
Genetic Diversity Analysis of Different Populations of Lutjanus kasmira Based on SNP Markers
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
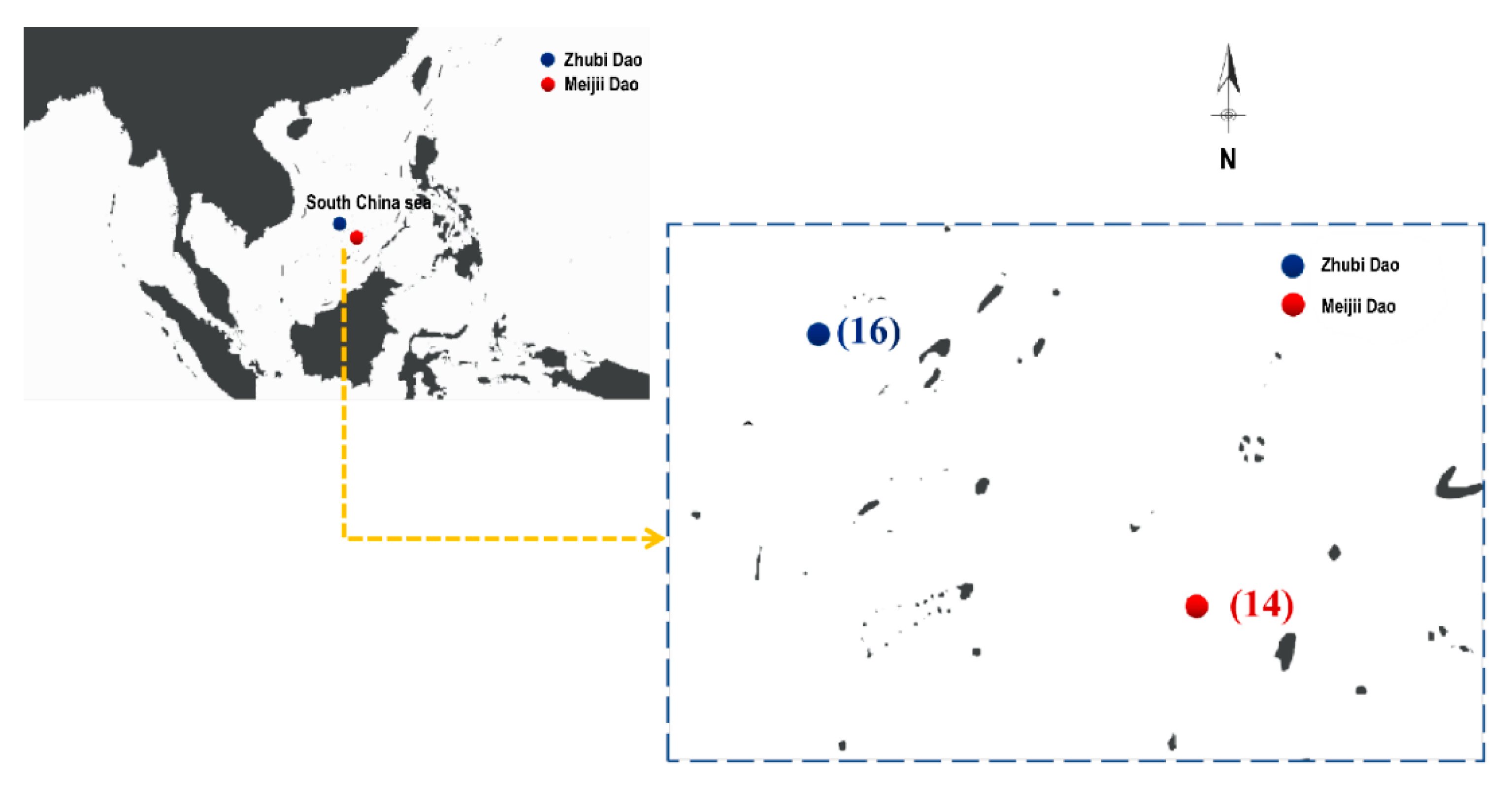
2.1. Sample Collection and DNA Extraction
2.2. RAD-Seq Library Construction and Sequencing
2.3. Sequence Alignment and SNP Genotyping
2.4. Genetic Diversity and Population Structure
3. Results
3.1. Genomic Data Statistics and SNP Discovery
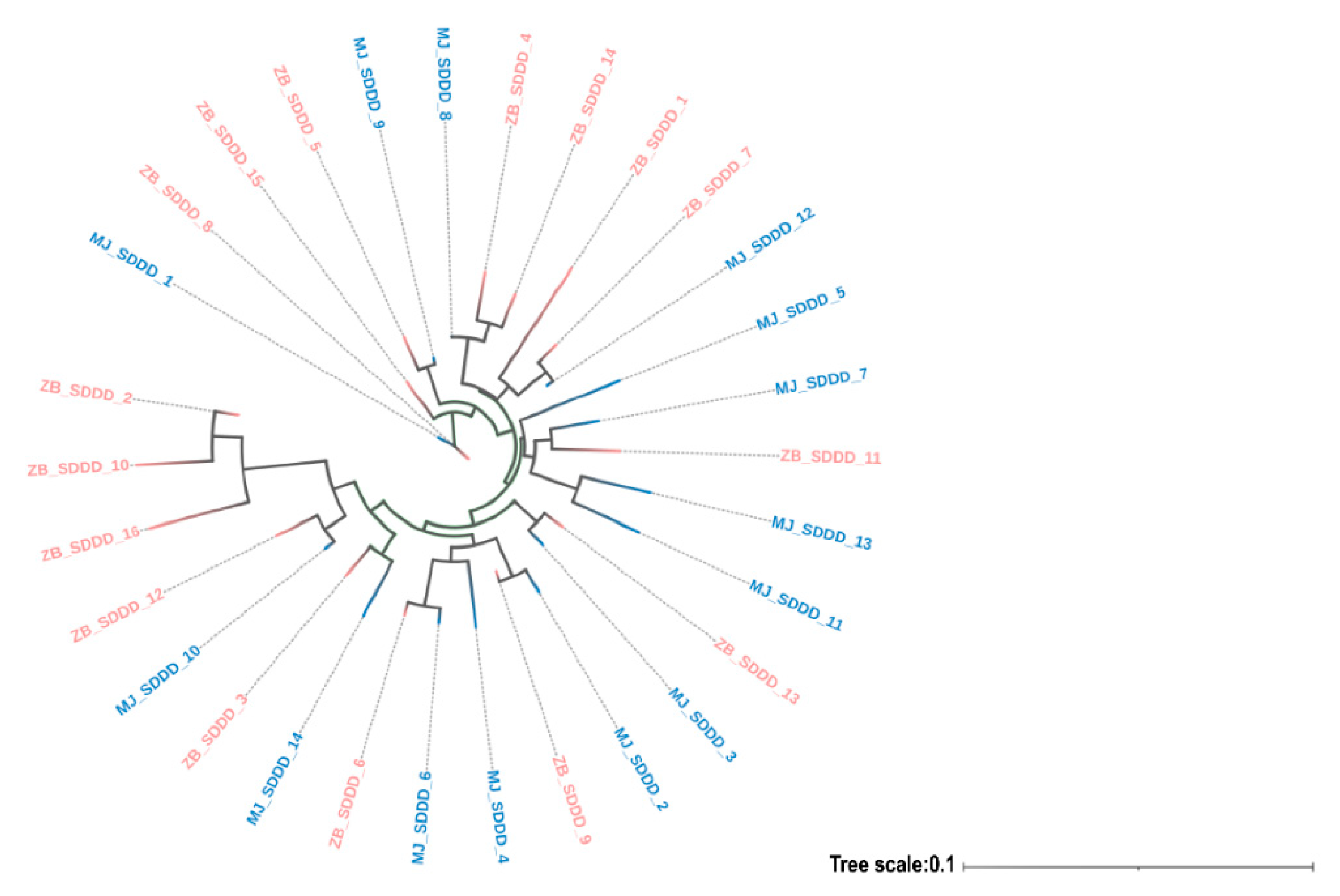
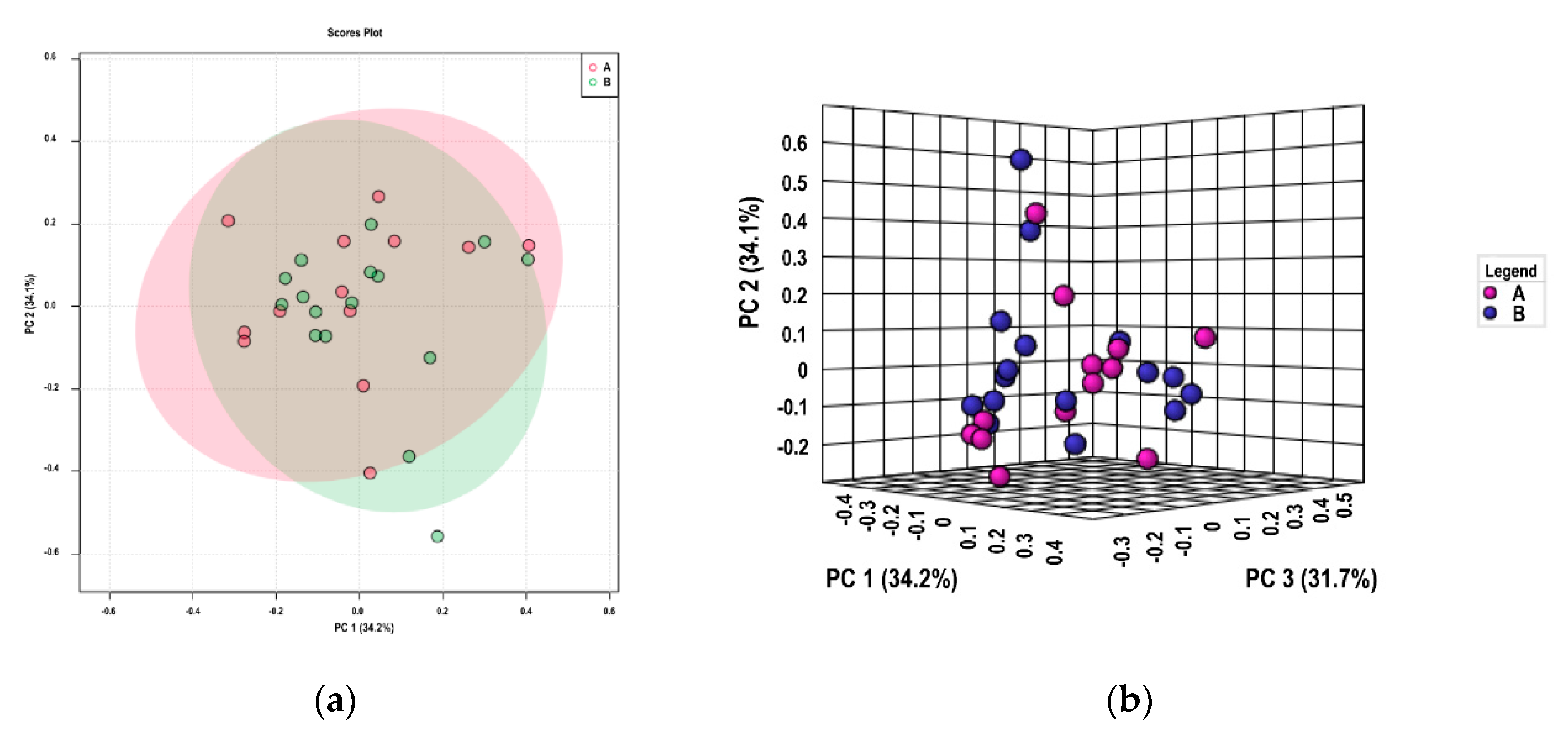
3.2. Genetic Diversity and Population Structure
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Niu, W.; Huang, Y.; Zhang, C.; Pu, T.; Lu, Y.; Jia, T.; You, Y.; Du, Y.; Mao, Y.; Ding, C. Development and Characterization of 31 SNP Markers for the Crested Ibis (Nipponia nippon). Conserv. Genet. Resour. 2021, 13, 5–7. [Google Scholar] [CrossRef]
- Li, Y.; Lou, F.; Song, P.; Liu, S.; Siyal, F.K.; Lin, L. New Perspective on the Genetic Structure and Habitat Adaptation of Pampus Minor off the Coast of China Based on RAD-Seq. Comp. Biochem. Physiol. Part D Genom. Proteom. 2021, 39, 100865. [Google Scholar] [CrossRef] [PubMed]
- Catanese, G.; Trotta, J.R.; Iriondo, M.; Grau, A.M.; Estonba, A. Discovery of SNP Markers of Red Shrimp Aristeus Antennatus for Population Structure in Western Mediterranean Sea. Conserv. Genet. Resour. 2021, 13, 21–25. [Google Scholar] [CrossRef]
- Chaves, C.L.; Blanc-Jolivet, C.; Sebbenn, A.M.; Mader, M.; Meyer-Sand, B.R.V.; Paredes-Villanueva, K.; Honorio Coronado, E.N.; Garcia-Davila, C.; Tysklind, N.; Troispoux, V.; et al. Nuclear and Chloroplastic SNP Markers for Genetic Studies of Timber Origin for Hymenaea Trees. Conserv. Genet. Resour. 2019, 11, 329–331. [Google Scholar] [CrossRef]
- Yue, L.; Zhang, S.; Zhang, L.; Liu, Y.; Cheng, F.; Li, G.; Zhang, S.; Zhang, H.; Sun, R.; Li, F. Heterotic Prediction of Hybrid Performance Based on Genome-Wide SNP Markers and the Phenotype of Parental Inbred Lines in Heading Chinese Cabbage (Brassica rapa L. Ssp. Pekinensis). Sci. Hortic. 2022, 296, 110907. [Google Scholar] [CrossRef]
- Vignon, M.; Morat, F.; Galzin, R.; Sasal, P. Evidence for Spatial Limitation of the Bluestripe Snapper Lutjanus Kasmira in French Polynesia from Parasite and Otolith Shape Analysis. J. Fish Biol. 2008, 73, 2305–2320. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, Z. Variation in the population characteristics of blue-striped snapper Lutjanus kasmira in the South China Sea in recent 20years. Oceanol. Limnol. Sin. 2020, 51, 114–124. [Google Scholar]
- Tan, W.; Wang, Z. Structure and Evolution of Complete Mitochondrial Genome of Lutjanus kasmira. In Proceedings of the 12th Annual Conference of China Association for Science and Technology; The Transformation of Economic Development Pattern and Independent Innovation, Fuzhou, China, 1–3 November 2010; Volume 3, pp. 1143–1150. [Google Scholar]
- Tang, C.; Xiao, L.; Zhang, Q.; Zhou, Q.; Xu, S.; Wang, Y. DNA barcoding of some Lutjanus species in China and its adjacent sea areas based on COI gene. Mar. Fish. 2019, 41, 129–137. [Google Scholar] [CrossRef]
- Davey, J.W.; Blaxter, M.L. RADSeq: Next-Generation Population Genetics. Brief. Funct. Genom. 2010, 9, 416–423. [Google Scholar] [CrossRef]
- Wang, Y.; Hu, Y. Current status and perspective of RAD-seq in genomic research. Hereditas 2014, 36, 41–49. [Google Scholar] [CrossRef]
- Catchen, J.M.; Amores, A.; Hohenlohe, P.; Cresko, W.; Postlethwait, J.H. Stacks: Building and Genotyping Loci De Novo from Short-Read Sequences. G3 Genes Genomes Genet. 2011, 1, 171–182. [Google Scholar] [CrossRef]
- Paris, J.R.; Stevens, J.R.; Catchen, J.M. Lost in Parameter Space: A Road Map for stacks. Methods Ecol. Evol. 2017, 8, 1360–1373. [Google Scholar] [CrossRef] [Green Version]
- Mastretta-Yanes, A.; Arrigo, N.; Alvarez, N.; Jorgensen, T.H.; Piñero, D.; Emerson, B.C. Restriction Site-Associated DNA Sequencing, Genotyping Error Estimation and de Novo Assembly Optimization for Population Genetic Inference. Mol. Ecol. Resour. 2015, 15, 28–41. [Google Scholar] [CrossRef]
- Rochette, N.C.; Catchen, J.M. Deriving Genotypes from RAD-Seq Short-Read Data Using Stacks. Nat. Protoc. 2017, 12, 2640–2659. [Google Scholar] [CrossRef]
- Sun, L.; Gan, J.; Jiang, L.; Wu, R. Recursive Test of Hardy-Weinberg Equilibrium in Tetraploids. Trends Genet. 2021, 37, 504–513. [Google Scholar] [CrossRef]
- Kamvar, Z.N.; Tabima, J.F.; Grunwald, N.J. Poppr: An R Package for Genetic Analysis of Populations with Clonal, Partially Clonal, and/or Sexual Reproduction. PeerJ 2014, 2, e281. [Google Scholar] [CrossRef] [Green Version]
- Schaub, F.X.; Dhankani, V.; Berger, A.C.; Trivedi, M.; Richardson, A.B.; Shaw, R.; Zhao, W.; Zhang, X.; Ventura, A.; Liu, Y.; et al. Pan-Cancer Alterations of the MYC Oncogene and Its Proximal Network across the Cancer Genome Atlas. Cell Syst. 2018, 6, 282–300.e2. [Google Scholar] [CrossRef] [Green Version]
- Kamvar, Z.N.; Larsen, M.M.; Kanaskie, A.M.; Hansen, E.M.; Grünwald, N.J. Spatial and Temporal Analysis of Populations of the Sudden Oak Death Pathogen in Oregon Forests. Phytopathology 2015, 105, 982–989. [Google Scholar] [CrossRef] [Green Version]
- Felsenstein, J. PHYLIP (Phylogeny Inference Package), version 3.6; Distributed by the author; Department of Genome Sciences, University of Washington: Seattle, DC, USA, 2005. [Google Scholar]
- Parry, R.M.; Wang, M.D. A Fast Least-Squares Algorithm for Population Inference. BMC Bioinform. 2013, 14, 28. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Du, W.; Tang, R.; Liu, Y.; Zou, X.; Yuan, D.; Wang, Z.; Liu, J.; Guo, J.; Yang, X.; et al. Genomic History and Forensic Characteristics of Sherpa Highlanders on the Tibetan Plateau Inferred from High-Resolution InDel Panel and Genome-Wide SNPs. Forensic Sci. Int. Genet. 2022, 56, 102633. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Z. Analysis of the Genetic Structure and Diversity of Upland Cotton Groups in Different Planting Areas Based on SNP Markers. Gene 2022, 809, 146042. [Google Scholar] [CrossRef]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A Tool for Genome-Wide Complex Trait Analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [Green Version]
- Lecaudey, L.A.; Schliewen, U.K.; Osinov, A.G.; Taylor, E.B.; Bernatchez, L.; Weiss, S.J. Inferring Phylogenetic Structure, Hybridization and Divergence Times within Salmoninae (Teleostei: Salmonidae) Using RAD-Sequencing. Mol. Phylogenet. Evol. 2018, 124, 82–99. [Google Scholar] [CrossRef]
- Zhou, W.; Ji, X.; Obata, S.; Pais, A.; Dong, Y.; Peet, R.; Xiang, Q.-Y.J. Resolving Relationships and Phylogeographic History of the Nyssa Sylvatica Complex Using Data from RAD-Seq and Species Distribution Modeling. Mol. Phylogenet. Evol. 2018, 126, 1–16. [Google Scholar] [CrossRef]
- Díaz-Arce, N.; Arrizabalaga, H.; Murua, H.; Irigoien, X.; Rodríguez-Ezpeleta, N. RAD-Seq Derived Genome-Wide Nuclear Markers Resolve the Phylogeny of Tunas. Mol. Phylogenet. Evol. 2016, 102, 202–207. [Google Scholar] [CrossRef] [Green Version]
- Singha, H.; Vorimore, F.; Saini, S.; Deshayes, T.; Saqib, M.; Tripathi, B.N.; Laroucau, K. Molecular Epidemiology of Burkholderia Mallei Isolates from India (2015–2016): New SNP Markers for Strain Tracing. Infect. Genet. Evol. 2021, 95, 105059. [Google Scholar] [CrossRef]
- Bao, W.; Ao, D.; Wuyun, T.; Wang, L.; Bai, S.; Bai, Y. Development and Characterization of 72 SNP Markers in Armeniaca Sibirica Based on Transcriptomics. Conserv. Genet. Resour. 2020, 12, 373–378. [Google Scholar] [CrossRef]
- Fukuda, S.; Nagano, Y.; Matsuguma, K.; Ishimoto, K.; Hiehata, N.; Nagano, A.J.; Tezuka, A.; Yamamoto, T. Construction of a High-Density Linkage Map for Bronze Loquat Using RAD-Seq. Sci. Hortic. 2019, 251, 59–64. [Google Scholar] [CrossRef]
- Palaiokostas, C.; Bekaert, M.; Khan, M.G.Q.; Taggart, J.B.; Gharbi, K.; McAndrew, B.J.; Penman, D.J. Mapping and Validation of the Major Sex-Determining Region in Nile Tilapia (Oreochromis niloticus L.) Using RAD Sequencing. PLoS ONE 2013, 8, e68389. [Google Scholar] [CrossRef]
- Li, P.; Wang, Z.; Guo, Y.; Liu, L.; Liu, C. Microsatellite analysis of Lutjanus bengalensis and L. Kasmira. J. Oceanogr. Taiwan Strait 2011, 30, 517–521. [Google Scholar]
- Park, S.-H.; Scheffler, J.A.; Ray, J.D.; Scheffler, B.E. Identification of Simple Sequence Repeat (SSR) and Single Nucleotide Polymorphism (SNP) That Are Associated with the Nectariless Trait of Gossypium hirsutum L. Euphytica 2021, 217, 78. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, Y.; Liu, X.; Yu, H.; Li, D.; Zhang, Y. Genetic Diversity of the Sichuan Snub-Nosed Monkey (Rhinopithecus roxellana) in Shennongjia National Park, China Using RAD-Seq Analyses. Genetica 2019, 147, 327–335. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Sample Location | Length (mm) | Sampling Date | Sample ID | Sample Location | Length (mm) | Sampling Date |
|---|---|---|---|---|---|---|---|
| ZB_SDDD_1 | Zhubi Dao | 136 | 2019 | MJ_SDDD_1 | Meiji Dao | 153 | 2019 |
| ZB_SDDD_2 | Zhubi Dao | 146 | 2019 | MJ_SDDD_2 | Meiji Dao | 179 | 2019 |
| ZB_SDDD_3 | Zhubi Dao | 150 | 2019 | MJ_SDDD_3 | Meiji Dao | 153 | 2019 |
| ZB_SDDD_4 | Zhubi Dao | 128 | 2019 | MJ_SDDD_4 | Meiji Dao | 164 | 2019 |
| ZB_SDDD_5 | Zhubi Dao | 133 | 2019 | MJ_SDDD_5 | Meiji Dao | 123 | 2019 |
| ZB_SDDD_6 | Zhubi Dao | 137 | 2019 | MJ_SDDD_6 | Meiji Dao | 154 | 2019 |
| ZB_SDDD_7 | Zhubi Dao | 146 | 2019 | MJ_SDDD_7 | Meiji Dao | 126 | 2019 |
| ZB_SDDD_8 | Zhubi Dao | 148 | 2019 | MJ_SDDD_8 | Meiji Dao | 137 | 2019 |
| ZB_SDDD_9 | Zhubi Dao | 152 | 2019 | MJ_SDDD_9 | Meiji Dao | 146 | 2019 |
| ZB_SDDD_10 | Zhubi Dao | 139 | 2019 | MJ_SDDD_10 | Meiji Dao | 125 | 2019 |
| ZB_SDDD_11 | Zhubi Dao | 120 | 2019 | MJ_SDDD_11 | Meiji Dao | 143 | 2019 |
| ZB_SDDD_12 | Zhubi Dao | 149 | 2019 | MJ_SDDD_12 | Meiji Dao | 135 | 2019 |
| ZB_SDDD_13 | Zhubi Dao | 132 | 2019 | MJ_SDDD_13 | Meiji Dao | 154 | 2019 |
| ZB_SDDD_14 | Zhubi Dao | 125 | 2019 | MJ_SDDD_14 | Meiji Dao | 144 | 2019 |
| ZB_SDDD_15 | Zhubi Dao | 165 | 2019 | ||||
| ZB_SDDD_16 | Zhubi Dao | 143 | 2019 |
| Sample ID | Number of Clean Reads | Bases (Q20%) | GC% | Sample ID | Number of Clean Reads | Bases (Q20%) | GC% |
|---|---|---|---|---|---|---|---|
| ZB_SDDD_1 | 7,974,070 | 99.32 | 39 | MJ_SDDD_1 | 9,265,891 | 95.32 | 39 |
| ZB_SDDD_2 | 6,867,444 | 96.12 | 40 | MJ_SDDD_2 | 13,500,657 | 96.75 | 39 |
| ZB_SDDD_3 | 11,342,742 | 93.64 | 39 | MJ_SDDD_3 | 9,120,598 | 99.30 | 39 |
| ZB_SDDD_4 | 6,831,672 | 95.40 | 40 | MJ_SDDD_4 | 12,363,573 | 99.64 | 39 |
| ZB_SDDD_5 | 10,947,352 | 96.32 | 40 | MJ_SDDD_5 | 8,022,139 | 98.20 | 39 |
| ZB_SDDD_6 | 8,673,002 | 98.89 | 39 | MJ_SDDD_6 | 10,972,365 | 97.67 | 39 |
| ZB_SDDD_7 | 8,040,810 | 96.63 | 39 | MJ_SDDD_7 | 14,935,273 | 94.03 | 39 |
| ZB_SDDD_8 | 7,251,357 | 97.60 | 39 | MJ_SDDD_8 | 10,483,102 | 96.43 | 39 |
| ZB_SDDD_9 | 8,440,580 | 96.30 | 40 | MJ_SDDD_9 | 9,245,798 | 94.60 | 39 |
| ZB_SDDD_10 | 7,441,155 | 94.56 | 39 | MJ_SDDD_10 | 6,732,763 | 98.75 | 39 |
| ZB_SDDD_11 | 8,497,499 | 93.21 | 39 | MJ_SDDD_11 | 9,673,994 | 95.30 | 39 |
| ZB_SDDD_12 | 6,672,063 | 95.63 | 39 | MJ_SDDD_12 | 9,046,087 | 98.00 | 39 |
| ZB_SDDD_13 | 9,912,874 | 99.11 | 39 | MJ_SDDD_13 | 6,814,092 | 95.19 | 39 |
| ZB_SDDD_14 | 7,340,574 | 98.65 | 39 | MJ_SDDD_14 | 7,280,377 | 96.32 | 39 |
| ZB_SDDD_15 | 21,514,652 | 99.30 | 39 | ||||
| ZB_SDDD_16 | 11,059,675 | 97.21 | 39 |
| Population ID | π | Ho | He | Average_Fis | Fst |
|---|---|---|---|---|---|
| ZB population | 0.02478 | 0.46834 | 0.33585 | −0.18729 | 0.00255102 |
| MJ population | 0.02154 | 0.23103 | 0.22098 | 0.03256 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, F.; Guo, L.; Zhang, N.; Yang, J.; Zhu, K.; Guo, H.; Liu, B.; Liu, B.; Zhang, D.; Jiang, S. Genetic Diversity Analysis of Different Populations of Lutjanus kasmira Based on SNP Markers. J. Mar. Sci. Eng. 2022, 10, 1547. https://doi.org/10.3390/jmse10101547
Zhao F, Guo L, Zhang N, Yang J, Zhu K, Guo H, Liu B, Liu B, Zhang D, Jiang S. Genetic Diversity Analysis of Different Populations of Lutjanus kasmira Based on SNP Markers. Journal of Marine Science and Engineering. 2022; 10(10):1547. https://doi.org/10.3390/jmse10101547
Chicago/Turabian StyleZhao, Fangcao, Liang Guo, Nan Zhang, Jingwen Yang, Kecheng Zhu, Huayang Guo, Baosuo Liu, Bo Liu, Dianchang Zhang, and Shigui Jiang. 2022. "Genetic Diversity Analysis of Different Populations of Lutjanus kasmira Based on SNP Markers" Journal of Marine Science and Engineering 10, no. 10: 1547. https://doi.org/10.3390/jmse10101547