

Diversity and Biogeography of Coral Mucus-Associated Bacterial Communities: The Case of Acropora formosa
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. PCR Amplification and 16S rRNA Gene Sequencing
2.3. Data Cleaning and Analyses
3. Results
3.1. Overview of the Sequencing Dataset and Data Preprocessing
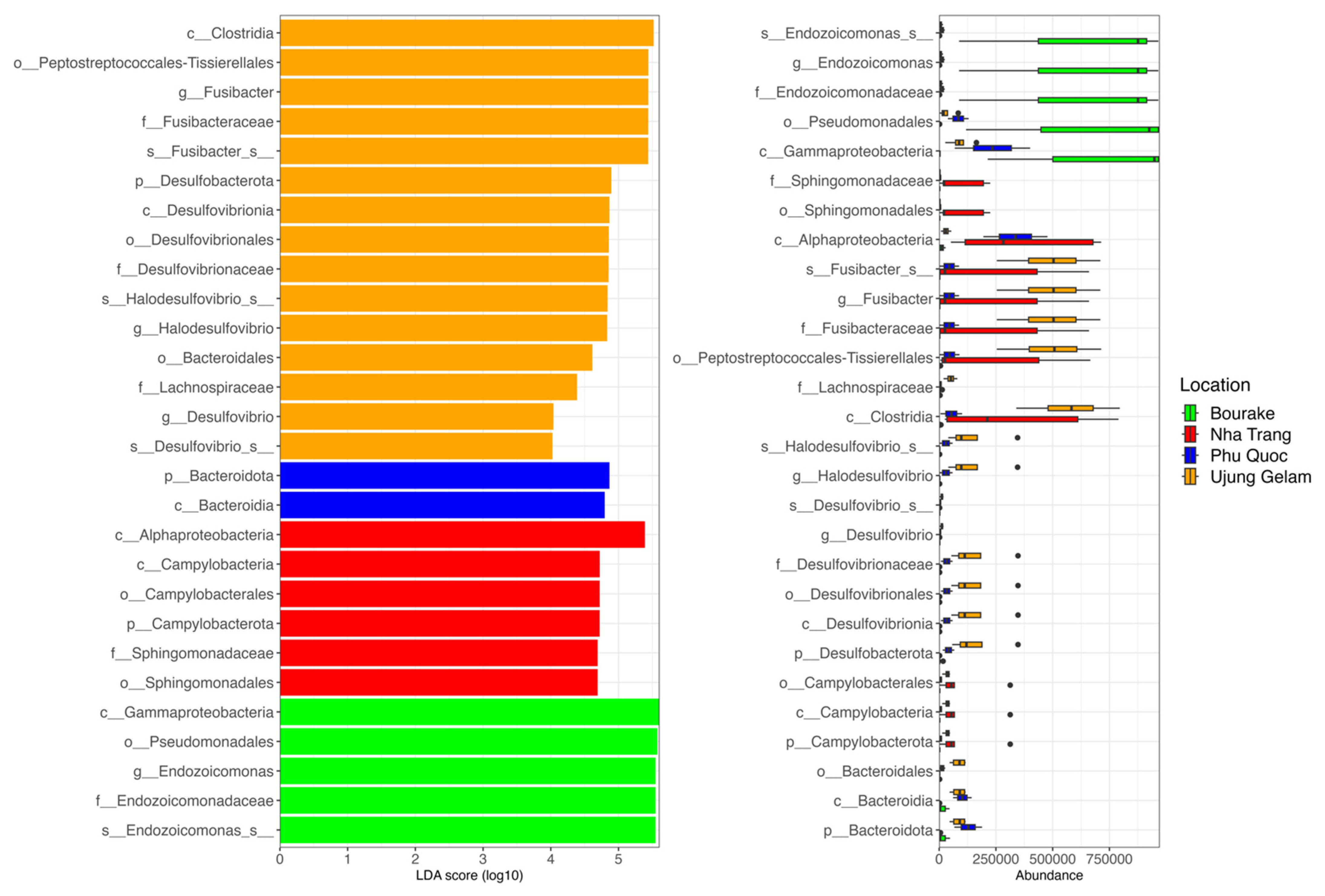
3.2. Taxonomic Assignment of Coral-Associated Bacterial Communities
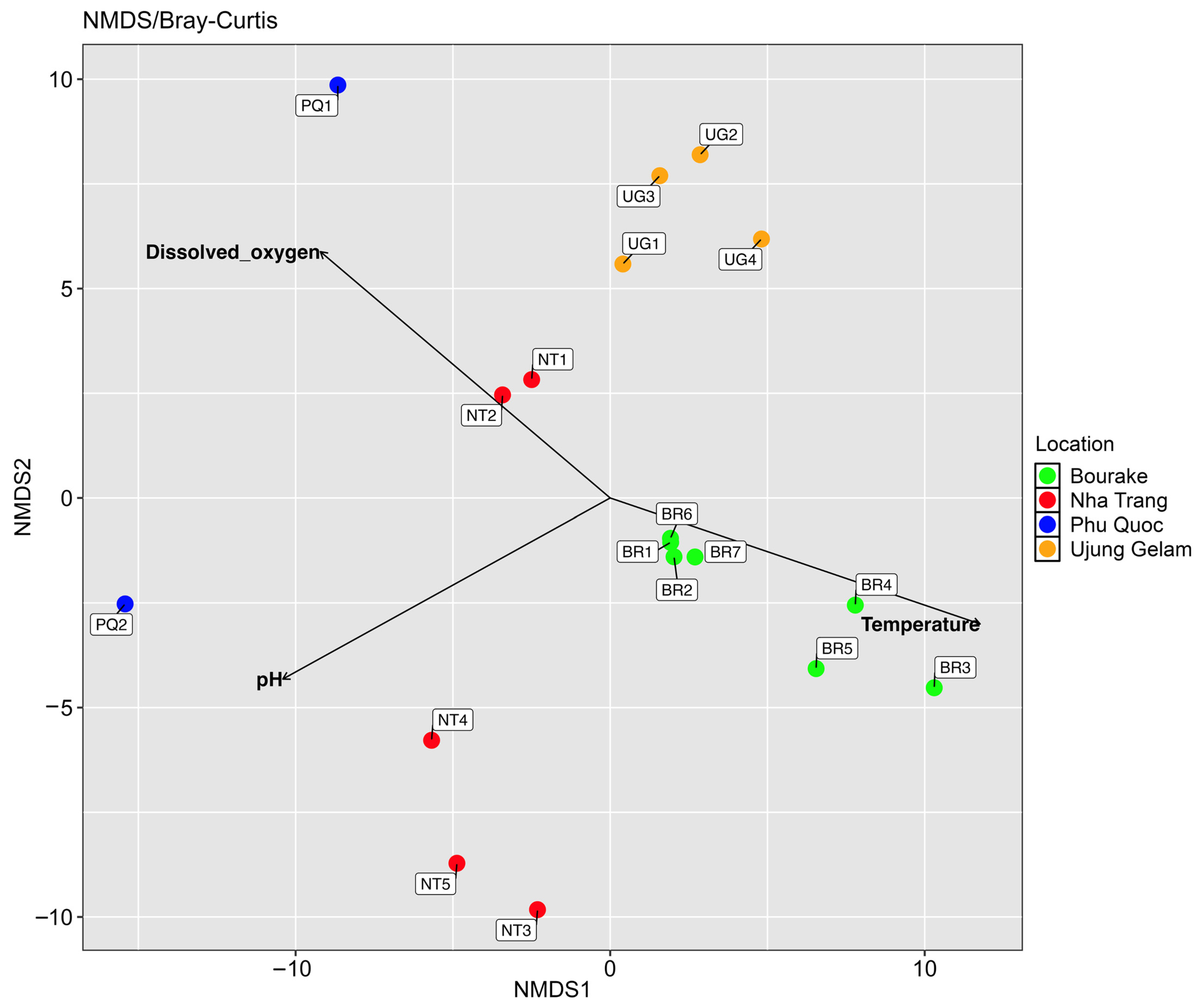
3.3. Diversity and Biogeography of Bacterial Communities
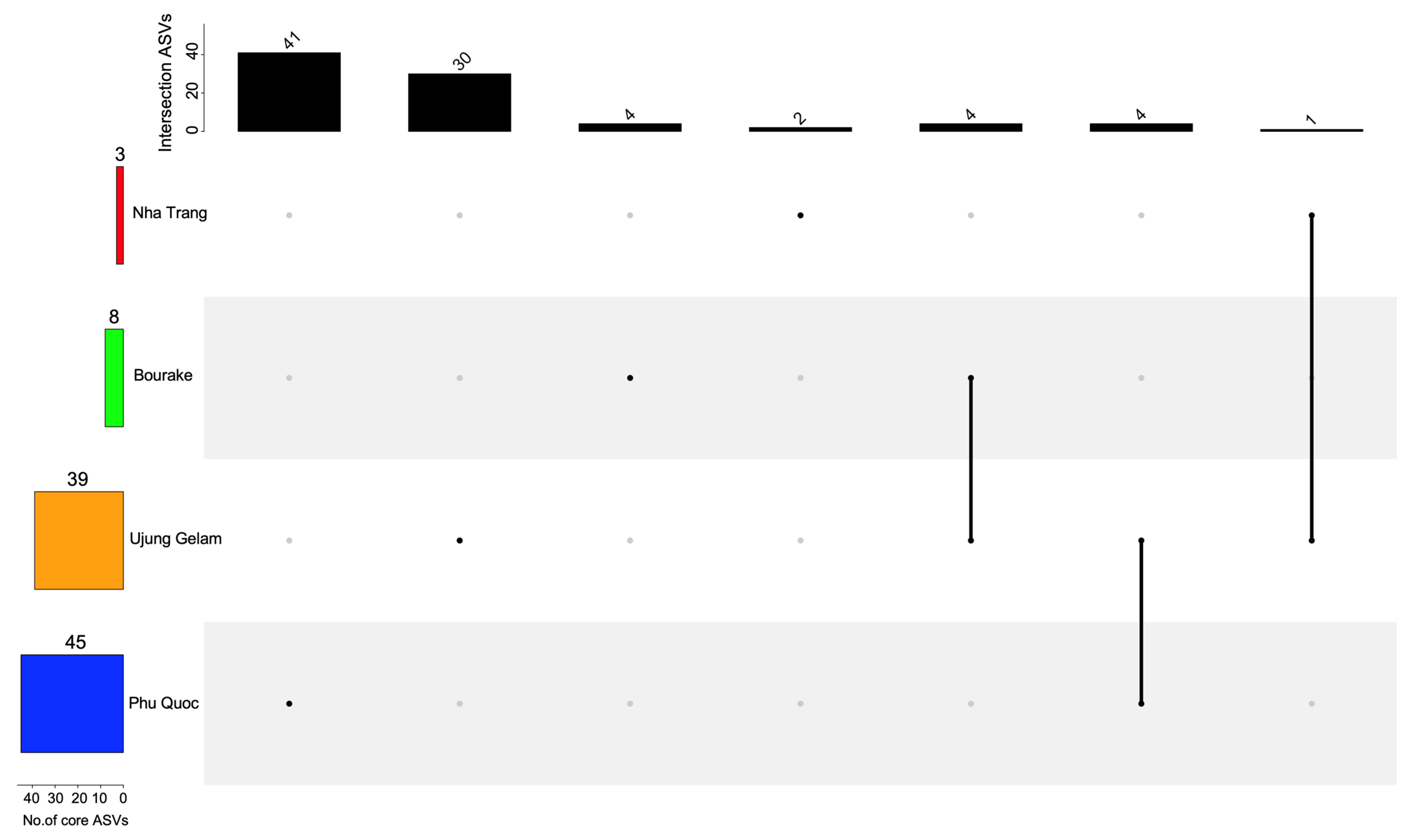
3.4. The Coral Core Microbiome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Woo, S.; Yang, S.-H.; Chen, H.-J.; Tseng, Y.-F.; Hwang, S.-J.; Palmas, S.D.; Denis, V.; Imahara, Y.; Iwase, F.; Yum, S.; et al. Geographical Variations in Bacterial Communities Associated with Soft Coral Scleronephthya Gracillimum. PLoS ONE 2017, 12, e0183663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osman, E.O.; Suggett, D.J.; Voolstra, C.R.; Pettay, D.T.; Clark, D.R.; Pogoreutz, C.; Sampayo, E.M.; Warner, M.E.; Smith, D.J. Coral Microbiome Composition along the Northern Red Sea Suggests High Plasticity of Bacterial and Specificity of Endosymbiotic Dinoflagellate Communities. Microbiome 2020, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Welle, P.D.; Small, M.J.; Doney, S.C.; Azevedo, I.L. Estimating the Effect of Multiple Environmental Stressors on Coral Bleaching and Mortality. PLoS ONE 2017, 12, e0175018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ezzat, L.; Lamy, T.; Maher, R.L.; Munsterman, K.S.; Landfield, K.M.; Schmeltzer, E.R.; Clements, C.S.; Vega Thurber, R.L.; Burkepile, D.E. Parrotfish Predation Drives Distinct Microbial Communities in Reef-Building Corals. Anim. Microbiome 2020, 2, 5. [Google Scholar] [CrossRef] [Green Version]
- Tong, H.; Cai, L.; Zhou, G.; Zhang, W.; Huang, H.; Qian, P.-Y. Correlations Between Prokaryotic Microbes and Stress-Resistant Algae in Different Corals Subjected to Environmental Stress in Hong Kong. Front. Microbiol. 2020, 11, 686. [Google Scholar] [CrossRef]
- Littman, R.A.; Willis, B.L.; Pfeffer, C.; Bourne, D.G. Diversities of Coral-Associated Bacteria Differ with Location, but Not Species, for Three Acroporid Corals on the Great Barrier Reef. FEMS Microbiol. Ecol. 2009, 68, 152–163. [Google Scholar] [CrossRef]
- Ritchie, K.B. Regulation of Microbial Populations by Coral Surface Mucus and Mucus-Associated Bacteria. Mar. Ecol. Prog. Ser. 2006, 322, 1–14. [Google Scholar] [CrossRef]
- Banin, E.; Israely, T.; Kushmaro, A.; Loya, Y.; Orr, E.; Rosenberg, E. Penetration of the Coral-Bleaching Bacterium Vibrio Shiloi into Oculina Patagonica. Appl. Environ. Microbiol. 2000, 66, 3031–3036. [Google Scholar] [CrossRef] [Green Version]
- Ben-Haim, Y.; Banim, E.; Kushmaro, A.; Loya, Y.; Rosenberg, E. Inhibition of Photosynthesis and Bleaching of Zooxanthellae by the Coral Pathogen Vibrio Shiloi. Environ. Microbiol. 1999, 1, 223–229. [Google Scholar] [CrossRef]
- Soler-Hurtado, M.M.; Sandoval-Sierra, J.V.; Machordom, A.; Diéguez-Uribeondo, J. Aspergillus Sydowii and Other Potential Fungal Pathogens in Gorgonian Octocorals of the Ecuadorian Pacific. PLoS ONE 2016, 11, e0165992. [Google Scholar] [CrossRef]
- Joyner, J.L.; Sutherland, K.P.; Kemp, D.W.; Berry, B.; Griffin, A.; Porter, J.W.; Amador, M.H.B.; Noren, H.K.G.; Lipp, E.K. Systematic Analysis of White Pox Disease in Acropora Palmata of the Florida Keys and Role of Serratia Marcescens. Appl. Environ. Microbiol. 2015, 81, 4451–4457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweet, M.J.; Bulling, M.T. On the Importance of the Microbiome and Pathobiome in Coral Health and Disease. Front. Mar. Sci. 2017, 4, 9. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-Y.; Ling, J.; Yang, Q.-S.; Wang, Y.-S.; Sun, C.-C.; Sun, H.-Y.; Feng, J.-B.; Jiang, Y.-F.; Zhang, Y.-Z.; Wu, M.-L.; et al. The Diversity of Coral Associated Bacteria and the Environmental Factors Affect Their Community Variation. Ecotoxicology 2015, 24, 1467–1477. [Google Scholar] [CrossRef] [PubMed]
- Bettarel, Y.; Halary, S.; Auguet, J.-C.; Mai, T.C.; Van Bui, N.; Bouvier, T.; Got, P.; Bouvier, C.; Monteil-Bouchard, S.; Christelle, D. Corallivory and the Microbial Debacle in Two Branching Scleractinians. ISME J. 2018, 12, 1109–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen-Kim, H.; Bouvier, T.; Bouvier, C.; Bui, V.N.; Le-Lan, H.; Bettarel, Y. Viral and Bacterial Epibionts in Thermally-Stressed Corals. J. Mar. Sci. Eng. 2015, 3, 1272–1286. [Google Scholar] [CrossRef]
- Gignoux-Wolfsohn, S.A.; Aronson, F.M.; Vollmer, S.V. Complex Interactions between Potentially Pathogenic, Opportunistic, and Resident Bacteria Emerge during Infection on a Reef-Building Coral. FEMS Microbiol. Ecol. 2017, 93, fix080. [Google Scholar] [CrossRef] [Green Version]
- Brown, B.E. Coral Bleaching: Causes and Consequences. Coral Reefs 1997, 16, S129–S138. [Google Scholar] [CrossRef]
- Mhuantong, W.; Nuryadi, H.; Trianto, A.; Sabdono, A.; Tangphatsornruang, S.; Eurwilaichitr, L.; Kanokratana, P.; Champreda, V. Comparative Analysis of Bacterial Communities Associated with Healthy and Diseased Corals in the Indonesian Sea. PeerJ 2019, 7, e8137. [Google Scholar] [CrossRef]
- Camp, E.F.; Suggett, D.J.; Pogoreutz, C.; Nitschke, M.R.; Houlbreque, F.; Hume, B.C.C.; Gardner, S.G.; Zampighi, M.; Rodolfo-Metalpa, R.; Voolstra, C.R. Corals Exhibit Distinct Patterns of Microbial Reorganisation to Thrive in an Extreme Inshore Environment. Coral Reefs 2020, 39, 701–716. [Google Scholar] [CrossRef]
- Blankenberg, D.; Gordon, A.; Von Kuster, G.; Coraor, N.; Taylor, J.; Nekrutenko, A. Manipulation of FASTQ Data with Galaxy. Bioinformatics 2010, 26, 1783–1785. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; Sankaran, K.; Fukuyama, J.A.; McMurdie, P.J.; Holmes, S.P. Bioconductor Workflow for Microbiome Data Analysis: From Raw Reads to Community Analyses. F1000Res 2016, 5, 1492. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package R Package Version 2.5-7; R Foundation for Statistical Computing: Vienna, Austria, 2020. Available online: https://cran.r-project.org/package=vegan (accessed on 27 August 2021).
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic Biomarker Discovery and Explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Agreda, A.; Gates, R.D.; Ainsworth, T.D. Defining the Core Microbiome in Corals’ Microbial Soup. Trends Microbiol. 2017, 25, 125–140. [Google Scholar] [CrossRef]
- Hester, E.R.; Barott, K.L.; Nulton, J.; Vermeij, M.J.; Rohwer, F.L. Stable and Sporadic Symbiotic Communities of Coral and Algal Holobionts. ISME J. 2016, 10, 1157–1169. [Google Scholar] [CrossRef] [Green Version]
- Lee, O.O.; Yang, J.; Bougouffa, S.; Wang, Y.; Batang, Z.; Tian, R.; Al-Suwailem, A.; Qian, P.-Y. Spatial and Species Variations in Bacterial Communities Associated with Corals from the Red Sea as Revealed by Pyrosequencing. Appl. Environ. Microbiol. 2012, 78, 7173–7184. [Google Scholar] [CrossRef] [Green Version]
- Pantos, O.; Bongaerts, P.; Dennis, P.G.; Tyson, G.W.; Hoegh-Guldberg, O. Habitat-Specific Environmental Conditions Primarily Control the Microbiomes of the Coral Seriatopora Hystrix. ISME J. 2015, 9, 1916–1927. [Google Scholar] [CrossRef]
- Meron, D.; Atias, E.; Iasur Kruh, L.; Elifantz, H.; Minz, D.; Fine, M.; Banin, E. The Impact of Reduced PH on the Microbial Community of the Coral Acropora Eurystoma. ISME J. 2011, 5, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Webster, N.S.; Negri, A.P.; Botté, E.S.; Laffy, P.W.; Flores, F.; Noonan, S.; Schmidt, C.; Uthicke, S. Host-Associated Coral Reef Microbes Respond to the Cumulative Pressures of Ocean Warming and Ocean Acidification. Sci. Rep. 2016, 6, 19324. [Google Scholar] [CrossRef] [Green Version]
- Huntley, N.; Brandt, M.E.; Becker, C.C.; Miller, C.A.; Meiling, S.S.; Correa, A.M.S.; Holstein, D.M.; Muller, E.M.; Mydlarz, L.D.; Smith, T.B.; et al. Experimental Transmission of Stony Coral Tissue Loss Disease Results in Differential Microbial Responses within Coral Mucus and Tissue. ISME Commun. 2022, 2, 1–11. [Google Scholar] [CrossRef]
- Becker, C.C.; Brandt, M.; Miller, C.A.; Apprill, A. Microbial Bioindicators of Stony Coral Tissue Loss Disease Identified in Corals and Overlying Waters Using a Rapid Field-Based Sequencing Approach. Environ. Microbiol. 2022, 24, 1166–1182. [Google Scholar] [CrossRef] [PubMed]
- Glasl, B.; Herndl, G.J.; Frade, P.R. The Microbiome of Coral Surface Mucus Has a Key Role in Mediating Holobiont Health and Survival upon Disturbance. ISME J. 2016, 10, 2280–2292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, J.L.; Gunasekera, S.P.; Scott, R.M.; Paul, V.J.; Teplitski, M. Microbiome Shifts and the Inhibition of Quorum Sensing by Black Band Disease Cyanobacteria. ISME J. 2016, 10, 1204–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neave, M.J.; Rachmawati, R.; Xun, L.; Michell, C.T.; Bourne, D.G.; Apprill, A.; Voolstra, C.R. Differential Specificity between Closely Related Corals and Abundant Endozoicomonas Endosymbionts across Global Scales. ISME J. 2017, 11, 186–200. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.-Y.; Shiu, J.-H.; Chen, W.-M.; Chiang, Y.-R.; Tang, S.-L. Genomic Insight into the Host–Endosymbiont Relationship of Endozoicomonas Montiporae CL-33T with Its Coral Host. Front. Microbiol. 2016, 7, 251. [Google Scholar] [CrossRef] [Green Version]
- Bourne, D.G.; Dennis, P.G.; Uthicke, S.; Soo, R.M.; Tyson, G.W.; Webster, N. Coral Reef Invertebrate Microbiomes Correlate with the Presence of Photosymbionts. ISME J. 2013, 7, 1452–1458. [Google Scholar] [CrossRef]
- Morrow, K.M.; Bourne, D.G.; Humphrey, C.; Botté, E.S.; Laffy, P.; Zaneveld, J.; Uthicke, S.; Fabricius, K.E.; Webster, N.S. Natural Volcanic CO2 Seeps Reveal Future Trajectories for Host–Microbial Associations in Corals and Sponges. ISME J. 2015, 9, 894–908. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, M.; Roik, A.; Porter, A.; Zubier, K.; Mudarris, M.S.; Ormond, R.; Voolstra, C.R. Coral Microbial Community Dynamics in Response to Anthropogenic Impacts near a Major City in the Central Red Sea. Mar. Pollut. Bull. 2016, 105, 629–640. [Google Scholar] [CrossRef] [Green Version]
- Seemann, P.; Gernert, C.; Schmitt, S.; Mebs, D.; Hentschel, U. Detection of Hemolytic Bacteria from Palythoa Caribaeorum (Cnidaria, Zoantharia) Using a Novel Palytoxin-Screening Assay. Antonie Van Leeuwenhoek 2009, 96, 405–411. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sampling Sites | Accessions Numbers | Species | Sample ID | pH | Temperature | Dissolved Oxygen |
|---|---|---|---|---|---|---|
| Phu Quoc (9°55′20.6″ N 104°01′16.4″ E | This study | Acropora formosa | PQ1 | 7.96 ± 0.01 | 27.8 ± 0.09 | 6.96 ± 0.11 |
| PQ2 | ||||||
| Nha Trang (Whale Island (12°39.1′ N, 109°23.9′ E) | SRP116049 [14] | Acropora formosa | NT1 | 8.19 ± 0.05 | 29.5 ± 0.4 | 5.92 ± 0.15 |
| NT2 | ||||||
| NT3 | ||||||
| NT4 | ||||||
| NT5 | ||||||
| Ujung Gelam (S 05°49′57.7″, E 110° 22′50.5″) | SRP071125 [18] | Acropora formosa | UG1 | 7.3 ± 0.1 | 30.1 ± 0.4 | 6.4 ± 0.6 |
| UG2 | ||||||
| UG3 | ||||||
| UG4 | ||||||
| Bourake-Lagoon (21°56.915 S; 165°59.577 E) | PRJNA510614 [19] | Acropora formosa | BR1 | 7.3 | 33 | 2.3 |
| BR2 | ||||||
| BR3 | ||||||
| BR4 | ||||||
| BR5 | ||||||
| BR6 | ||||||
| BR7 |
| Sampling Sites | Species | Sample ID | No. of Raw Sequences | No. of Sequences after QC | Observed ASVs | Chao1 | Shannon |
|---|---|---|---|---|---|---|---|
| Phu Quoc | Acropora formosa | PQ1 | 31,064 | 21,432 | 231 | 322.22 | 4.79 |
| PQ2 | 33,409 | 6008 | 174 | 210.67 | 4.38 | ||
| Nha Trang | Acropora formosa | NT1 | 49,936 | 15,729 | 85 | 95.20 | 3.65 |
| NT2 | 52,510 | 28,100 | 77 | 96.46 | 3.33 | ||
| NT3 | 75,709 | 60,117 | 37 | 37.50 | 3.02 | ||
| NT4 | 65,007 | 52,221 | 54 | 54.50 | 3.47 | ||
| NT5 | 45,634 | 7970 | 123 | 143.32 | 3.67 | ||
| Ujung Gelam | Acropora formosa | UG1 | 186,435 | 133,877 | 169 | 250.00 | 3.89 |
| UG2 | 195,464 | 128,950 | 236 | 426.83 | 4.56 | ||
| UG3 | 232,120 | 175,525 | 228 | 436.41 | 4.44 | ||
| UG4 | 145,104 | 75,380 | 142 | 266.25 | 3.81 | ||
| Bourake | Acropora formosa | BR1 | 65,875 | 52,843 | 28 | 28.60 | 2.29 |
| BR2 | 55,164 | 43,544 | 37 | 41.00 | 2.62 | ||
| BR3 | 32,452 | 28,524 | 157 | 262.64 | 3.19 | ||
| BR4 | 10,782 | 9288 | 81 | 114.06 | 2.37 | ||
| BR5 | 4635 | 3764 | 65 | 70.35 | 2.55 | ||
| BR6 | 76,145 | 62,827 | 31 | 35.02 | 2.32 | ||
| BR7 | 33,266 | 28,381 | 24 | 29.00 | 2.05 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bui, V.N.; Nguyen, D.H.; Chu, N.H.; Bettarel, Y.; Auguet, J.-C.; Bouvier, T.; Chu, H.H. Diversity and Biogeography of Coral Mucus-Associated Bacterial Communities: The Case of Acropora formosa. J. Mar. Sci. Eng. 2023, 11, 74. https://doi.org/10.3390/jmse11010074
Bui VN, Nguyen DH, Chu NH, Bettarel Y, Auguet J-C, Bouvier T, Chu HH. Diversity and Biogeography of Coral Mucus-Associated Bacterial Communities: The Case of Acropora formosa. Journal of Marine Science and Engineering. 2023; 11(1):74. https://doi.org/10.3390/jmse11010074
Chicago/Turabian StyleBui, Van Ngoc, Duong Huy Nguyen, Nhat Huy Chu, Yvan Bettarel, Jean-Christophe Auguet, Thierry Bouvier, and Ha Hoang Chu. 2023. "Diversity and Biogeography of Coral Mucus-Associated Bacterial Communities: The Case of Acropora formosa" Journal of Marine Science and Engineering 11, no. 1: 74. https://doi.org/10.3390/jmse11010074