
RNA-TGGE, a Tool for Assessing the Potential for Bioremediation in Impacted Marine Ecosystems

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Nucleic Acid Extraction
2.3. DNase Treatment of RNA Samples
2.4. cDNA Synthesis
2.5. Bacterial cDNA Amplification
2.6. TGGE (Temperature Gradient Gel Electrophoresis)
2.7. Identification of Bacterial Species from TGGE Gel
2.8. Statistical Analyses
3. Results and Discussion
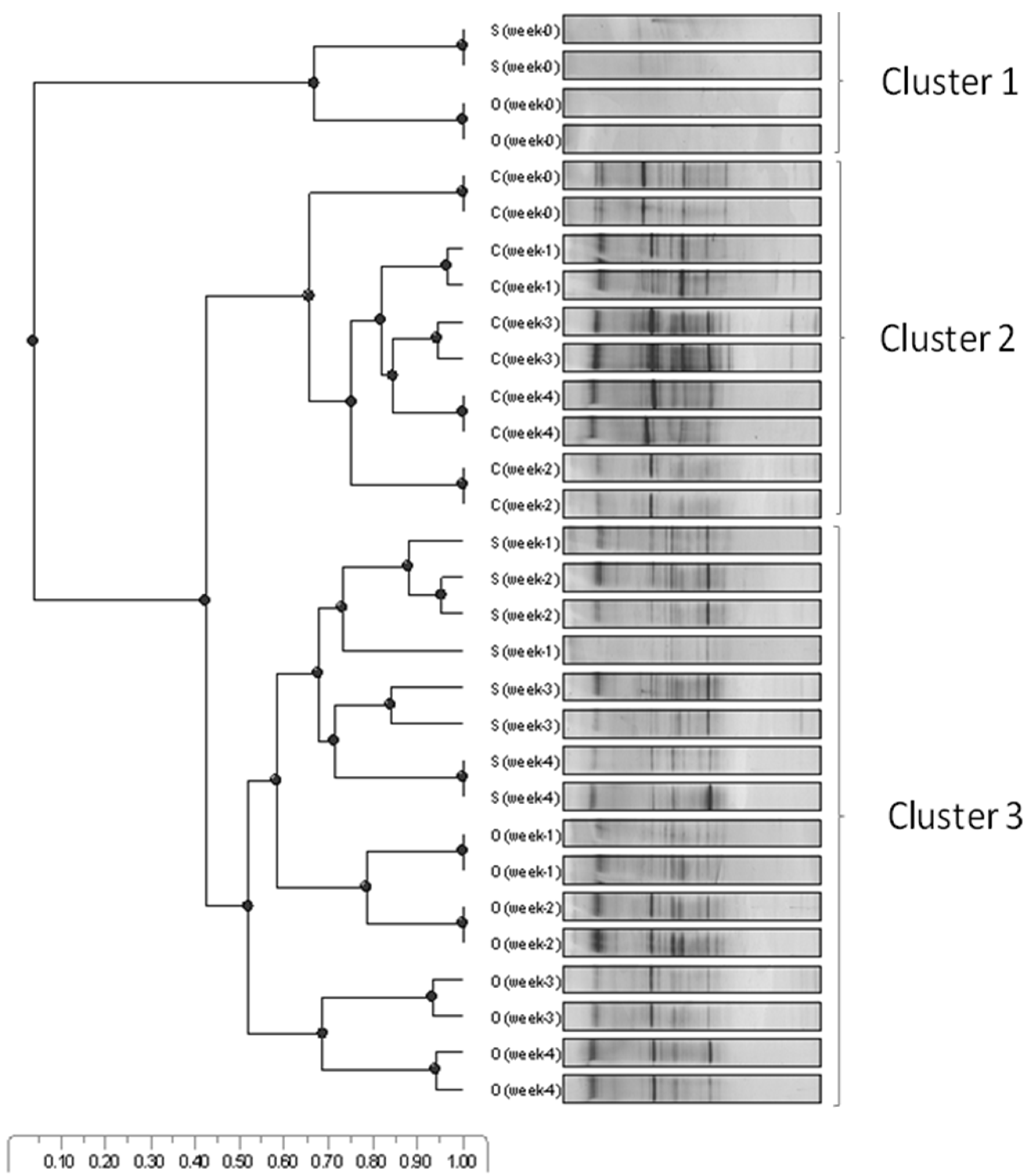
3.1. Microbial Community Dynamics

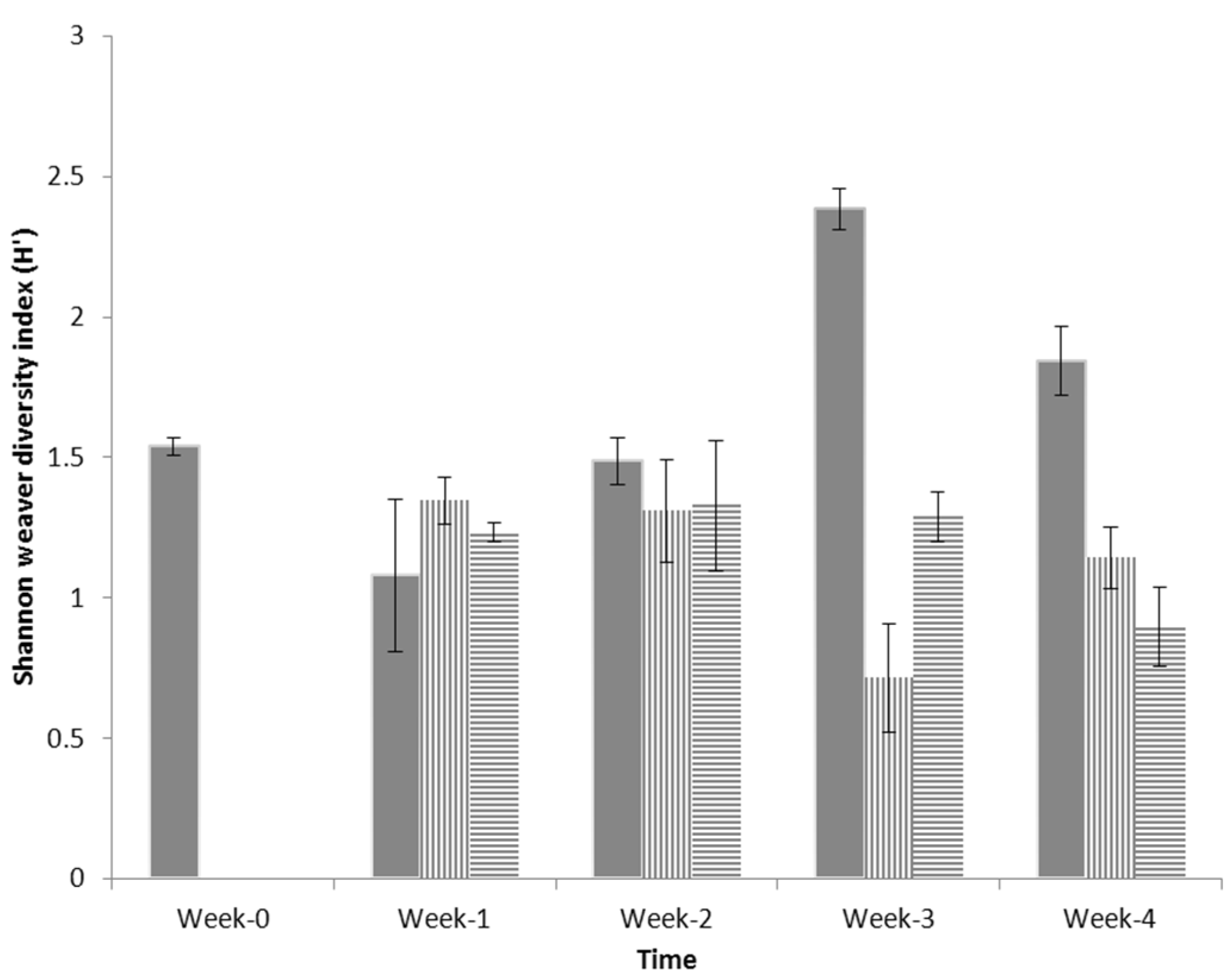
3.2. Shannon Weaver Diversity
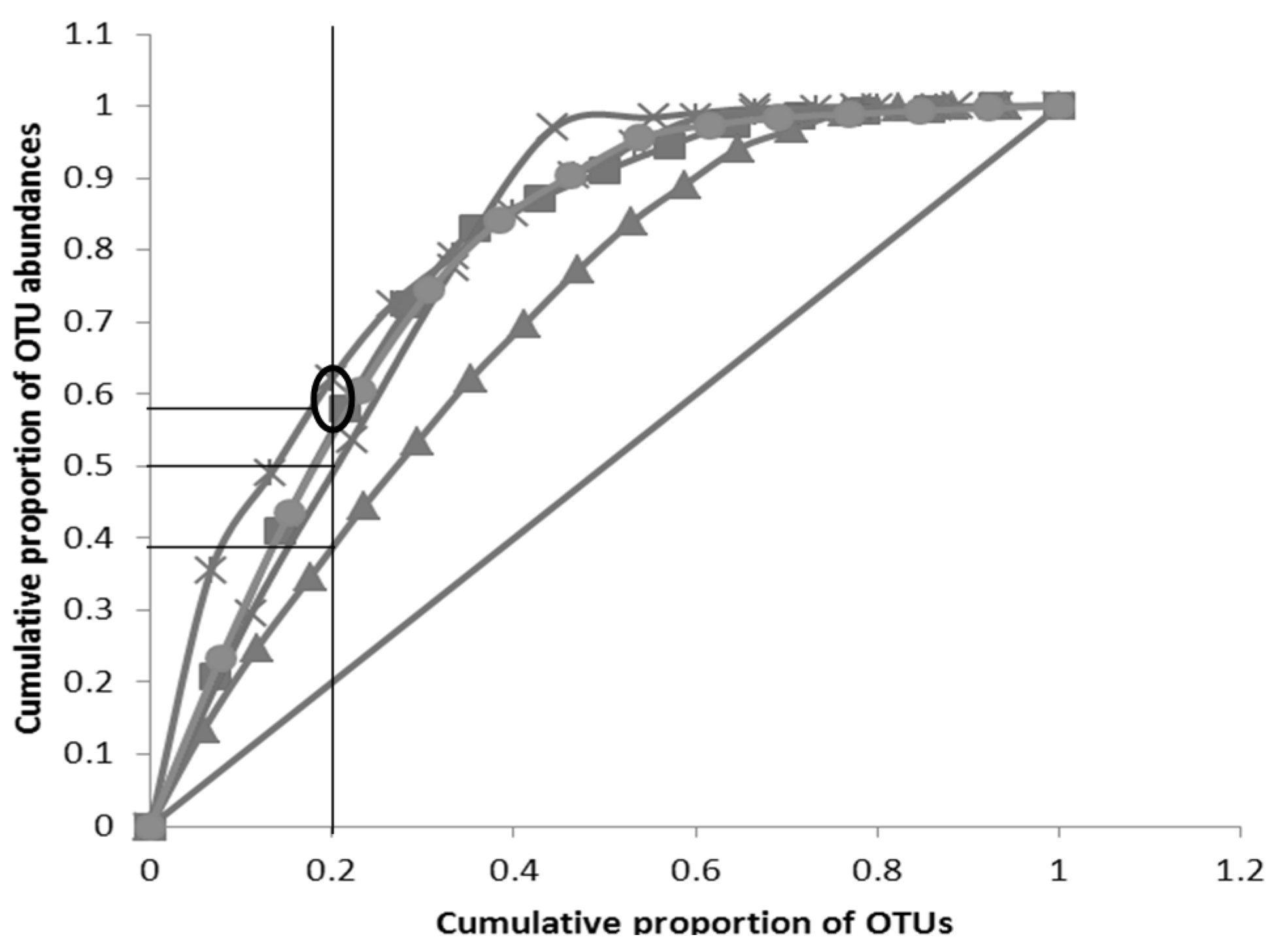
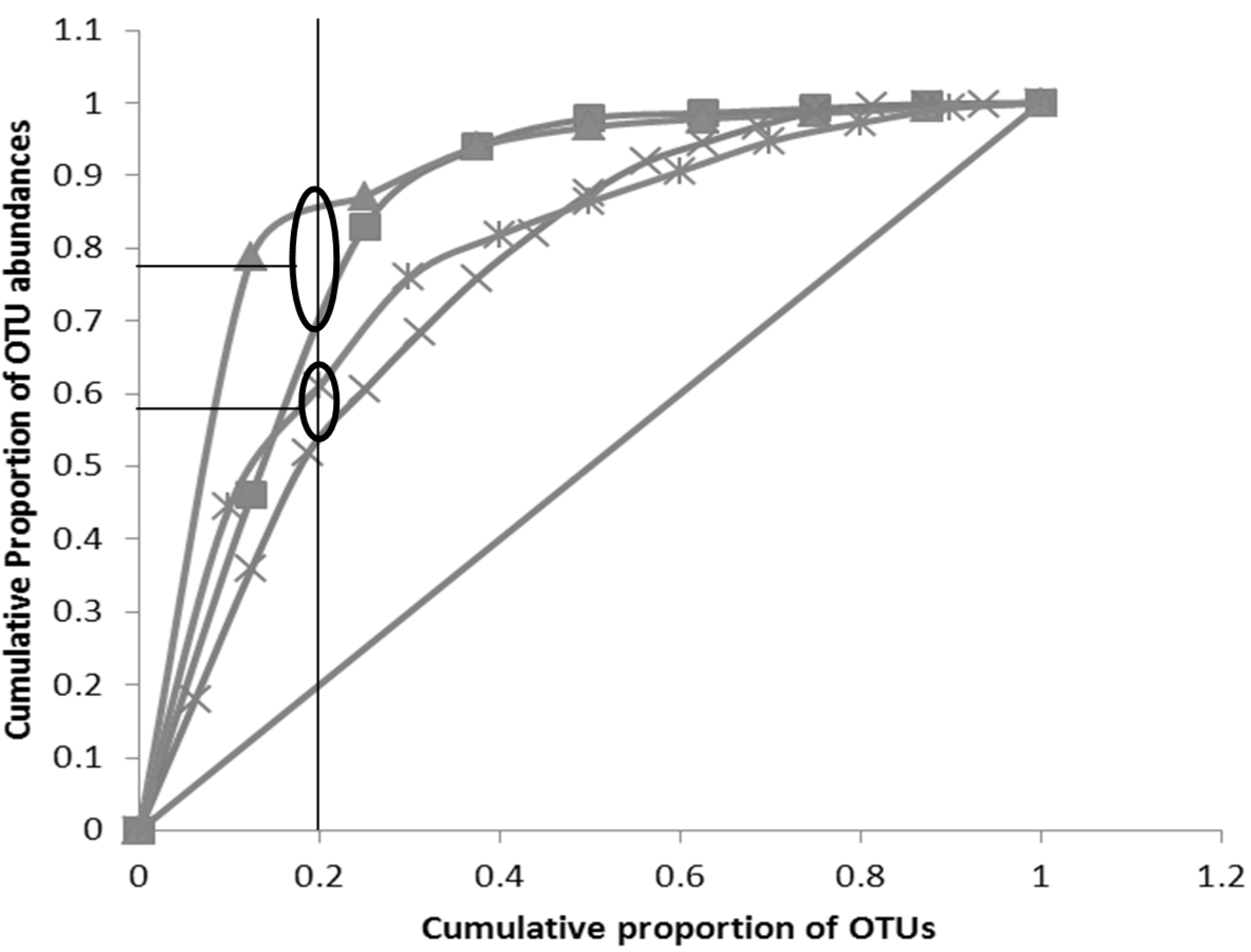
3.3. Pareto-Lorenz Curve
 ” C (seawater + BH medium + weathered crude oil + consortia); “
” C (seawater + BH medium + weathered crude oil + consortia); “  ” O (seawater + BH medium + weathered crude oil); and “
” O (seawater + BH medium + weathered crude oil); and “  ” S (seawater + BH medium) from Week 0 to Week 4. Duplicate data (n = 2) were analysed.
” C (seawater + BH medium + weathered crude oil + consortia); “ ” O (seawater + BH medium + weathered crude oil); and “ ” S (seawater + BH medium) from Week 0 to Week 4. Duplicate data (n = 2) were analysed.
” S (seawater + BH medium) from Week 0 to Week 4. Duplicate data (n = 2) were analysed.
” C (seawater + BH medium + weathered crude oil + consortia); “ ” O (seawater + BH medium + weathered crude oil); and “ ” S (seawater + BH medium) from Week 0 to Week 4. Duplicate data (n = 2) were analysed.
 ” Perfect evenness line; “
” Perfect evenness line; “  ” C (Week 4); “
” C (Week 4); “  ” C (Week 3); “
” C (Week 3); “  ” C (Week 2);“
” C (Week 2);“  ” C (Week 1);“
” C (Week 1);“  ” C (Week 0)).Oval represents a grouping of curves (more than one) and horizontal line without oval represents asingle PL curve.
” Perfect evenness line; “ ” C (Week 4); “ ” C (Week 3); “ ” C (Week 2);“ ” C (Week 1);“ ” C (Week 0)).Oval represents a grouping of curves (more than one) and horizontal line without oval represents asingle PL curve.
” C (Week 0)).Oval represents a grouping of curves (more than one) and horizontal line without oval represents asingle PL curve.
” Perfect evenness line; “ ” C (Week 4); “ ” C (Week 3); “ ” C (Week 2);“ ” C (Week 1);“ ” C (Week 0)).Oval represents a grouping of curves (more than one) and horizontal line without oval represents asingle PL curve. ” Perfect evenness line; “ ” O (Week 4); “ ” O (Week 3); “ ” O (Week 2);“ ” O (Week 1)).
” Perfect evenness line; “ ” O (Week 4); “ ” O (Week 3); “ ” O (Week 2);“ ” O (Week 1)).
” Perfect evenness line; “ ” O (Week 4); “ ” O (Week 3); “ ” O (Week 2);“ ” O (Week 1)).
” Perfect evenness line; “ ” O (Week 4); “ ” O (Week 3); “ ” O (Week 2);“ ” O (Week 1)). ” Perfect evenness line; “ ” S (Week 4); “ ” S (Week 3);“ ” S (Week 2); “ ” S (Week 1)).
” Perfect evenness line; “ ” S (Week 4); “ ” S (Week 3);“ ” S (Week 2); “ ” S (Week 1)).
” Perfect evenness line; “ ” S (Week 4); “ ” S (Week 3);“ ” S (Week 2); “ ” S (Week 1)).
” Perfect evenness line; “ ” S (Week 4); “ ” S (Week 3);“ ” S (Week 2); “ ” S (Week 1)).
3.4. RNA-TGGE Bands

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Identified Species | Treatment | Accession No. | Similarity (%) |
|---|---|---|---|
| Pseudomonas sp. (Gammaproteobacteria) | C | JF778683.1 | 96 |
| Uncultured marine bacterium | C | FM211075.1 | 93 |
| Uncultured Alcanivorax sp. (Gammaproteobacteria) | C | JF979266.1 | 84 |
| Uncultured bacterium clone | C | HQ827507.1 | 100 |
| Uncultured organism clone | C | JN528201.1 | 88 |
| Uncultured bacterium clone | C | HM580751.1 | 99 |
| Uncultured bacterium clone | C | JN178389.1 | 100 |
| Alcanivoraxborkuumensis (Gammaproteobacteria) | C | FJ218422.1 | 94 |
| Alcanivorax sp. (Gammaproteobacteria) | C | AB681673.1 | 91 |
| Alcanivoracaceae bacterium (Gammaproteobacteria) | O | HQ537302.1 | 97 |
| Uncultured betaproteobacteruim | O | GQ274246.1 | 88 |
| Alcanivorax sp. (Gammaproteobacteria) | O | HE586882.1 | 96 |
| Uncultured bacterium | O | EU255846.1 | 81 |
| Uncultured bacterium | S | DQ861039.1 | 91 |
| Alcanivorax sp. (Gammaproteobacteria) | S | HM171217.1 | 95 |
| Alcanivorax sp. (Gammaproteobacteria) | S | HM171217.1 | 98 |
| Marinobactermobilis strain (Gammaproteobacteria) | S | NR_044456.1 | 95 |
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ogino, O.; Koshikawa, H.; Nakahara, T.; Uchiyama, H. Succession of microbial communities during a biostimulation process as evaluated by DGGE and clone library analysis. J. Appl. Microbiol. 2001, 91, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Kadali, K.K.; Simons, K.L.; Skuza, P.P.; Moore, R.B.; Ball, A.S. A complementary approach to identifying and assessing the remediation potential of hydrocarbonoclastic bacteria. J. Microbiol. Methods 2012, 88, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Roose-Amsaleg, C.L.; Garnier-Sillam, E.; Harry, M. Extraction and purification of microbial DNA from soil and sediment samples. Appl. Soil Ecol. 2001, 18, 47–60. [Google Scholar] [CrossRef]
- Widada, J.; Nojiri, H.; Omori, T. Recent developments in molecular techniques for identification and monitoring of Xenobiotic-degrading bacteria and catabolic genes in bioremediation. Appl. Microbiol. Biotechnol. 2002, 60, 45–49. [Google Scholar] [PubMed]
- Connon, S.A.; Giovannoni, S.J. High-throughput methods for culturing microorganisms in very-low-nutrient media yield diverse new marine isolates. Appl. Environ. Microbiol. 2002, 68, 3878–3885. [Google Scholar] [CrossRef] [PubMed]
- Frias-Lopez, J.; Shi, Y.; Tyson, G.; Coleman, M.L.; Schuster, S.C.; Chisholm, S.W.; Delong, E.F. Microbial community gene expression in ocean surface waters. Proc. Natl. Acad. Sci. USA 2008, 105, 3805–3810. [Google Scholar] [CrossRef] [PubMed]
- Muyzer, G.; de Waal, E.C.; Uitterlinden, A.G. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl. Environ. Microbiol. 1993, 59, 695–700. [Google Scholar] [PubMed]
- Nocker, A.; Burr, M.; Camper, A.K. Genotypic microbial community profiling: A critical technical review. Microb. Ecol. 2007, 54, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Muyzer, G. DGGE/TGGE a method for identifying genes from natural ecosystems. Curr. Opin. Microbiol. 1999, 2, 317–322. [Google Scholar] [CrossRef]
- Muyzer, G.; Smalla, K. Application of denaturing gradient gel electrophoresis (DGGE) and temperature gradient gel electrophoresis (TGGE) in microbial ecology. Antonie Van Leeuwenhoek 1998, 73, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, P.J.J.; Illman, W.A. Bioremediation and Natural Attenuation; John Wiley and Sons, Inc.: Hoboken, NJ, USA, 2006. [Google Scholar]
- Felske, A.; Akkermans, A.D.L.; de vos, W.M. Quantification of 16S rRNAs in complex bacterial communities by multiple competitive reverse transcription-PCR in temperature gradient gel electrophoresis fingerprints. Appl. Environ. Microbiol. 1998, 64, 4581–4587. [Google Scholar] [PubMed]
- Cheung, P.; Kinkle, B.K. Mycobacterium diversity and pyrine mineralization in petroleum-contaminated soils. Appl. Environ. Microbiol. 2001, 67, 2222–2229. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Villalba, B.; Calvo, C.; Vilchez, R.; Gonzalez-Lopez, J.; Rodelas, B. TGGE analysis of the diversity of ammonia-oxidizing and denitrifying bacteria in submerged filter biofilms for the treatment of urban wastewater. Appl. Microbiol. Biotechnol. 2006, 72, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Beier, S.; Witzel, K.; Marxsen, J. Bacterial community composition in central European running waters examined by temperature gradient gel electrophoresis and sequence analysis of 16S rRNA genes. Appl. Environ. Microbiol. 2008, 74, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Kaartokallio, H.; Tuomainen, J.; Kuosa, H.; Kuparinen, J.; Martikainen, P.J.; Servomaa, K. Succession of sea-ice bacterial communities in the Baltic sea fast ice. Polar Biol. 2008, 31, 783–793. [Google Scholar] [CrossRef]
- Felske, A.; Wolterink, A.; Lis, R.V.; Akkermans, A.D.L. Phylogeny of the main bacterial 16S rRNA sequences in Drentse A grassland soils (The Netherlands). Appl. Environ. Microbiol. 1998, 64, 871–879. [Google Scholar] [PubMed]
- Panas, P.; McMullan, G.; Dooley, J.S.G. RT-TGGE as a guide for the successful isolation of phosphonoacetate degrading bacteria. J. Appl. Microbiol. 2007, 103, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Nocker, A.; Camper, A.K. Novel approaches toward preferential detection of viable cells using nucleic acid amplification techniques. FEMS Microbiol. Lett. 2009, 291, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Wagner, R. The regulation of ribosomal RNA synthesis and bacterial cell growth. Arch. Microbiol. 1994, 161, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Boon, N.; Top, E.M.; Verstraete, W.; Siciliano, S.D. Bioaugmentation as a tool to protect the structure and function of an activated-sludge microbial community against a 3-chloroaniline shock load. Appl. Environ. Microbiol. 2003, 69, 1511–1520. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Nolan, T.; Pfaffl, M.W. Quantitative real-time RT-PCR—A perspective. J. Mol. Endocrinol. 2005, 34, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.J.; Osborn, A.M. Advantages and limitations of quantitative PCR (Q-PCR)-based approaches in microbial ecology. FEMS Microbiol. Ecol. 2009, 67, 6–20. [Google Scholar] [CrossRef] [PubMed]
- Zoetendal, E.G.; Akkermans, A.D.L.; de vos, W.M. Temperature gradient gel electrophoresis analysis of 16S rRNA from human fecal samples reveals stable and host-specific communities of active bacteria. Appl. Environ. Microbiol. 1998, 64, 3854–3859. [Google Scholar] [PubMed]
- Cappello, S.; Denaro, R.; Genovese, M.; Giuliano, L.; Yakimov, M.M. Predominant growth of Alcanivorax during experiments on “oil spill bioremediation” in mesocosms. Microbiol. Res. 2007, 162, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Kadali, K.K.; Simons, K.L.; Sheppard, P.J.; Ball, A.S. Mineralisation of weathered crude oil by a hydrocarbonoclastic consortia in marine mesocosms. Water Air Soil Pollut. 2012, 223, 4283–4295. [Google Scholar] [CrossRef]
- Girvan, M.S.; Bullimore, J.; Pretty, J.N.; Osborn, A.M.; Ball, A.S. Soil type is the primary determinant of the composition of the total and active bacterial communities in Arable soils. Appl. Environ. Microbiol. 2003, 69, 1800–1809. [Google Scholar] [CrossRef] [PubMed]
- Marzorati, M.; Wittebolle, L.; Boon, N.; Daffonchio, D.; Verstraete, W. How to get more out of molecular fingerprints: Practical tools for microbial ecology. Environ. Microbiol. 2008, 10, 1571–1581. [Google Scholar] [CrossRef] [PubMed]
- Ovreas, L. Population and community level approaches for analysing microbial diversity in natural environments. Ecol. Lett. 2000, 3, 236–251. [Google Scholar] [CrossRef]
- Romantschuk, M.; Sarand, I.; Petanen, T.; Peltola, R.; Jonsson-Vihanne, M.; Koivula, T.; Yrjala, K.; Haahtela, K. Means to improve the effect of in situ bioremediation of contaminated soil: An overview of novel approaches. Environ. Pollut. 2000, 107, 179–185. [Google Scholar] [CrossRef]
- Martinez-Gomez, C.; Vethaak, A.D.; Hylland, K.; Burgeot, T.; Kohler, A.; Lyons, B.P.; Thain, J.; Gubbins, M.J.; Davies, I.M. A guide to toxicity assessment and monitoring effects at lower levels of biological organization following marine oil spill in European waters. ICES J. Mar. Sci. 2010, 67, 1–14. [Google Scholar] [CrossRef]
- Head, I.M.; Jones, D.M.; Roling, W.F.M. Marine microorganisms make a meal of oil. Nat. Rev. 2006, 4, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Onwurah, I.N.E.; Ogugua, V.N.; Onyike, N.B.; Ochonogor, A.E.; Otitoju, O.F. Crude oil spill in environment, effects and some innovative clean-up biotechnologies. Int. J. Environ. Res. 2007, 1, 307–320. [Google Scholar]
- Rojo, F. Degradation of alkanes by bacteria. Environ. Microbiol. 2009, 11, 2477–2490. [Google Scholar] [CrossRef] [PubMed]
- Yuste, L.; Corbella, M.E.; Turiegano, M.J.; Karlson, U.; Puyet, A.; Rojo, F. Characterization of bacterial strains able to degrade to grow on high molecular mass residues from crude oil processing. FEMS Microbiol. Ecol. 2000, 32, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Heuer, H.; Hartung, K.; Wieland, G.; Kramer, I.; Smalla, K. Polynucleotide probes that target a hypervariable region of 16S rRNA genes to identify bacterial isolates corresponding to bands of community fingerprints. Appl. Environ. Microbiol. 1999, 65, 1045–1049. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kadali, K.K.; Shahsavari, E.; Simons, K.L.; Sheppard, P.J.; Ball, A.S. RNA-TGGE, a Tool for Assessing the Potential for Bioremediation in Impacted Marine Ecosystems. J. Mar. Sci. Eng. 2015, 3, 968-980. https://doi.org/10.3390/jmse3030968
Kadali KK, Shahsavari E, Simons KL, Sheppard PJ, Ball AS. RNA-TGGE, a Tool for Assessing the Potential for Bioremediation in Impacted Marine Ecosystems. Journal of Marine Science and Engineering. 2015; 3(3):968-980. https://doi.org/10.3390/jmse3030968
Chicago/Turabian StyleKadali, Krishna K., Esmaeil Shahsavari, Keryn L. Simons, Petra J. Sheppard, and Andrew S. Ball. 2015. "RNA-TGGE, a Tool for Assessing the Potential for Bioremediation in Impacted Marine Ecosystems" Journal of Marine Science and Engineering 3, no. 3: 968-980. https://doi.org/10.3390/jmse3030968