
Uncovering the Prokaryotic Diversity of the Bathyal Waters above the Kuril–Kamchatka Trench
 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
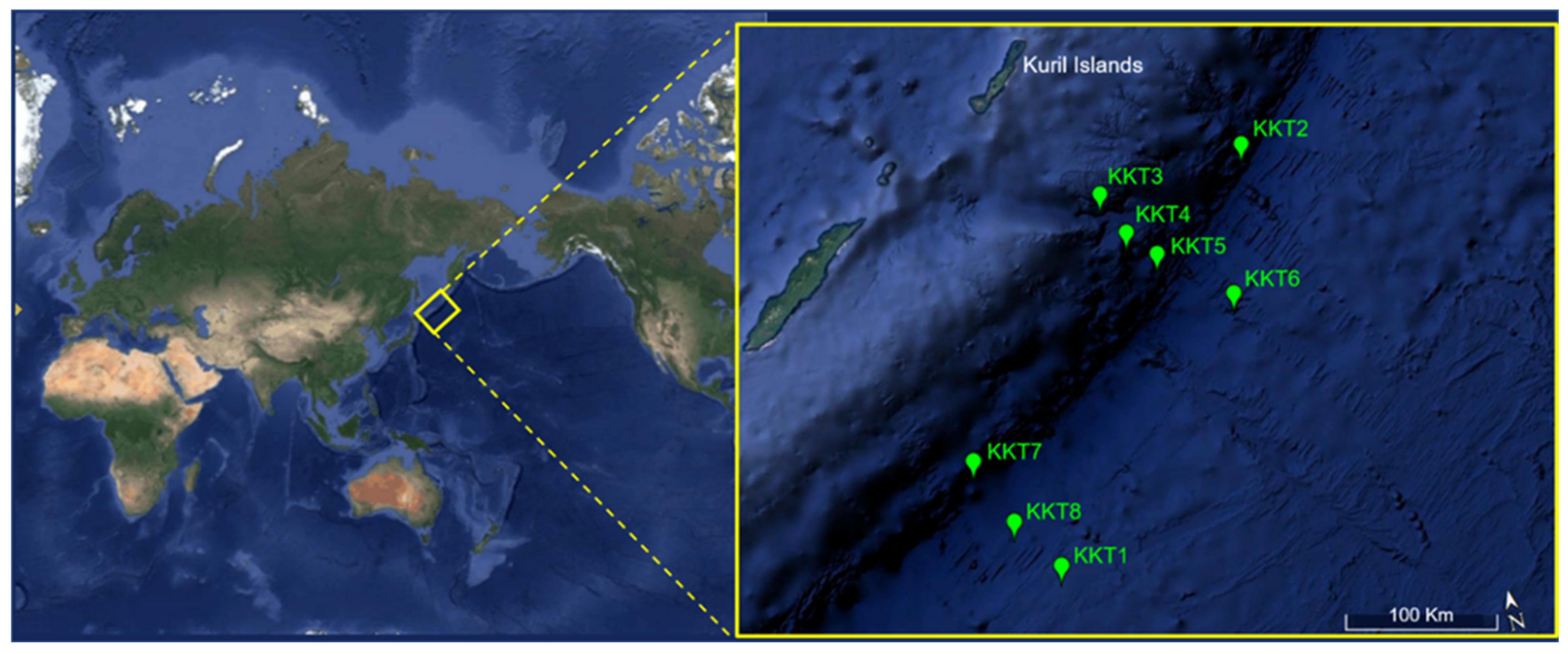
2.1. Collection of Samples and Their Characterization
2.2. Extraction of Total DNA, Preparation and Sequencing of the Amplicon Libraries
2.3. Processing of Sequences and Data Analysis
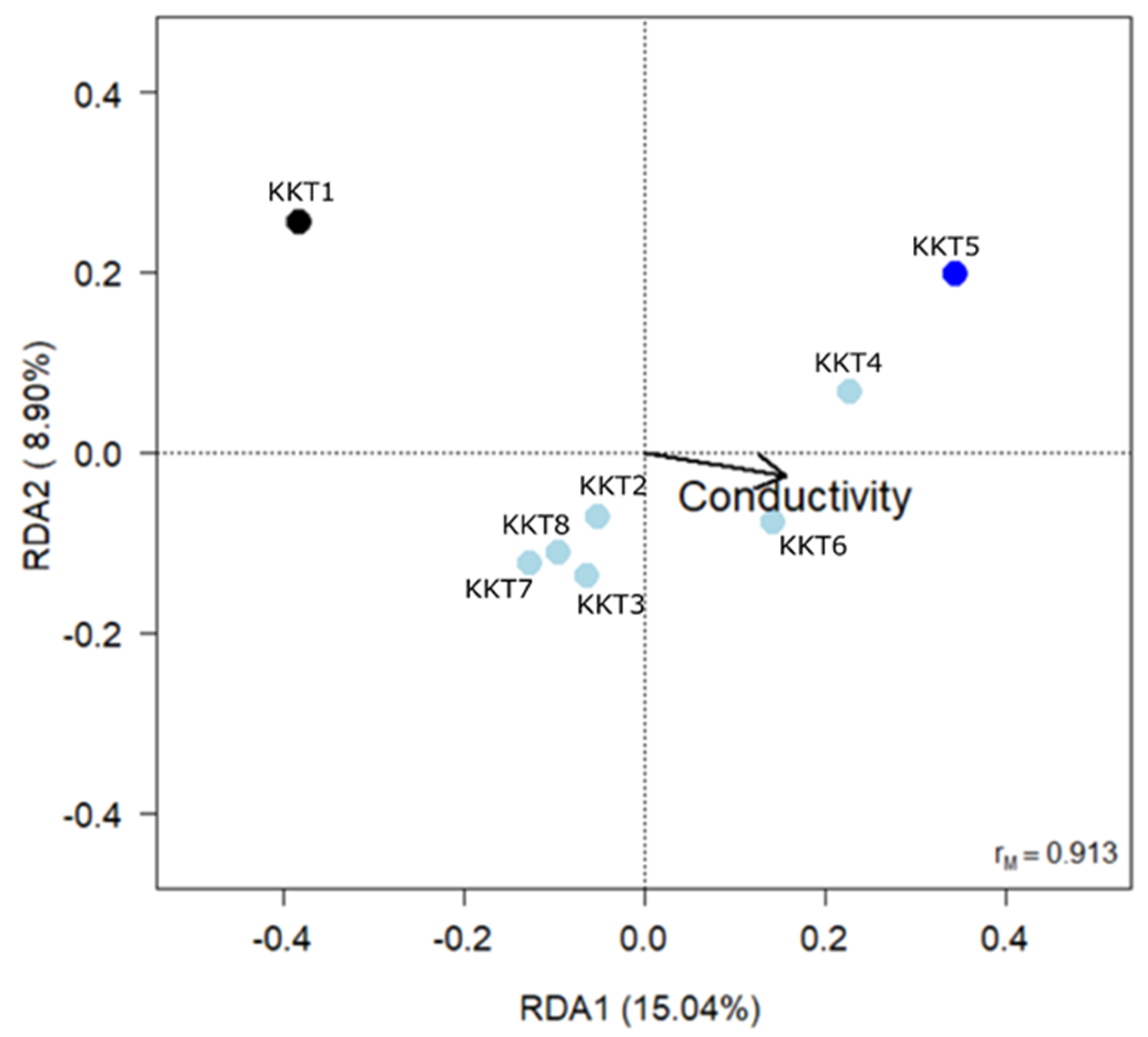
2.4. Statistical Analyses
3. Results and Discussions
3.1. α- and β-Diversity of the KKT Prokaryotic Communities
3.2. Bacterial Community Profiling
3.3. Archaeal Community Profiling
3.4. Bacterial and Archaeal Ecological Function Prediction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kennish, M.J. Practical Handbook of Marine Science, 4th ed.; CRC Press: Boca Raton, FL, USA, 2019; ISBN 9781351654104. [Google Scholar]
- Speight, M.R.; Henderson, P.A. Marine Ecology: Concepts and Applications; John Wiley & Sons: Oxford, UK, 2013; ISBN 978-1-118-68731-4. [Google Scholar]
- Xue, C.-X.; Liu, J.; Lea-Smith, D.J.; Rowley, G.; Lin, H.; Zheng, Y.; Zhu, X.-Y.; Liang, J.; Ahmad, W.; Todd, J.D.; et al. Insights into the Vertical Stratification of Microbial Ecological Roles across the Deepest Seawater Column on Earth. Microorganisms 2020, 8, 1309. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Wang, L.; Wei, Y.; Fang, J. The Hadal Biosphere: Recent Insights and New Directions. In Deep Sea Research Part I: Oceanographic Research Papers; Elsevier: Amsterdam, The Netherlands, 2018; Volume 155, pp. 11–18. [Google Scholar] [CrossRef]
- Nunoura, T.; Takaki, Y.; Hirai, M.; Shimamura, S.; Makabe, A.; Koide, O.; Kikuchi, T.; Miyazaki, J.; Koba, K.; Yoshida, N.; et al. Hadal Biosphere: Insight into the Microbial Ecosystem in the Deepest Ocean on Earth. Proc. Natl. Acad. Sci. USA 2015, 112, 1230–1236. [Google Scholar] [CrossRef] [PubMed]
- Tarn, J.; Peoples, L.M.; Hardy, K.; Cameron, J.; Bartlett, D.H. Identification of Free-Living and Particle-Associated Microbial Communities Present in Hadal Regions of the Mariana Trench. Front. Microbiol. 2016, 7, 665. [Google Scholar] [CrossRef] [PubMed]
- Peoples, L.M.; Donaldson, S.; Osuntokun, O.; Xia, Q.; Nelson, A.; Blanton, J.; Allen, E.E.; Church, M.J.; Bartlett, D.H. Vertically Distinct Microbial Communities in the Mariana and Kermadec Trenches. PLoS ONE 2018, 13, e0195102. [Google Scholar] [CrossRef]
- Peoples, L.M.; Grammatopoulou, E.; Pombrol, M.; Xu, X.; Osuntokun, O.; Blanton, J.; Allen, E.E.; Nunnally, C.C.; Drazen, J.C.; Mayor, D.J.; et al. Microbial Community Diversity within Sediments from Two Geographically Separated Hadal Trenches. Front. Microbiol. 2019, 10, 347. [Google Scholar] [CrossRef]
- Liu, J.; Zheng, Y.; Lin, H.; Wang, X.; Li, M.; Liu, Y.; Yu, M.; Zhao, M.; Pedentchouk, N.; Lea-Smith, D.J.; et al. Proliferation of Hydrocarbon-Degrading Microbes at the Bottom of the Mariana Trench. Microbiome 2019, 7, 1–13. [Google Scholar] [CrossRef]
- Liu, R.; Wang, Z.; Wang, L.; Li, Z.; Fang, J.; Wei, X.; Wei, W.; Cao, J.; Wei, Y.; Xie, Z. Bulk and Active Sediment Prokaryotic Communities in the Mariana and Mussau Trenches. Front. Microbiol. 2020, 11, 1521. [Google Scholar] [CrossRef]
- Gao, Z.; Huang, J.; Cui, G.; Li, W.; Li, J.; Wei, Z.; Chen, J.; Xin, Y.; Cai, D.; Zhang, A.; et al. In Situ Meta-omic Insights into the Community Compositions and Ecological Roles of Hadal Microbes in the Mariana Trench. Environ. Microbiol. 2019, 21, 4092–4108. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, Z.M.; Li, J.; He, L.S.; Cui, G.J.; Li, W.L.; Chen, J.; Xin, Y.Z.; Cai, D.S.; Zhang, A.Q. Hadal Water Sampling by in Situ Microbial Filtration and Fixation (ISMIFF) Apparatus. In Deep Sea Research Part I: Oceanographic Research Papers; Elsevier: Amsterdam, The Netherlands, 2019; Volume 144, pp. 132–137. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, L.; Liu, R.; Li, Z.; Wu, J.X.; Wei, X.; Wei, W.; Fang, J.; Cao, J.; Wei, Y.; et al. Community Structure and Activity Potentials of Archaeal Communities in Hadal Sediments of the Mariana and Mussau Trenches. Mar. Life Sci. Technol. 2022, 4, 150–161. [Google Scholar] [CrossRef]
- Hiraoka, S.; Hirai, M.; Matsui, Y.; Makabe, A.; Minegishi, H.; Tsuda, M.; Juliarni; Rastelli, E.; Danovaro, R.; Corinaldesi, C.; et al. Microbial Community and Geochemical Analyses of Trans-Trench Sediments for Understanding the Roles of Hadal Environments. ISME J. 2019, 14, 740–756. [Google Scholar] [CrossRef]
- Eloe, E.A.; Fadrosh, D.W.; Novotny, M.; Allen, L.; Kim, M.; Lombardo, M.J.; Yee-Greenbaum, J.; Yooseph, S.; Allen, E.E.; Lasken, R.; et al. Going Deeper: Metagenome of a Hadopelagic Microbial Community. PLoS ONE 2011, 6, e20388. [Google Scholar] [CrossRef] [PubMed]
- Eloe, E.A.; Shulse, C.N.; Fadrosh, D.W.; Williamson, S.J.; Allen, E.E.; Bartlett, D.H. Compositional Differences in Particle-Associated and Free-Living Microbial Assemblages from an Extreme Deep-Ocean Environment. Environ. Microbiol. Rep. 2011, 3, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Nunoura, T.; Hirai, M.; Yoshida-Takashima, Y.; Nishizawa, M.; Kawagucci, S.; Yokokawa, T.; Miyazaki, J.; Koide, O.; Makita, H.; Takaki, Y.; et al. Distribution and Niche Separation of Planktonic Microbial Communities in the Water Columns from the Surface to the Hadal Waters of the Japan Trench under the Eutrophic Ocean. Front. Microbiol. 2016, 7, 1261. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Wang, L.; Liu, Q.; Wang, Z.; Li, Z.; Fang, J.; Zhang, L.; Luo, M. Depth-Resolved Distribution of Particle-Attached and Free-Living Bacterial Communities in the Water Column of the New Britain Trench. Front. Microbiol. 2018, 9, 625. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, W.; Liu, Y.; Cai, M.; Luo, Z.; Li, M. Metagenomics Reveals Microbial Diversity and Metabolic Potentials of Seawater and Surface Sediment from a Hadal Biosphere at the Yap Trench. Front. Microbiol. 2018, 9, 2402. [Google Scholar] [CrossRef]
- Zhao, X.; Luo, H.; He, S.; Yang, B.; Wei, T.; Hu, Y.; Wang, Z.; Li, X. Vertical Distribution of Size-Fractionated Bacterial Communities in the Water Column of the Atacama Trench. Reg. Stud. Mar. Sci. 2022, 55, 102470. [Google Scholar] [CrossRef]
- Brandt, A.; Brix, S.; Riehl, T.; Malyutina, M. Biodiversity and Biogeography of the Abyssal and Hadal Kuril-Kamchatka Trench and Adjacent NW Pacific Deep-Sea Regions. Prog. Oceanogr. 2020, 181, 102232. [Google Scholar] [CrossRef]
- Stern, R.J. Ocean Trenches. In Encyclopedia of Geology, 2nd ed.; Alderton, D., Elias, S.A., Eds.; Academic Press: Cambridge, UK, 2021; Volume 2, pp. 845–854. [Google Scholar]
- Jamieson, A.J.; Stewart, H.A. Hadal Zones of the Northwest Pacific Ocean. Prog. Oceanogr. 2021, 190, 102477. [Google Scholar] [CrossRef]
- Kamenev, G.M.; Mordukhovich, V.V.; Alalykina, I.L.; Chernyshev, A.V.; Maiorova, A.S. Macrofauna and Nematode Abundance in the Abyssal and Hadal Zones of Interconnected Deep-Sea Ecosystems in the Kuril Basin (Sea of Okhotsk) and the Kuril-Kamchatka Trench (Pacific Ocean). Front. Mar. Sci. 2022, 9, 34. [Google Scholar] [CrossRef]
- Mitnik, L.M.; Khazanova, E.S.; Dubina, V.A. Mesoscale and Synoptic Scale Dynamic Phenomena in the Oyashio Current Region Observed in SAR Imagery. Int. J. Remote Sens. 2019, 41, 5861–5883. [Google Scholar] [CrossRef]
- Fuhr, M.; Laukert, G.; Yu, Y.; Nürnberg, D.; Frank, M. Tracing Water Mass Mixing From the Equatorial to the North Pacific Ocean With Dissolved Neodymium Isotopes and Concentrations. Front. Mar. Sci. 2021, 7, 1261. [Google Scholar] [CrossRef]
- Andreev, A.; Pipko, I. Water Circulation, Temperature, Salinity, and pCO2 Distribution in the Surface Layer of the East Kamchatka Current. J. Mar. Sci. Eng. 2022, 10, 1787. [Google Scholar] [CrossRef]
- Wolff, T. The Hadal Community, an Introduction. Deep. Sea Res. (1953) 1959, 6, 95–124. [Google Scholar] [CrossRef]
- Bogorov, V.G. Fauna of the Kurile–Kamchatka Trench and Its Environment, Based on Data of the 38th Cruise of the R/V “Vityaz”; Israel Program for Scientific Translations: Jerusalem, Israel, 1972. [Google Scholar]
- Zenkevich, L.A. Biology of the Seas of the USSR; George Allen & Unwin Ltd.: London, UK, 1963. [Google Scholar]
- Krylova, E.M.; Drozdov, A.; Mironov, A. Presence of Bacteria in Gills of Hadal Bivalve “Vesicomya” Sergeevi Filatova, 1971. Ruthenica 2000, 10, 76–79. [Google Scholar]
- Karaseva, N.P.; Gantsevich, M.M.; Obzhirov, A.I.; Shakirov, R.B.; Starovoytov, A.V.; Smirnov, R.V.; Malakhov, V.V. Siboglinids (Annelida, Siboglinidae) as Possible Indicators of Carbohydrates on the Case of the Sea of Okhotsk. Dokl. Akad. Nauk. 2019, 486, 127–130. [Google Scholar] [CrossRef]
- Li, Y.; Jing, H.; Xia, X.; Cheung, S.; Suzuki, K.; Liu, H. Metagenomic Insights into the Microbial Community and Nutrient Cycling in the Western Subarctic Pacific Ocean. Front. Microbiol. 2018, 9, 623. [Google Scholar] [CrossRef]
- Jing, H.; Cheung, S.; Xia, X.; Suzuki, K.; Nishioka, J.; Liu, H. Geographic Distribution of Ammonia-Oxidizing Archaea along the Kuril Islands in the Western Subarctic Pacific. Front. Microbiol. 2017, 8, 1247. [Google Scholar] [CrossRef]
- Zenkevitch, L.A.; Birstein, Y.A.; Belyaev, G.M. Study of the Bottom Fauna of the Kuril-Kamchatka Basin. Tr. Inst. Okeanol. im. P. P. Shirshova, Akad. Nauk SSSR 1955, 12, 345–381. [Google Scholar]
- Malyutina, M.v.; Brandt, A. Munnopsidae (Crustacea, Isopoda, Asellota) from the Kuril–Kamchatka Trench with a Regional and Inter-Ocean Comparison of Their Biogeographic and Richness Patterns. Prog. Oceanogr. 2020, 183, 102289. [Google Scholar] [CrossRef]
- Brandt, A.; Malyutina, M.V. The German–Russian Deep-Sea Expedition KuramBio (Kurile Kamchatka Biodiversity Studies) on Board of the RV Sonne in 2012 Following the Footsteps of the Legendary Expeditions with RV Vityaz. In Deep Sea Research Part II: Topical Studies in Oceanography; Elsevier: Amsterdam, The Netherlands, 2015; Volume 111, pp. 1–9. [Google Scholar] [CrossRef]
- Gorrasi, S.; Franzetti, A.; Brandt, A.; Minzlaff, U.; Pasqualetti, M.; Fenice, M. Insights into the Prokaryotic Communities of the Abyssal-Hadal Benthic-Boundary Layer of the Kuril Kamchatka Trench. Environ. Microbiome 2023, 18, 1–18. [Google Scholar] [CrossRef]
- Huber, J.A.; Mark Welch, D.B.; Morrison, H.G.; Huse, S.M.; Neal, P.R.; Butterfield, D.A.; Sogin, M.L. Microbial Population Structures in the Deep Marine Biosphere. Science 2007, 318, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qian, P.Y. Conservative Fragments in Bacterial 16S RRNA Genes and Primer Design for 16S Ribosomal DNA Amplicons in Metagenomic Studies. PLoS ONE 2009, 4, e7401. [Google Scholar] [CrossRef] [PubMed]
- Takai, K.; Horikoshi, K. Rapid Detection and Quantification of Members of the Archaeal Community by Quantitative PCR Using Fluorogenic Probes. Appl. Environ. Microbiol. 2000, 66, 5066–5072. [Google Scholar] [CrossRef] [PubMed]
- Baker, G.C.; Smith, J.J.; Cowan, D.A. Review and Re-Analysis of Domain-Specific 16S Primers. J. Microbiol. Methods 2003, 55, 541–555. [Google Scholar] [CrossRef]
- Gorrasi, S.; Pasqualetti, M.; Franzetti, A.; Gonzalez-Martinez, A.; Gonzalez-Lopez, J.; Muñoz-Palazon, B.; Fenice, M. Persistence of Enterobacteriaceae Drawn into a Marine Saltern (Saline Di Tarquinia, Italy) from the Adjacent Coastal Zone. Water 2021, 13, 1443. [Google Scholar] [CrossRef]
- Gorrasi, S.; Franzetti, A.; Ambrosini, R.; Pittino, F.; Pasqualetti, M.; Fenice, M. Spatio-Temporal Variation of the Bacterial Communities along a Salinity Gradient within a Thalassohaline Environment (Saline Di Tarquinia Salterns, Italy). Molecules 2021, 26, 1338. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly Accurate OTU Sequences from Microbial Amplicon Reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian Classifier for Rapid Assignment of RRNA Sequences into the New Bacterial Taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Claesson, M.J.; O’Sullivan, O.; Wang, Q.; Nikkilä, J.; Marchesi, J.R.; Smidt, H.; de Vos, W.M.; Ross, R.P.; O’Toole, P.W. Comparative Analysis of Pyrosequencing and a Phylogenetic Microarray for Exploring Microbial Community Structures in the Human Distal Intestine. PLoS ONE 2009, 4, e6669. [Google Scholar] [CrossRef]
- Good, I.J. The population frequencies of species and the estimation of population parameters. Biometrika 1953, 40, 237–264. [Google Scholar] [CrossRef]
- Chao, A. Nonparametric estimation of the number of classes in a population. Scand. J. Statist. 1984, 11, 265–270. [Google Scholar]
- Shannon, C.E. A Mathematical Theory of Communication. Bell. Sys. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef]
- Gini, C. Variabilità e Mutabilità. Contributo Allo Studio Delle Distribuzioni e Delle Relazioni Statistiche; Tipografia di Paolo Cuppini: Bologna, Italy, 1912. [Google Scholar]
- McMurdie, P.J.; Holmes, S. Waste Not, Want Not: Why Rarefying Microbiome Data Is Inadmissible. PLoS Comput. Biol. 2014, 10, e1003531. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Yekutieli, D. The Control of the False Discovery Rate in Multiple Testing under Dependency. Ann. Stat. 2001, 29, 1165–1188. [Google Scholar] [CrossRef]
- Louca, S.; Parfrey, L.W.; Doebeli, M. Decoupling Function and Taxonomy in the Global Ocean Microbiome. Science 2016, 353, 1272–1277. [Google Scholar] [CrossRef]
- Salazar, G.; Cornejo-Castillo, F.M.; Benítez-Barrios, V.; Fraile-Nuez, E.; Álvarez-Salgado, X.A.; Duarte, C.M.; Gasol, J.M.; Acinas, S.G. Global Diversity and Biogeography of Deep-Sea Pelagic Prokaryotes. ISME J. 2015, 10, 596–608. [Google Scholar] [CrossRef]
- Wei, Z.F.; Li, W.L.; Huang, J.M.; Wang, Y. Metagenomic Studies of SAR202 Bacteria at the Full-Ocean Depth in the Mariana Trench. In Deep Sea Research Part I: Oceanographic Research Papers; Elsevier: Amsterdam, The Netherlands, 2020; Volume 165, p. 103396. [Google Scholar] [CrossRef]
- Jing, H.; Xia, X.; Suzuki, K.; Liu, H. Vertical Profiles of Bacteria in the Tropical and Subarctic Oceans Revealed by Pyrosequencing. PLoS ONE 2013, 8, e79423. [Google Scholar] [CrossRef]
- Tseng, C.H.; Chiang, P.W.; Lai, H.C.; Shiah, F.K.; Hsu, T.C.; Chen, Y.L.; Wen, L.S.; Tseng, C.M.; Shieh, W.Y.; Saeed, I.; et al. Prokaryotic Assemblages and Metagenomes in Pelagic Zones of the South China Sea. BMC Genomics 2015, 16, 219. [Google Scholar] [CrossRef]
- Tian, J.; Fan, L.; Liu, H.; Liu, J.; Li, Y.; Qin, Q.; Gong, Z.; Chen, H.; Sun, Z.; Zou, L.; et al. A Nearly Uniform Distributional Pattern of Heterotrophic Bacteria in the Mariana Trench Interior. In Deep Sea Research Part I: Oceanographic Research Papers; Elsevier: Amsterdam, The Netherlands, 2018; Volume 142, pp. 116–126. [Google Scholar] [CrossRef]
- Herndl, G.J.; Bayer, B.; Baltar, F.; Reinthaler, T. Prokaryotic life in the deep ocean’s water column. Annu. Rev. Mar. Sci. 2023, 15, 461–483. [Google Scholar] [CrossRef]
- Mestre, M.; Borrull, E.; Sala, M.M.; Gasol, J.M. Patterns of bacterial diversity in the marine planktonic particulate matter continuum. ISME J. 2017, 11, 999–1010. [Google Scholar] [CrossRef]
- Milici, M.; Vital, M.; Tomasch, J.; Badewien, T.H.; Giebel, H.A.; Plumeier, I.; Wang, H.; Pieper, D.H.; Wagner-Döbler, I.; Simon, M. Diversity and Community Composition of Particle-Associated and Free-Living Bacteria in Mesopelagic and Bathypelagic Southern Ocean Water Masses: Evidence of Dispersal Limitation in the Bransfield Strait. Limnol. Oceanogr. 2017, 62, 1080–1095. [Google Scholar] [CrossRef]
- Qin, J.; Feng, Y.; Lü, X.; Zong, Z. Precise Species Identification for Acinetobacter: A Genome-Based Study with Description of Two Novel Acinetobacter Species. mSystems 2021, 6, e0023721. [Google Scholar] [CrossRef] [PubMed]
- Nemec, A.; Radolfová-Křížová, L.; Maixnerová, M.; Nemec, M.; Shestivska, V.; Španělová, P.; Kyselková, M.; Wilharm, G.; Higgins, P.G. Acinetobacter smyesii sp. nov., Widespread in the Soil and Water Environment and Animals. Int. J. Syst. Evol. Microbiol. 2022, 72, 005642. [Google Scholar] [CrossRef] [PubMed]
- Pandolfo, E.; Caracciolo, A.B.; Rolando, L. Recent Advances in Bacterial Degradation of Hydrocarbons. Water 2023, 15, 375. [Google Scholar] [CrossRef]
- Ma, M.; Gao, W.; Li, Q.; Han, B.; Zhu, A.; Yang, H.; Zheng, L. Biodiversity and Oil Degradation Capacity of Oil-Degrading Bacteria Isolated from Deep-Sea Hydrothermal Sediments of the South Mid-Atlantic Ridge. Mar. Pollut. Bull. 2021, 171, 112770. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, J.; Zhou, S.; Zheng, Y.; Wu, Y.; Kogure, K.; Zhang, X.H. Diversity of Culturable Heterotrophic Bacteria from the Mariana Trench and Their Ability to Degrade Macromolecules. Mar. Life Sci. Technol. 2020, 2, 181–193. [Google Scholar] [CrossRef]
- Nitahara, S.; Kato, S.; Usui, A.; Urabe, T.; Suzuki, K.; Yamagishi, A. Archaeal and Bacterial Communities in Deep-Sea Hydrogenetic Ferromanganese Crusts on Old Seamounts of the Northwestern Pacific. PLoS ONE 2017, 12, e0173071. [Google Scholar] [CrossRef]
- Zhang, J.; Sun, Q.L.; Zeng, Z.G.; Chen, S.; Sun, L. Microbial Diversity in the Deep-Sea Sediments of Iheya North and Iheya Ridge, Okinawa Trough. Microbiol. Res. 2015, 177, 43–52. [Google Scholar] [CrossRef]
- Maruyama, A.; Honda, D.; Yamamoto, H.; Kitamura, K.; Higashihara, T. Phylogenetic Analysis of Psychrophilic Bacteria Isolated from the Japan Trench, Including a Description of the Deep-Sea Species Psychrobacter pacificensis sp. nov. Int. J. Syst. Evol. Microbiol. 2000, 50, 835–846. [Google Scholar] [CrossRef]
- Durand, P.; Benyagoub, A.; Prieur, D. Numerical Taxonomy of Heterotrophic Sulfur-Oxidizing Bacteria Isolated from Southwestern Pacific Hydrothermal Vents. Can. J. Microbiol. 2011, 40, 690–697. [Google Scholar] [CrossRef]
- Zhang, L.; Kang, M.; Xu, J.; Xu, J.; Shuai, Y.; Zhou, X.; Yang, Z.; Ma, K. Bacterial and Archaeal Communities in the Deep-Sea Sediments of Inactive Hydrothermal Vents in the Southwest India Ridge. Sci. Rep. 2016, 6, 25982. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.D.; Calomiris, J.J.; Herbert, T.L.; Colwell, R.R. Petroleum Hydrocarbons: Degradation and Growth Potential for Atlantic Ocean Sediment Bacteria. Mar. Biol. 1976, 34, 1–9. [Google Scholar] [CrossRef]
- Rappé, M.S.; Connon, S.A.; Vergin, K.L.; Giovannoni, S.J. Cultivation of the ubiquitous SAR11 marine bacterioplankton clade. Nature 2002, 418, 630–633. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, S.J. SAR11 Bacteria: The Most Abundant Plankton in the Oceans. Annu. Rev. Mar. Sci. 2017, 9, 231–255. [Google Scholar] [CrossRef] [PubMed]
- Moore, E.R.; Davie-Martin, C.L.; Giovannoni, S.J.; Halsey, K.H. Pelagibacter Metabolism of Diatom-Derived Volatile Organic Compounds Imposes an Energetic Tax on Photosynthetic Carbon Fixation. Environ. Microbiol. 2020, 22, 1720–1733. [Google Scholar] [CrossRef]
- Jin, L.; Huy, H.; Kim, K.K.; Lee, H.G.; Kim, H.S.; Ahn, C.Y.; Oh, H.M. Aquihabitans daechungensis gen. nov., sp. nov., an Actinobacterium Isolated from Reservoir Water. Int. J. Syst. Evol. Microbiol. 2013, 63, 2970–2974. [Google Scholar] [CrossRef]
- Tan, G.; Liu, Y.; Peng, S.; Yin, H.; Meng, D.; Tao, J.; Gu, Y.; Li, J.; Yang, S.; Xiao, N.; et al. Soil Potentials to Resist Continuous Cropping Obstacle: Three Field Cases. Environ. Res. 2021, 200, 111319. [Google Scholar] [CrossRef]
- Xue, Y.; Zhou, J.; Chen, X.; Liao, M.; Zhang, L.; Xie, J.; Sun, C. Microbiota in Monocultured Litopenaeus vannamei vs. Polyculture with Trachinotus ovatus. Isr. J. Aquac.-Bamidgeh 2023, 75, 1–14. [Google Scholar] [CrossRef]
- Liu, H.; Yan, C.; Hao, C.; Wang, D.; Liu, Y.; Luo, Z.B.; Han, S.Z.; Wang, J.X.; Li, D.; Zhu, J.; et al. Dynamic Changes in Intestinal Microbiota and Metabolite Composition of Pre-Weaned Beef Calves. Microb. Pathog. 2023, 175, 105991. [Google Scholar] [CrossRef]
- Yu, B.; Liu, C.; Wang, S.; Wang, W.; Zhao, S.; Zhu, G. Applying Constructed Wetland-Microbial Electrochemical System to Enhance NH4+ Removal at Low Temperature. Sci. Total Environ. 2020, 724, 138017. [Google Scholar] [CrossRef]
- Nogi, Y.; Hosoya, S.; Kato, C.; Horikoshi, K. Colwellia piezophila sp. nov., a Novel Piezophilic Species from Deep-Sea Sediments of the Japan Trench. Int. J. Syst. Evol. Microbiol. 2004, 54, 1627–1631. [Google Scholar] [CrossRef] [PubMed]
- Peoples, L.M.; Bartlett, D.H. Ecogenomics of Deep-Ocean Microbial Bathytypes. In Microbial Ecology of Extreme Environments; Chénard, C., Lauro, F.M., Eds.; Springer: Cham, Switzerland, 2017; pp. 7–50. [Google Scholar]
- Zhang, M.; Li, A.; Yao, Q.; Wu, Q.; Zhu, H. Nitrogen Removal Characteristics of a Versatile Heterotrophic Nitrifying-Aerobic Denitrifying Bacterium, Pseudomonas bauzanensis DN13-1, Isolated from Deep-Sea Sediment. Bioresour. Technol. 2020, 305, 122626. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Sun, J.; Hao, J. Spatial Variability of Bacterial Community Compositions in the Mariana Trench. Can. J. Microbiol. 2022, 68, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Fujinami, S.; Oikawa, Y.; Araki, T.; Shinmura, Y.; Midorikawa, R.; Ishizaka, H.; Kato, C.; Horikoshi, K.; Ito, M.; Tamegai, H. Genome Sequence of the Deep-Sea Denitrifier Pseudomonas sp. Strain MT-1, Isolated from the Mariana Trench. Genome Announc. 2014, 2, e131314. [Google Scholar] [CrossRef] [PubMed]
- Peoples, L.M.; Kyaw, T.S.; Ugalde, J.A.; Mullane, K.K.; Chastain, R.A.; Yayanos, A.A.; Kusube, M.; Methé, B.A.; Bartlett, D.H. Distinctive Gene and Protein Characteristics of Extremely Piezophilic Colwellia. BMC Genom. 2020, 21, 692. [Google Scholar] [CrossRef]
- Appolinario, L.R.; Tschoeke, D.; Paixão, R.V.S.; Venas, T.; Calegario, G.; Leomil, L.; Silva, B.S.; Thompson, C.C.; Thompson, F.L. Metagenomics Sheds Light on the Metabolic Repertoire of Oil-Biodegrading Microbes of the South Atlantic Ocean. Environ. Pollut. 2019, 249, 295–304. [Google Scholar] [CrossRef]
- Zheng, M.L.; Jiao, J.Y.; Dong, L.; Han, M.X.; Li, L.H.; Xiao, M.; Chen, C.; Qu, P.H.; Li, W.J. Pseudofrancisella aestuarii gen. nov., sp. nov., a Novel Member of the Family Francisellaceae Isolated from Estuarine Seawater. Anton. Leeuw. Int. J. 2019, 112, 877–886. [Google Scholar] [CrossRef]
- Shade, A.; Handelsman, J. Beyond the Venn Diagram: The Hunt for a Core Microbiome. Environ. Microbiol. 2012, 14, 4–12. [Google Scholar] [CrossRef]
- Falagán, C.; Johnson, D.B. Acidibacter ferrireducens gen. nov., sp. nov.: An Acidophilic Ferric Iron-Reducing Gammaproteobacterium. Extremophiles 2014, 18, 1067–1073. [Google Scholar] [CrossRef]
- Lu, H.; Deng, T.; Liu, F.; Wang, Y.; Yang, X.; Xu, M. Duganella albus sp. nov., Duganella aquatilis sp. nov., Duganella pernnla sp. nov. and Duganella levis sp. nov., Isolated from Subtropical Streams in China. Int. J. Syst. Evol. Microbiol. 2020, 70, 3801–3808. [Google Scholar] [CrossRef]
- Li, W.L.; Huang, J.M.; Zhang, P.W.; Cui, G.J.; Wei, Z.F.; Wu, Y.Z.; Gao, Z.M.; Han, Z.; Wang, Y. Periodic and Spatial Spreading of Alkanes and Alcanivorax Bacteria in Deep Waters of the Mariana Trench. Appl. Environ. Microbiol. 2019, 85, e02089-18. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.E.; Richter, R.A.; Dupont, C.L. Planktonic Marine Archaea. Annu. Rev. Mar. Sci. 2019, 11, 131–158. [Google Scholar] [CrossRef] [PubMed]
- Evans, P.N.; Boyd, J.A.; Leu, A.O.; Woodcroft, B.J.; Parks, D.H.; Hugenholtz, P.; Tyson, G.W. An Evolving View of Methane Metabolism in the Archaea. Nat. Rev. Microbiol. 2019, 17, 219–232. [Google Scholar] [CrossRef]
- Liu, X.; Li, M.; Castelle, C.J.; Probst, A.J.; Zhou, Z.; Pan, J.; Liu, Y.; Banfield, J.F.; Gu, J.D. Insights into the Ecology, Evolution, and Metabolism of the Widespread Woesearchaeotal Lineages. Microbiome 2018, 6, 1–16. [Google Scholar] [CrossRef]
- Huang, W.C.; Liu, Y.; Zhang, X.; Zhang, C.J.; Zou, D.; Zheng, S.; Xu, W.; Luo, Z.; Liu, F.; Li, M. Comparative Genomic Analysis Reveals Metabolic Flexibility of Woesearchaeota. Nat. Commun. 2021, 12, 5281. [Google Scholar] [CrossRef] [PubMed]
- Gugliandolo, C.; Maugeri, T.L. Phylogenetic Diversity of Archaea in Shallow Hydrothermal Vents of Eolian Islands, Italy. Diversity 2019, 11, 156. [Google Scholar] [CrossRef]
- Zhong, H.; Zhong, H.; Lehtovirta-Morley, L.; Liu, J.; Liu, J.; Zheng, Y.; Lin, H.; Song, D.; Todd, J.D.; Tian, J.; et al. Novel Insights into the Thaumarchaeota in the Deepest Oceans: Their Metabolism and Potential Adaptation Mechanisms. Microbiome 2020, 8, 1–16. [Google Scholar] [CrossRef]
- Bayer, B.; Hansman, R.L.; Bittner, M.J.; Noriega-Ortega, B.E.; Niggemann, J.; Dittmar, T.; Herndl, G.J. Ammonia-Oxidizing Archaea Release a Suite of Organic Compounds Potentially Fueling Prokaryotic Heterotrophy in the Ocean. Environ. Microbiol. 2019, 21, 4062–4075. [Google Scholar] [CrossRef]
- Kim, J.G.; Gazi, K.S.; Awala, S.I.; Jung, M.Y.; Rhee, S.K. Ammonia-Oxidizing Archaea in Biological Interactions. J. Microbiol. 2021, 59, 298–310. [Google Scholar] [CrossRef]
- Kitzinger, K.; Marchant, H.K.; Bristow, L.A.; Herbold, C.W.; Padilla, C.C.; Kidane, A.T.; Littmann, S.; Daims, H.; Pjevac, P.; Stewart, F.J.; et al. Single Cell Analyses Reveal Contrasting Life Strategies of the Two Main Nitrifiers in the Ocean. Nat. Commun. 2020, 11, 767. [Google Scholar] [CrossRef]
- Shi, J.; Wang, H.; Zeng, Y.; Fan, Y.; Chen, H.; Yuan, C.; Li, Y.; Huang, M.; Shi, X.; He, P. Diversity and Distribution of Archaeal and Bacterial Nitrifiers in Deep Oceans. J. Sea Res. 2023, 193, 102389. [Google Scholar] [CrossRef]
- Gutierrez, T. Occurrence and Roles of the Obligate Hydrocarbonoclastic Bacteria in the Ocean When There Is No Obvious Hydrocarbon Contamination. In Taxonomy, Genomics and Ecophysiology of Hydrocarbon-Degrading Microbes; McGenity, T., Ed.; Springer: Cham, Switzerland, 2019; pp. 337–352. [Google Scholar]
- Yakimov, M.M.; Bargiela, R.; Golyshin, P.N. Calm and Frenzy: Marine Obligate Hydrocarbonoclastic Bacteria Sustain Ocean Wellness. Curr. Opin. Biotechnol. 2022, 73, 337–345. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Station | Bottom Depth (m) | Coordinates (Latitude; Longitude) | Sampling Depth (m) | Temperature (°C) | Conductivity (S/m) | Oxygen (V) |
|---|---|---|---|---|---|---|---|
| KKT1 | St.1 | 5144.4 | 43° 49.20′ N; 151° 45.60′ E | 2000 | 1.70 | 3.12 | 1.89 |
| KKT2 | St.2 | 8126.8 | 46° 04.62′ N; 153° 58.20′ E | 1000 | 2.72 | 3.14 | 1.04 |
| KKT3 | St.3 | 5958.3 | 45° 57.73′ N; 152° 39.91′ E | 1000 | 2.79 | 3.14 | 0.99 |
| KKT4 | St.4 | 6040.1 | 45° 41.58′ N; 152° 49.49′ E | 1000 | 2.94 | 3.15 | 1.01 |
| KKT5 | St.5 | 7757.6 | 45° 31.35′ N; 153° 02.82′ E | 1500 | 2.33 | 3.14 | 1.12 |
| KKT6 | St.6 | 59526 | 45° 11.00′ N; 153° 36.99′ E | 1000 | 2.72 | 3.14 | 0.92 |
| KKT7 | St.7 | 8392.0 | 44° 31.50′ N; 151° 11.59′ E | 1000 | 2.54 | 3.13 | 1.03 |
| KKT8 | St.8 | 6547.0 | 44° 07.59′ N; 151° 26.58′ E | 1000 | 2.54 | 3.13 | 1.04 |
| Bacteria | Archaea | |||||||
|---|---|---|---|---|---|---|---|---|
| Sample | N. OTUs | Chao1 Index | Shannon Index | Gini Index | N. OTUs | Chao1 Index | Shannon Index | Gini Index |
| KKT1 | 950 | 1165.458 | 3.442 | 0.938 | 327 | 367.167 | 3.774 | 0.942 |
| KKT2 | 1055 | 1228.257 | 4.474 | 0.908 | 441 | 488.264 | 4.090 | 0.914 |
| KKT3 | 994 | 1266.739 | 4.122 | 0.922 | 417 | 437.638 | 3.868 | 0.928 |
| KKT4 | 902 | 1107.271 | 3.938 | 0.937 | 267 | 313.600 | 3.155 | 0.964 |
| KKT5 | 867 | 1031.864 | 3.932 | 0.940 | 252 | 342.417 | 3.243 | 0.966 |
| KKT6 | 1019 | 1248.700 | 3.969 | 0.922 | 340 | 397.122 | 3.380 | 0.955 |
| KKT7 | 944 | 1175.207 | 3.910 | 0.936 | 401 | 475.094 | 3.816 | 0.927 |
| KKT8 | 709 | 1035.237 | 2.693 | 0.972 | 395 | 458.133 | 3.902 | 0.927 |
| Variable | df | Variance | F | p |
|---|---|---|---|---|
| Conductivity | 1 | 0.0322 | 2.455 | 0.027 |
| Depth | 1 | 0.0211 | 1.613 | 0.147 |
| Residuals | 5 | |||
| F2,5 = 2.102, p = 0.032, R2adj = 0.239 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorrasi, S.; Brandt, A.; Pittino, F.; Franzetti, A.; Pasqualetti, M.; Muñoz-Palazon, B.; Novello, G.; Fenice, M. Uncovering the Prokaryotic Diversity of the Bathyal Waters above the Kuril–Kamchatka Trench. J. Mar. Sci. Eng. 2023, 11, 2145. https://doi.org/10.3390/jmse11112145
Gorrasi S, Brandt A, Pittino F, Franzetti A, Pasqualetti M, Muñoz-Palazon B, Novello G, Fenice M. Uncovering the Prokaryotic Diversity of the Bathyal Waters above the Kuril–Kamchatka Trench. Journal of Marine Science and Engineering. 2023; 11(11):2145. https://doi.org/10.3390/jmse11112145
Chicago/Turabian StyleGorrasi, Susanna, Angelika Brandt, Francesca Pittino, Andrea Franzetti, Marcella Pasqualetti, Barbara Muñoz-Palazon, Giorgia Novello, and Massimiliano Fenice. 2023. "Uncovering the Prokaryotic Diversity of the Bathyal Waters above the Kuril–Kamchatka Trench" Journal of Marine Science and Engineering 11, no. 11: 2145. https://doi.org/10.3390/jmse11112145