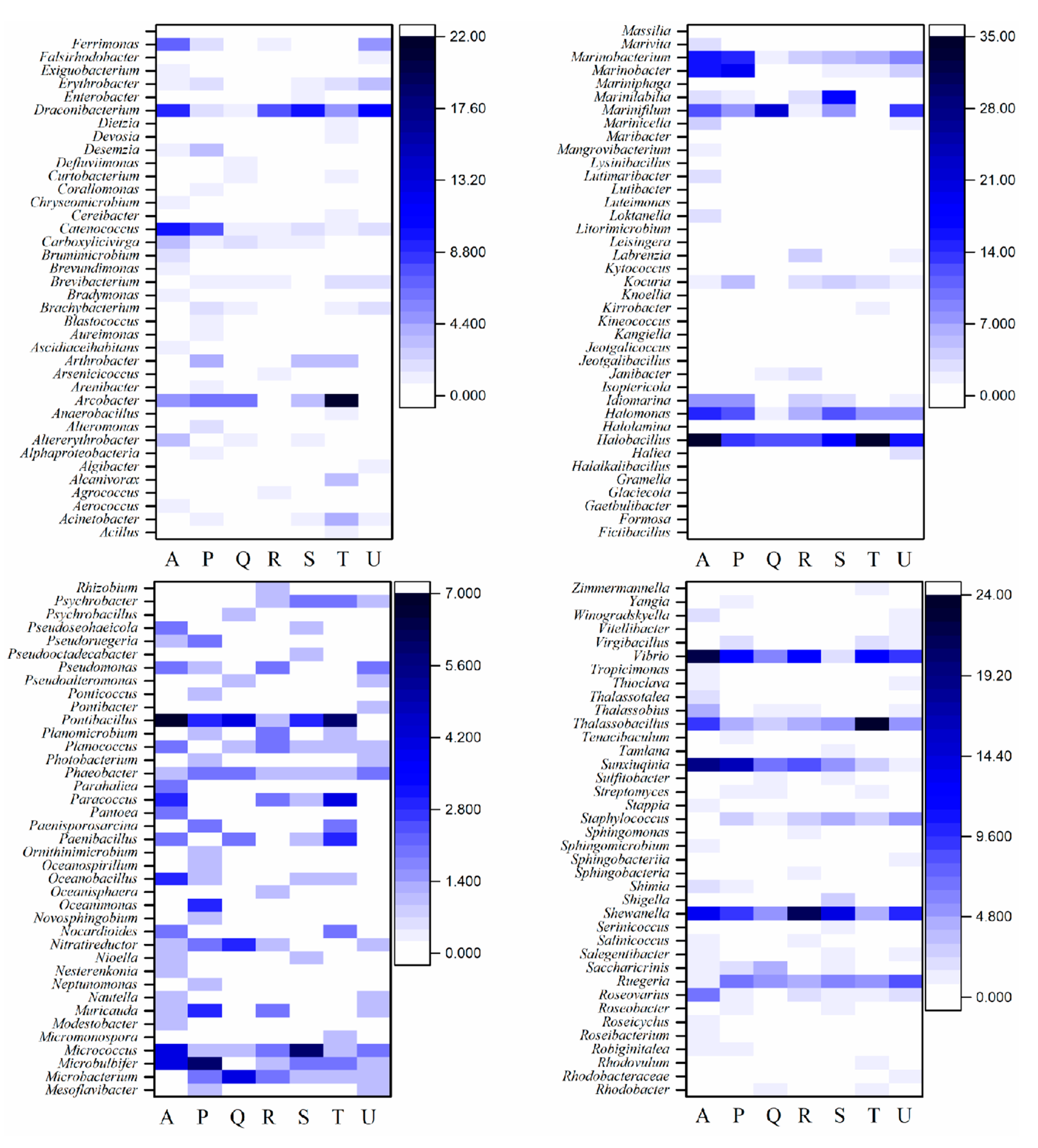
3.2. Taxonomic Composition of Cultivable Bacteria
3.3. Microbial Community Characteristics Based on the High-Throughput 16S rRNA Gene Amplicon Sequencing
3.3.1. Diversity and Richness of Microbial Community
3.3.2. Impact of Antibiotic on the Microbial Community at Phylum Level
3.3.3. Impact of Antibiotic on the Microbial Community at Genus Level
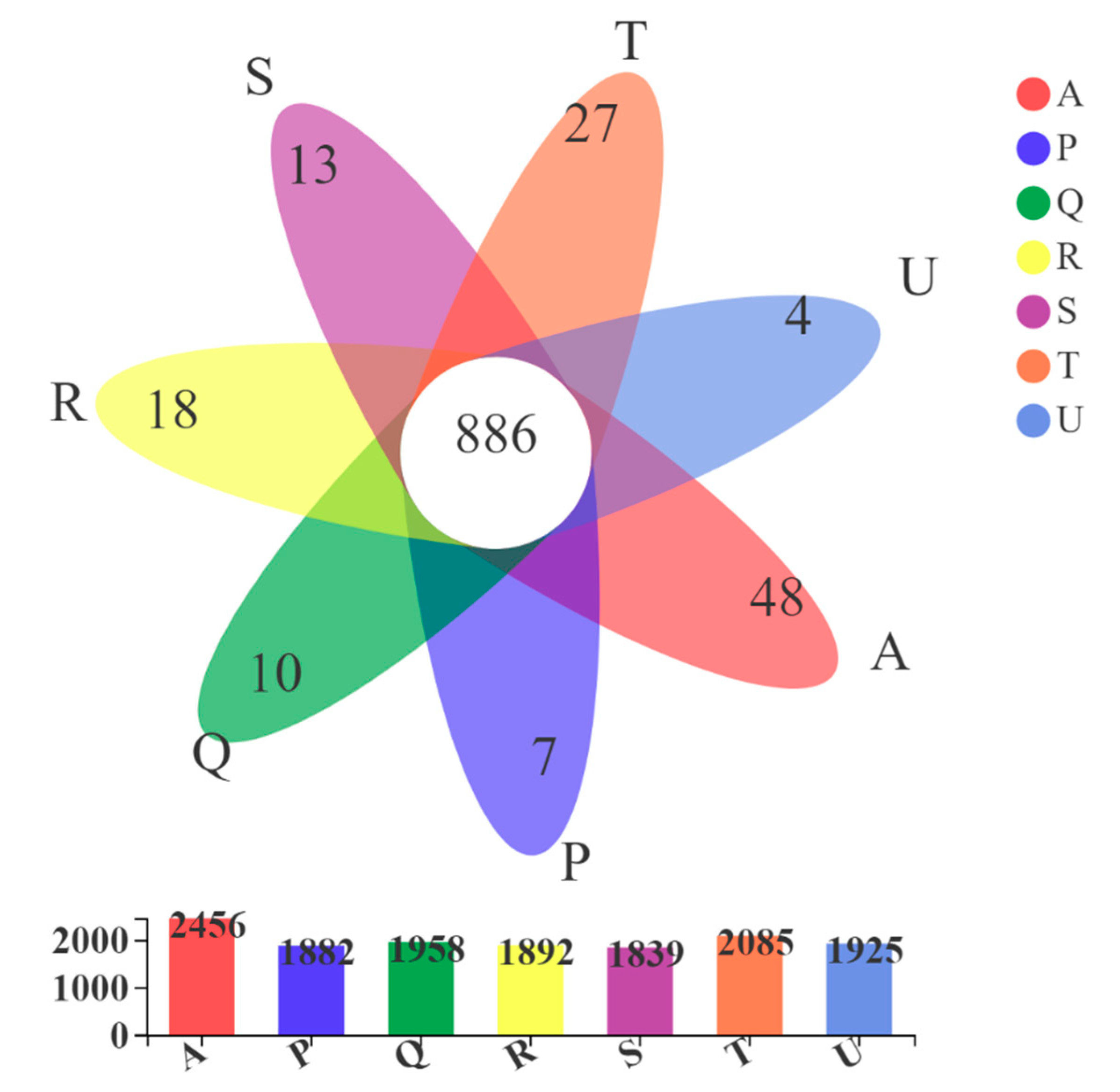
3.3.4. Comparative Analysis of Microbial Community Structures under Different Antibiotic Selective Pressures
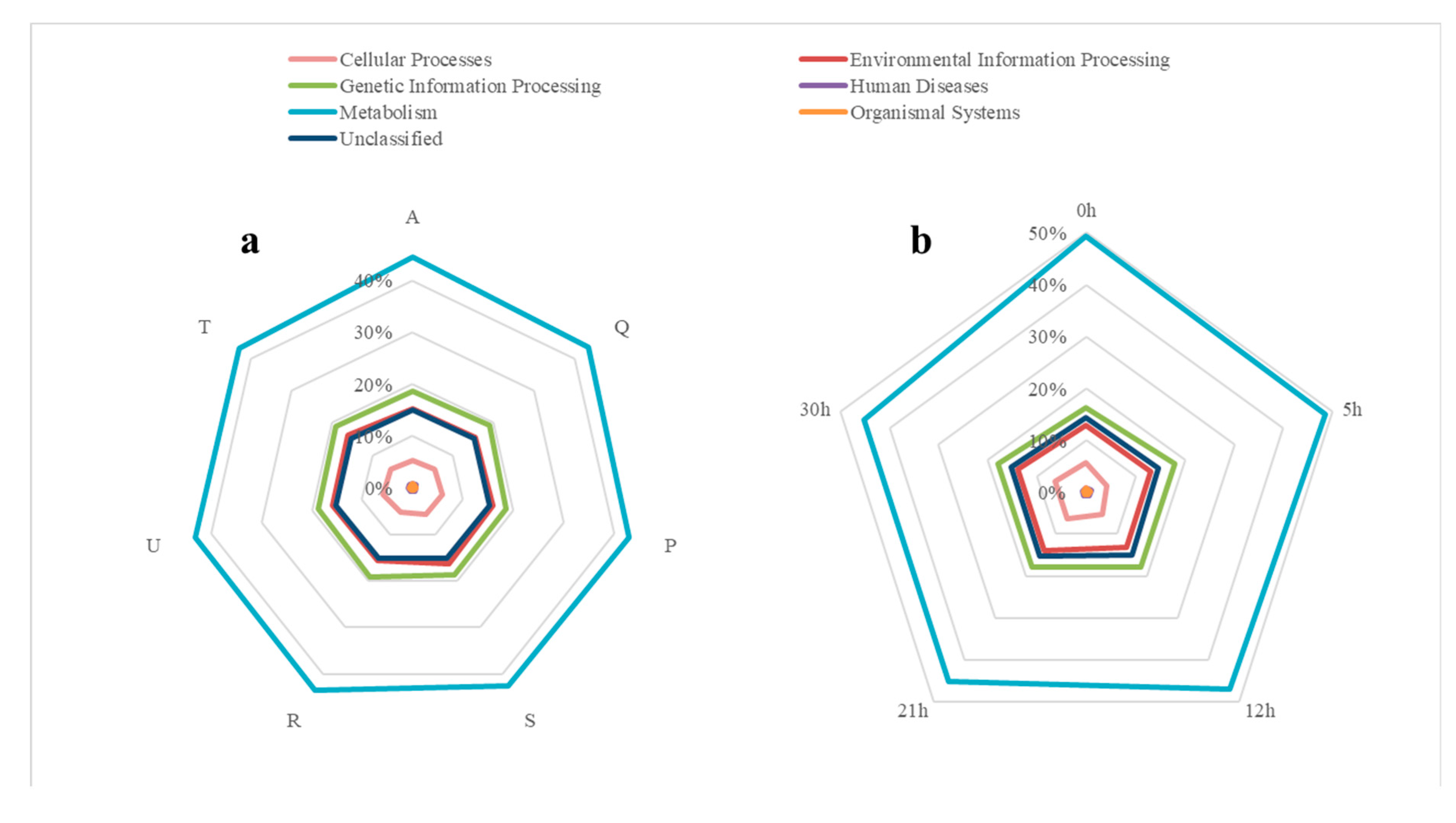
3.4. Analysis of Bacterial Functions
3.5. Antibiotic Resistance Genes Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sharma, V.K.; Johnson, N.; Cizmas, L.; McDonald, T.J.; Kim, H. A review of the influence of treatment strategies on antibiotic resistant bacteria and antibiotic resistance genes. Chemosphere 2016, 150, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Chen, H.; Li, J. Sources, distribution and potential risks of pharmaceuticals and personal care products in Qingshan Lake basin, Eastern China. Ecotoxicol. Environ. Saf. 2013, 96, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, V.O.; Duffy, B. Use of antibiotics in plant agriculture. Rev. Sci. Tech. 2012, 31, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Cabello, F.C. Heavy use of prophylactic antibiotics in aquaculture: A growing problem for human and animal health and for the environment. Environ. Microbiol. 2006, 8, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Lu, S.; Guo, W.; Xi, B.; Wang, W. Antibiotics in the aquatic environments: A review of lakes, China. Sci. Total Environ. 2018, 627, 1195–1208. [Google Scholar] [CrossRef]
- Klein, E.Y.; Van Boeckel, T.P.; Martinez, E.M.; Pant, S.; Gandra, S.; Levin, S.A.; Goossens, H.; Laxminarayan, R. Global increase and geographic convergence in antibiotic consumption between 2000 and 2015. Proc. Natl. Acad. Sci. USA 2018, 115, E3463–E3470. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.J.; Ying, G.G.; Liu, S.; Zhao, J.L.; Yang, B.; Chen, Z.F.; Lai, H.J. Occurrence and fate of eleven classes of antibiotics in two typical wastewater treatment plants in South China. Sci. Total Environ. 2013, 452, 365–376. [Google Scholar] [CrossRef]
- Dolliver, H.; Gupta, S. Antibiotic Losses in Leaching and Surface Runoff from Manure-Amended Agricultural Land. J. Environ. Qual. 2008, 37, 1227. [Google Scholar] [CrossRef]
- Hanna, N.; Sun, P.; Sun, Q.; Li, X.; Yang, X.; Ji, X.; Zou, H.; Ottoson, J.; Nilsson, L.E.; Berglund, B.; et al. Presence of antibiotic residues in various environmental compartments of Shandong province in eastern China: Its potential for resistance development and ecological and human risk. Environ. Int. 2018, 114, 131–142. [Google Scholar] [CrossRef]
- Andremont, A.; Walsh, T.R. The role of sanitation in the development and spread of antimicrobial resistance. AMR Control 2015, 67, 68–73. [Google Scholar]
- Qiao, M.; Ying, G.; Singer, A.C.; Zhu, Y. Review of antibiotic resistance in China and its environment. Environ. Int. 2018, 110, 160–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Yang, Y.; Lu, D.; Niu, Z.; Feng, J.; Chen, Y.; Tou, F.; Garner, E.; Xu, J.; Liu, M.; et al. Biofilms as a sink for antibiotic resistance genes (ARGs) in the Yangtze Estuary. Water Res. 2018, 129, 277–286. [Google Scholar]
- Allen, H.K.; Donato, J.; Wang, H.H.; Cloud-Hansen, K.A.; Davies, J.; Handelsman, J. Call of the wild: Antibiotic resistance genes in natural environments. Nat. Rev. Microbiol. 2010, 8, 251–259. [Google Scholar] [CrossRef]
- Näslund, J.; Hedman, J.E.; Agestrand, C. Effects of the antibiotic ciprofloxacin on the bacterial community structure and degradation of pyrene in marine sediment. Aquat. Toxicol. 2008, 90, 223–227. [Google Scholar] [CrossRef]
- Guan, Y.; Jia, J.; Wu, L.; Xue, X.; Zhang, G.; Wang, Z. Analysis of bacterial community characteristics, abundance of antibiotics and antibiotic resistance genes along a pollution gradient of Ba river in Xi’an, China. Front. Microbiol. 2018, 9, 3191. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Zhu, Y.; Ke, M.; Peijnenburg, W.J.G.M.; Zhang, M.; Wang, T.; Chen, J.; Qian, H. Evaluation of the taxonomic and functional variation of freshwater plankton communities induced by trace amounts of the antibiotic ciprofloxacin. Environ. Int. 2019, 126, 268–278. [Google Scholar] [CrossRef]
- Semedo, M.; Song, B.; Sparrer, T.; Phillips, R.L. Antibiotic effects on microbial communities responsible for denitrification and N2O production in grassland soils. Front. Microbiol. 2018, 9, 2121. [Google Scholar] [CrossRef] [Green Version]
- Costanzo, S.D.; Murby, J.; Bates, J. Ecosystem response to antibiotics entering the aquatic environment. Mar. Pollut. Bull. 2005, 51, 218–223. [Google Scholar] [CrossRef]
- Thiele-Bruhn, S.; Beck, I. Effects of sulfonamide and tetracycline antibiotics on soil microbial activity and microbial biomass. Chemosphere 2005, 59, 457–465. [Google Scholar] [CrossRef]
- Bell, T.; Newman, J.A.; Silverman, B.W.; Turner, S.L.; Lilley, A.K. The contribution of species richness and composition to bacterial services. Nature 2005, 436, 1157–1160. [Google Scholar] [CrossRef]
- Cabello, F.C.; Godfrey, H.P.; Buschmann, A.H.; Dolz, H.J. Aquaculture as yet another environmental gateway to the development and globalisation of antimicrobial resistance. Lancet Infect. Dis. 2016, 16, e127–e133. [Google Scholar] [CrossRef]
- Henriksson, P.J.G.; Rico, A.; Troell, M.; Klinger, D.H.; Buschmann, A.H.; Saksida, S.; Chadag, M.V.; Zhang, W. Unpacking factors influencing antimicrobial use in global aquaculture and their implication for management: A review from a systems perspective. Sustain. Sci. 2018, 13, 1105–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rico, A.; Satapornvanit, K.; Haque, M.M.; Min, J.; Nguyen, P.T.; Telfer, T.C.; van den Brink, P.J. Use of chemicals and biological products in Asian aquaculture and their potential environmental risks: A critical review. Rev. Aquac. 2012, 4, 75–93. [Google Scholar] [CrossRef]
- Deng, Y.; Wu, Y.; Tan, A.; Huang, Y.; Jiang, L.; Xue, H.; Wang, W.; Luo, L.; Zhao, F. Analysis of Antimicrobial Resistance Genes in Aeromonas spp. Isolated from Cultured Freshwater Animals in China. Microb. Drug Resist. 2014, 20, 350–356. [Google Scholar] [CrossRef]
- Aedo, S.; Ivanova, L.; Tomova, A.; Cabello, F.C. Plasmid-Related Quinolone Resistance Determinants in Epidemic Vibrio parahaemolyticus, Uropathogenic Escherichia coli, and Marine Bacteria from an Aquaculture Area in Chile. Microb. Ecol. 2014, 68, 324–328. [Google Scholar] [CrossRef]
- Di Cesare, A.; Luna, G.M.; Vignaroli, C.; Pasquaroli, S.; Tota, S.; Paroncini, P.; Biavasco, F. Aquaculture can promote the presence and spread of antibiotic-resistant Enterococci in marine sediments. PLoS ONE 2013, 8, e62838. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Zhu, D.; Sheng, G.D.; O’Connor, P.; Zhu, Y. Soil oxytetracycline exposure alters the microbial community and enhances the abundance of antibiotic resistance genes in the gut of Enchytraeus crypticus. Sci. Total Environ. 2019, 673, 357–366. [Google Scholar] [CrossRef]
- Shi, L.; Ge, B.; Liu, B.; Liu, X.; Jiang, M.; Zhang, K. Impact of Wuyiencin application on the soil microbial community and fate of typical antibiotic resistance genes. Sci. Rep. 2019, 9, 4016. [Google Scholar] [CrossRef] [Green Version]
- Huerta, B.; Marti, E.; Gros, M.; López, P.; Pompêo, M.; Armengol, J.; Barceló, D.; Balcázar, J.L.; Rodríguez-Mozaz, S.; Marcé, R. Exploring the links between antibiotic occurrence, antibiotic resistance, and bacterial communities in water supply reservoirs. Sci. Total Environ. 2013, 456, 161–170. [Google Scholar] [CrossRef]
- Zhao, R.; Feng, J.; Liu, J.; Fu, W.; Li, X.; Li, B. Deciphering of microbial community and antibiotic resistance genes in activated sludge reactors under high selective pressure of different antibiotics. Water Res. 2019, 151, 388–402. [Google Scholar]
- Zhang, Y.; Geng, J.; Ma, H.; Ren, H.; Xu, K.; Ding, L. Characterization of microbial community and antibiotic resistance genes in activated sludge under tetracycline and sulfamethoxazole selection pressure. Sci. Total Environ. 2016, 571, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yang, Y.; Sun, H.; Zhao, L.; Liu, Y. Effect of tetracycline on microbial community structure associated with enhanced biological N&P removal in sequencing batch reactor. Bioresour. Technol. 2018, 256, 414–420. [Google Scholar] [PubMed]
- Zou, S.; Xu, W.; Zhang, R.; Tang, J.; Chen, Y.; Zhang, G. Occurrence and distribution of antibiotics in coastal water of the Bohai Bay, China: Impacts of river discharge and aquaculture activities. Environ. Pollut. 2011, 159, 2913–2920. [Google Scholar] [CrossRef]
- Xie, H.; Wang, X.; Chen, J.; Li, X.; Jia, G.; Zou, Y.; Zhang, Y.; Cui, Y. Occurrence, distribution and ecological risks of antibiotics and pesticides in coastal waters around Liaodong Peninsula, China. Sci. Total Environ. 2019, 656, 946–951. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Zhao, H.; Liu, S.; Xie, H.; Wang, Y.; Chen, J. Antibiotics in the coastal water of the South Yellow Sea in China: Occurrence, distribution and ecological risks. Sci. Total Environ. 2017, 595, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Tang, J.; Li, J.; Zheng, Q.; Liu, D.; Chen, Y.; Zou, Y.; Chen, X.; Luo, C.; Zhang, G. Antibiotics in the offshore waters of the Bohai Sea and the Yellow Sea in China: Occurrence, distribution and ecological risks. Environ. Pollut. 2013, 174, 71–77. [Google Scholar] [CrossRef]
- Turnidge, J.; Christiansen, K. Antibiotic use and resistance—Proving the obvious. Lancet 2005, 365, 548–549. [Google Scholar] [CrossRef]
- Mu, D.S.; Liang, Q.Y.; Wang, X.M.; Lu, D.C.; Shi, M.J.; Chen, G.J.; Du, Z.J. Metatranscriptomic and comparative genomic insights into resuscitation mechanisms during enrichment culturing. Microbiome 2018, 6, 230. [Google Scholar] [CrossRef]
- Li, X.; Xiao, H.; Zhang, W.; Li, Y.; Tang, X.; Duan, J.; Yang, Z.; Wang, J.; Guan, F.; Ding, G. Analysis of cultivable aerobic bacterial community composition and screening for facultative sulfate-reducing bacteria in marine corrosive steel. J. Oceanol. Limnol. 2019, 37, 600–614. [Google Scholar] [CrossRef]
- Liu, Q.Q.; Wang, Y.; Li, J.; Du, Z.J.; Chen, G.J. Saccharicrinis carchari sp. nov., isolated from a shark, and emended descriptions of the genus Saccharicrinis and Saccharicrinis fermentans. Int. J. Syst. Evol. Microbiol. 2014, 64, 2204–2209. [Google Scholar] [CrossRef] [Green Version]
- Song, C.; Wang, B.; Tan, J.; Zhu, L.; Lou, D.; Cen, X. Comparative analysis of the gut microbiota of black bears in China using high-throughput sequencing. Mol. Genet. Genom. 2017, 292, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, J.; Han, Y.; Chen, J.; Liu, G.; Lu, H.; Yan, B.; Chen, S. Nutrients, heavy metals and microbial communities co-driven distribution of antibiotic resistance genes in adjacent environment of mariculture. Environ. Pollut. 2017, 220, 909–918. [Google Scholar] [CrossRef]
- Wang, S.; Cui, Y.; Li, A.; Wang, D.; Zhang, W.; Chen, Z. Seasonal dynamics of bacterial communities associated with antibiotic removal and sludge stabilization in three different sludge treatment wetlands. J. Environ. Manag. 2019, 240, 231–237. [Google Scholar] [CrossRef]
- Xu, Z.; Jiang, Y.; Te, S.; He, Y.; Gin, K. The Effects of Antibiotics on Microbial Community Composition in an Estuary Reservoir during Spring and Summer Seasons. Water 2018, 10, 154. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Yuan, L.; Li, Z.; Zhang, H.; Sheng, G. Tetracycline exposure shifted microbial communities and enriched antibiotic resistance genes in the aerobic granular sludge. Environ. Int. 2019, 130, 104902. [Google Scholar] [CrossRef]
- Zhang, H.; He, H.; Chen, S.; Huang, T.; Lu, K.; Zhang, Z.; Wang, R.; Zhang, X.; Li, H. Abundance of antibiotic resistance genes and their association with bacterial communities in activated sludge of wastewater treatment plants: Geographical distribution and network analysis. J. Environ. Sci. China 2019, 82, 24–38. [Google Scholar] [CrossRef]
- Tzeng, Y.; Ambrose, K.D.; Zughaier, S.M.; Zhou, X.; Miller, Y.K.; Shafer, W.M.; Stephens, D.S. Cationic Antimicrobial Peptide Resistance in Neisseria meningitidis. J. Bacteriol. 2005, 187, 5387–5396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Yang, Y.; Liang, X.; Yu, K.; Zhang, T.; Li, X. Metagenomic Profiles of Antibiotic Resistance Genes (ARGs) between Human Impacted Estuary and Deep Ocean Sediments. Environ. Sci. Technol. 2013, 47, 12753–12760. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Yang, Y.; Pruden, A. Effect of temperature on removal of antibiotic resistance genes by anaerobic digestion of activated sludge revealed by metagenomic approach. Appl. Microbiol. Biotechnol. 2015, 99, 7771–7779. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Designated as | Treatment | Concentration (mg/L) | Incubation Time (days) |
|---|---|---|---|---|
| A | A0 | Control | - | 0 |
| A1 | Control | - | 5 | |
| A2 | Control | - | 12 | |
| A3 | Control | - | 21 | |
| A4 | Control | - | 30 | |
| P | P0 | zinc bacitracin | 50 | 0 |
| P1 | zinc bacitracin | 50 | 5 | |
| P2 | zinc bacitracin | 50 | 12 | |
| P3 | zinc bacitracin | 50 | 21 | |
| P4 | zinc bacitracin | 50 | 30 | |
| JP3 | zinc bacitracin, re-added on Day 12 | 50 + 50 | 21 | |
| JP4 | zinc bacitracin, re-added on Day 12 | 50 + 50 | 30 | |
| Q | Q0 | ciprofloxacin | 30 | 0 |
| Q1 | ciprofloxacin | 30 | 5 | |
| Q2 | ciprofloxacin | 30 | 12 | |
| Q3 | ciprofloxacin | 30 | 21 | |
| Q4 | ciprofloxacin | 30 | 30 | |
| JQ3 | Ciprofloxacin, re-added on Day 12 | 30 + 30 | 21 | |
| JQ4 | Ciprofloxacin, re-added on Day 12 | 30 + 30 | 30 | |
| R | R0 | ampicillin sodium | 100 | 0 |
| R1 | ampicillin sodium | 100 | 5 | |
| R2 | ampicillin sodium | 100 | 12 | |
| R3 | ampicillin sodium | 100 | 21 | |
| R4 | ampicillin sodium | 100 | 30 | |
| JR3 | ampicillin sodium, re-added on Day 12 | 100 + 100 | 21 | |
| JR4 | ampicillin sodium, re-added on Day 12 | 100 + 100 | 30 | |
| S | S0 | chloramphenicol | 30 | 0 |
| S1 | chloramphenicol | 30 | 5 | |
| S2 | chloramphenicol | 30 | 12 | |
| S3 | chloramphenicol | 30 | 21 | |
| S4 | chloramphenicol | 30 | 30 | |
| JS3 | chloramphenicol, re-added on Day 12 | 30 + 30 | 21 | |
| JS4 | chloramphenicol, re-added on Day 12 | 30 + 30 | 30 | |
| T | T0 | tylosin | 50 | 0 |
| T1 | tylosin | 50 | 5 | |
| T2 | tylosin | 50 | 12 | |
| T3 | tylosin | 50 | 21 | |
| T4 | tylosin | 50 | 30 | |
| JT3 | tylosin, re-added on Day 12 | 50 + 50 | 21 | |
| JT4 | tylosin, re-added on Day 12 | 50 + 50 | 30 | |
| U | U0 | tetracycline | 30 | 0 |
| U1 | tetracycline | 30 | 5 | |
| U2 | tetracycline | 30 | 12 | |
| U3 | tetracycline | 30 | 21 | |
| U4 | tetracycline | 30 | 30 | |
| JU3 | tetracycline, re-added on Day 12 | 30 + 30 | 21 | |
| JU4 | tetracycline, re-added on Day 12 | 30 + 30 | 30 |
| Sample No. | Enrichment Time (d) | Number of Strains | Taxonomic Composition | ||
|---|---|---|---|---|---|
| Phyla | Genera | Species | |||
| Control | 372 | 4 | 76 | 145 | |
| A0 | 0 | 55 | 4 | 28 | 43 |
| A1 | 5 | 58 | 4 | 18 | 32 |
| A2 | 12 | 82 | 4 | 27 | 44 |
| A3 | 21 | 98 | 4 | 35 | 63 |
| A4 | 30 | 79 | 4 | 31 | 55 |
| Exposure to zinc bacitracin (P) | 254 | 4 | 67 | 137 | |
| P0 | 0 | 35 | 2 | 19 | 28 |
| P1 | 5 | 16 | 4 | 12 | 14 |
| P2 | 12 | 28 | 4 | 16 | 20 |
| P3 | 21 | 50 | 4 | 24 | 36 |
| JP3 | 21 | 44 | 4 | 22 | 35 |
| P4 | 30 | 21 | 3 | 13 | 21 |
| JP4 | 30 | 60 | 4 | 24 | 40 |
| Exposure to ciprofloxacin (Q) | 134 | 4 | 37 | 67 | |
| Q0 | 0 | 7 | 1 | 2 | 2 |
| Q1 | 5 | 16 | 3 | 5 | 11 |
| Q2 | 12 | 34 | 4 | 17 | 23 |
| Q3 | 21 | 25 | 4 | 13 | 18 |
| JQ3 | 21 | 24 | 4 | 12 | 18 |
| Q4 | 30 | 14 | 4 | 9 | 10 |
| JQ4 | 30 | 14 | 4 | 13 | 14 |
| Exposure to ampicillin sodium (R) | 156 | 4 | 46 | 83 | |
| R0 | 0 | 15 | 3 | 12 | 13 |
| R1 | 5 | 17 | 2 | 7 | 11 |
| R2 | 12 | 31 | 4 | 11 | 19 |
| R3 | 21 | 29 | 4 | 15 | 23 |
| JR3 | 21 | 26 | 4 | 15 | 18 |
| R4 | 30 | 19 | 3 | 14 | 17 |
| JR4 | 30 | 19 | 4 | 13 | 18 |
| Exposure to chloramphenicol (S) | 190 | 4 | 48 | 85 | |
| S0 | 0 | 5 | 2 | 3 | 3 |
| S1 | 5 | 17 | 3 | 6 | 10 |
| S2 | 12 | 39 | 4 | 17 | 27 |
| S3 | 21 | 31 | 4 | 16 | 24 |
| JS3 | 21 | 58 | 4 | 23 | 36 |
| S4 | 30 | 20 | 4 | 15 | 17 |
| JS4 | 30 | 20 | 3 | 12 | 15 |
| Exposure to tylosin (T) | 250 | 4 | 58 | 117 | |
| T0 | 0 | 5 | 1 | 2 | 34 |
| T1 | 5 | 22 | 4 | 10 | 14 |
| T2 | 12 | 31 | 2 | 11 | 23 |
| T3 | 21 | 39 | 3 | 12 | 25 |
| JT3 | 21 | 59 | 3 | 27 | 43 |
| T4 | 30 | 44 | 4 | 18 | 26 |
| JT4 | 30 | 50 | 4 | 21 | 37 |
| Exposure to tetracycline (U) | 193 | 4 | 51 | 93 | |
| U0 | 0 | 42 | 4 | 25 | 33 |
| U1 | 5 | 19 | 4 | 8 | 9 |
| U2 | 12 | 25 | 4 | 12 | 19 |
| U3 | 21 | 30 | 3 | 12 | 17 |
| JU3 | 21 | 39 | 4 | 19 | 28 |
| U4 | 30 | 10 | 4 | 10 | 10 |
| JU4 | 30 | 28 | 4 | 15 | 21 |
| Treatment | Sample ID | OTUs Number | Sobs | Shannon | Simpson | Ace | Chao | Coverage |
|---|---|---|---|---|---|---|---|---|
| Control (A) | A0 | 1972 | 1972 | 5.966149 | 0.007343 | 2190.212 | 2169.367 | 0.991121 |
| A1 | 850 | 850 | 3.488607 | 0.080376 | 1869.999 | 1467.281 | 0.990808 | |
| A2 | 987 | 987 | 3.530338 | 0.082274 | 1879.859 | 1570.807 | 0.990654 | |
| A3 | 1335 | 1335 | 4.175985 | 0.057538 | 2343.172 | 1923.443 | 0.991536 | |
| A4 | 1110 | 1110 | 3.934633 | 0.08665 | 1898.589 | 1602.39 | 0.989918 | |
| Zinc bacitracin (P) | P1 | 787 | 787 | 3.465812 | 0.076074 | 1802.525 | 1404.236 | 0.990751 |
| P2 | 946 | 946 | 3.290489 | 0.106069 | 1908.711 | 1466.348 | 0.991175 | |
| P3 | 894 | 894 | 3.602227 | 0.09099 | 1979.937 | 1553.758 | 0.989684 | |
| JP3 | 1094 | 896 | 3.45138 | 0.10768 | 1867.263 | 1485.578 | 0.991722 | |
| P4 | 896 | 1094 | 3.521291 | 0.116067 | 1636.944 | 1644.864 | 0.991047 | |
| JP4 | 806 | 806 | 3.2706 | 0.119642 | 1719.658 | 1370.8 | 0.992001 | |
| Ciprofloxacin (Q) | Q1 | 712 | 712 | 2.778247 | 0.138909 | 1695.499 | 1234.447 | 0.991287 |
| Q2 | 1254 | 1254 | 3.623877 | 0.109462 | 2287.941 | 1910.64 | 0.989922 | |
| Q3 | 884 | 884 | 3.248321 | 0.1079 | 1773.738 | 1409.007 | 0.991538 | |
| JQ3 | 1058 | 1058 | 3.047219 | 0.130086 | 2114.434 | 1634.563 | 0.992718 | |
| Q4 | 941 | 941 | 2.855148 | 0.175216 | 1936.621 | 1600.519 | 0.99218 | |
| JQ4 | 1030 | 1030 | 3.279126 | 0.122727 | 1858.773 | 1586.065 | 0.99266 | |
| Ampicillin sodium (R) | R1 | 830 | 830 | 2.853027 | 0.166461 | 1729.973 | 1333.879 | 0.991071 |
| R2 | 885 | 885 | 3.427011 | 0.073872 | 2260.472 | 1544.065 | 0.990153 | |
| R3 | 1133 | 1133 | 3.584233 | 0.072941 | 2253.065 | 1797.837 | 0.992216 | |
| JR3 | 762 | 762 | 3.175357 | 0.115644 | 1755.871 | 1290.186 | 0.993518 | |
| R4 | 793 | 793 | 3.473673 | 0.070351 | 2043.293 | 1423.629 | 0.991592 | |
| JR4 | 856 | 856 | 3.860091 | 0.048022 | 1680.427 | 1397.525 | 0.991285 | |
| Chloramphenicol (S) | S1 | 705 | 705 | 2.904588 | 0.129851 | 2197.763 | 1459.063 | 0.992152 |
| S2 | 982 | 982 | 3.618378 | 0.069773 | 1780.536 | 1528.923 | 0.992284 | |
| S3 | 776 | 776 | 3.318475 | 0.099976 | 1577.62 | 1306.472 | 0.992443 | |
| JS3 | 826 | 826 | 3.72379 | 0.066223 | 1766.24 | 1423.3 | 0.990486 | |
| S4 | 1056 | 1056 | 3.737706 | 0.074712 | 1965.486 | 1692 | 0.99179 | |
| JS4 | 709 | 709 | 3.23018 | 0.09656 | 1629.057 | 1183 | 0.992223 | |
| Tylosin (T) | T1 | 1240 | 1240 | 3.111999 | 0.156664 | 2279.861 | 1936.444 | 0.987293 |
| T2 | 1519 | 1519 | 4.406583 | 0.045868 | 1989.912 | 1937.915 | 0.988868 | |
| T3 | 700 | 700 | 2.380128 | 0.187307 | 1584.71 | 1165.124 | 0.9944 | |
| JT3 | 656 | 656 | 2.568263 | 0.231294 | 1831.557 | 1269.085 | 0.992333 | |
| T4 | 1138 | 1138 | 3.452094 | 0.077866 | 1690.838 | 1601.896 | 0.992349 | |
| JT4 | 706 | 706 | 2.336018 | 0.204594 | 2000.348 | 1395.545 | 0.99165 | |
| Tetracycline (U) | U1 | 1021 | 1021 | 3.614356 | 0.069014 | 2029.165 | 1648.687 | 0.988888 |
| U2 | 1070 | 1070 | 3.699218 | 0.067774 | 1978.579 | 1598.746 | 0.990355 | |
| U3 | 945 | 945 | 3.048431 | 0.145983 | 1429.909 | 1384.833 | 0.993129 | |
| JU3 | 901 | 901 | 2.861111 | 0.169415 | 1873.211 | 1480.333 | 0.993019 | |
| U4 | 730 | 730 | 2.761487 | 0.147617 | 1719.237 | 1280.225 | 0.99187 | |
| JU4 | 811 | 811 | 2.829413 | 0.137191 | 1585.21 | 1257.515 | 0.993866 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, M.-Q.; Chen, G.-J.; Du, Z.-J. Effects of Antibiotics on the Bacterial Community, Metabolic Functions and Antibiotic Resistance Genes in Mariculture Sediments during Enrichment Culturing. J. Mar. Sci. Eng. 2020, 8, 604. https://doi.org/10.3390/jmse8080604
Ye M-Q, Chen G-J, Du Z-J. Effects of Antibiotics on the Bacterial Community, Metabolic Functions and Antibiotic Resistance Genes in Mariculture Sediments during Enrichment Culturing. Journal of Marine Science and Engineering. 2020; 8(8):604. https://doi.org/10.3390/jmse8080604
Chicago/Turabian StyleYe, Meng-Qi, Guan-Jun Chen, and Zong-Jun Du. 2020. "Effects of Antibiotics on the Bacterial Community, Metabolic Functions and Antibiotic Resistance Genes in Mariculture Sediments during Enrichment Culturing" Journal of Marine Science and Engineering 8, no. 8: 604. https://doi.org/10.3390/jmse8080604