Liver Steatosis, Gut-Liver Axis, Microbiome and Environmental Factors. A Never-Ending Bidirectional Cross-Talk

 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Risk Factors, Clinical Features, and Pathophysiology of NAFLD
2.1. Definition of NAFLD
2.2. Risk Factors
2.3. Clinical Aspects
2.4. Pathophysiology of NAFLD
- (a)
- Influx of non-esterified fatty acids (NEFA): This step accounts for ~60% of total FFAs in the liver and brings to enrichment of the FFA pool in hepatocytes. NEFA originates from lipolysis of triglycerides in adipocyte under the control of insulin [46]. Once in the liver, FFAs have different fates:
- As fatty acyl-CoA, FFAs can enter the mitochondria under the control of carnitine palmitoyltransferase (CPT)-1 and undergo β-oxidation to acetyl-CoA joining the tricarbossilic acid cycle with production of ATP;
- FFAs are also esterified to TG (no more than 5% in the hepatocyte) via the key enzymes diaglyceride acyltransferase (DGAT)1 and DGAT2. This step is also controlled by insulin and excess TG can be stored as lipid droplets or exported to blood as very-low-density lipoprotein [47].
- (b)
- De novo lipogenesis (DNL) of FAs: This step accounts for ~25% of total FFAs in the liver and originate from dietary sugars. With DNL, hepatocytes converts excess glucose and fructose to FAs. Insulin mediates the transport of absorbed dietary carbohydrates to its target tissues (skeletal muscle and liver for storage as glycogen). Glucose in hepatocytes is partly metabolized to pyruvic acid via glycolysis and then to acetyl-CoA, to generate ATP in the tricarboxylic acid cycle and promote gluconeogenesis during hypoglycemia. The first step of DNL is the synthesis of malonyl-CoA from cytosolic acetyl-CoA by acetyl-CoA carboxylase (ACC). Malonyl-CoA serves as a substrate to form saturated FA via FA synthase (FAS). Stearoyl CoA desaturase (SCD)1 is an endoplasmic reticulum (ER) enzyme responsible for the formation of monounsaturated FAs from saturated FAs [41].
- (c)
- Influx of dietary FAs: Influx of FAs from diet is ~15% of all amount of FFAs in the liver [48]. Bile acids (BA) hydrolyze intestinal dietary TGs to form nascent chylomicrons. The FAs made from TG hydrolysis are taken up by adipose tissue and liver [49]. At the same time, BA acts as a potent metabolic regulator in the terminal ileum by activating the farnesoid X receptor (FXR) plus the pregnane X receptor (PXR), as well as the G-protein-coupled bile acid receptor-1 (GPBAR-1). These interactions produce effects in the liver and in the muscle, in adipocytes and brown adipose tissue for energy expenditure [12,50].
3. Beyond NAFLD: The Gut Liver-Axis, the Gut Barrier, and the Liver Barrier
3.1. Intestinal Microbiome
3.2. Intestinal Mucus
3.3. Gastrointestinal Motility, Secretions, and Enterohepatic Circulation of BAs
3.4. Intestinal Monocellular Layer
3.5. Immunological Gut Barrier
- Th17 cells release IL17-A, IL17-F, and IL-22 (which contributes to strengthen tight junction molecules and stimulate epithelial cell regeneration [176]); IgA production; and the expression of pIgR, which allows their translocation towards the gut lumen [177,178]. These cells accumulate if segmented filamentous bacteria reaches the intestinal epithelium [178,179,180].
- T regulatory cells are thymic derived cells (which recognize self-antigens and control the function of autoreactive T cells) and peripheral derived T regs (which recognize food antigens in the small intestine or microbial antigens in the large intestine), where they control tolerance to innocuous non self-antigens [181].
3.6. Gut Vascular Barrier
3.7. The Liver Barrier
4. The Main Constituents of the Ongoing Liver Damage
4.1. Inflammation
4.2. Oxidative Stress
4.3. The Intestinal Microbiota
4.4. Altered Intestinal Permeability
4.5. Products of Microbiota
5. The Microbiota as a Target of Environmental Factors Involved in NAFLD
5.1. Dietary Habits and Microbiota
5.2. Antibiotics, Probiotics, and Other Pharmacological Agents
5.3. Food Contamination with Chemicals of Environmental Origin, Microbiota, and NAFLD
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver; European Association for the Study of Diabetes; European Association for the Study of Obesity. EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Blissett, D.; Blissett, R.; Henry, L.; Stepanova, M.; Younossi, Y.; Racila, A.; Hunt, S.; Beckerman, R. The economic and clinical burden of nonalcoholic fatty liver disease in the United States and Europe. Hepatology 2016, 64, 1577–1586. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.D.; Stengel, J.; Asike, M.I.; Torres, D.M.; Shaw, J.; Contreras, M.; Landt, C.L.; Harrison, S.A. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: A prospective study. Gastroenterology 2011, 140, 124–131. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Angulo, P. Obesity and Nonalcoholic Fatty Liver Disease. Nutr. Rev. 2007, 65, S57–S63. [Google Scholar] [CrossRef]
- Liu, C.J. Prevalence and risk factors for non-alcoholic fatty liver disease in Asian people who are not obese. J. Gastroenterol. Hepatol. 2012, 27, 1555–1560. [Google Scholar] [CrossRef]
- Lindenmeyer, C.C.; McCullough, A.J. The natural history of nonalcoholic fatty liver disease—An evolving view. Clin. Liver Dis. 2018, 22, 11–21. [Google Scholar] [CrossRef]
- Rinella, M.E.; Sanyal, A.J. Management of NAFLD: A stage-based approach. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 196–205. [Google Scholar] [CrossRef]
- Tripathi, A.; Debelius, J.; Brenner, D.A.; Karin, M.; Loomba, R.; Schnabl, B.; Knight, R. The gut-liver axis and the intersection with the microbiome. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Di Ciaula, A.; Garruti, G.; Lunardi Baccetto, R.; Molina-Molina, E.; Bonfrate, L.; Wang, D.Q.; Portincasa, P. Bile acid physiology. Ann. Hepatol. 2017, 16, s4–s14. [Google Scholar] [CrossRef] [PubMed]
- Park, C.C.; Nguyen, P.; Hernandez, C.; Bettencourt, R.; Ramirez, K.; Fortney, L.; Hooker, J.; Sy, E.; Savides, M.T.; Alquiraish, M.H.; et al. Magnetic resonance elastography vs. transient elastography in detection of fibrosis and noninvasive measurement of steatosis in patients with biopsy-proven nonalcoholic fatty liver disease. Gastroenterology 2017, 152, 598–607.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boursier, J.; Vergniol, J.; Guillet, A.; Hiriart, J.B.; Lannes, A.; Le Bail, B.; Michalak, S.; Chermak, F.; Bertrais, S.; Foucher, J.; et al. Diagnostic accuracy and prognostic significance of blood fibrosis tests and liver stiffness measurement by FibroScan in non-alcoholic fatty liver disease. J. Hepatol. 2016, 65, 570–578. [Google Scholar] [CrossRef]
- Ludwig, J.; Viggiano, T.R.; McGill, D.B.; Oh, B.J. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin. Proc. Mayo Clin. 1980, 55, 434–438. [Google Scholar]
- Caldwell, S.H.; Oelsner, D.H.; Iezzoni, J.C.; Hespenheide, E.E.; Battle, E.H.; Driscoll, C.J. Cryptogenic cirrhosis: Clinical characterization and risk factors for underlying disease. Hepatology 1999, 29, 664–669. [Google Scholar] [CrossRef]
- Browning, J.D.; Kumar, K.S.; Saboorian, M.H.; Thiele, D.L. Ethnic differences in the prevalence of cryptogenic cirrhosis. Am. J. Gastroenterol. 2004, 99, 292–298. [Google Scholar] [CrossRef]
- Nasr, P.; Ignatova, S.; Kechagias, S.; Ekstedt, M. Natural history of nonalcoholic fatty liver disease: A prospective follow-up study with serial biopsies. Hepatol. Commun. 2018, 2, 199–210. [Google Scholar] [CrossRef] [Green Version]
- Mittal, S.; El-Serag, H.B.; Sada, Y.H.; Kanwal, F.; Duan, Z.; Temple, S.; May, S.B.; Kramer, J.R.; Richardson, P.A.; Davila, J.A. Hepatocellular carcinoma in the absence of cirrhosis in united states veterans is associated with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2016, 14, 124–131.e1. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Qin, H.; Qiu, S.; Chen, G.; Chen, Y. Correlation of triglyceride to high-density lipoprotein cholesterol ratio with nonalcoholic fatty liver disease among the non-obese Chinese population with normal blood lipid levels: A retrospective cohort research. Lipids Health Dis. 2019, 18, 162. [Google Scholar] [CrossRef] [Green Version]
- Ren, X.Y.; Shi, D.; Ding, J.; Cheng, Z.Y.; Li, H.Y.; Li, J.S.; Pu, H.Q.; Yang, A.M.; He, C.L.; Zhang, J.P.; et al. Total cholesterol to high-density lipoprotein cholesterol ratio is a significant predictor of nonalcoholic fatty liver: Jinchang cohort study. Lipids Health Dis. 2019, 18, 47. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Sanyal, A.J.; George, J.; International Consensus Panel. MAFLD: A consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef] [PubMed]
- Loomis, A.K.; Kabadi, S.; Preiss, D.; Hyde, C.; Bonato, V.; St Louis, M.; Desai, J.; Gill, J.M.; Welsh, P.; Waterworth, D.; et al. Body mass index and risk of nonalcoholic fatty liver disease: Two electronic health record prospective studies. J. Clin. Endocrinol. Metab. 2016, 101, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Younes, R.; Bugianesi, E. NASH in lean individuals. Semin. Liver Dis. 2019, 39, 86–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NCD Risk Factor Collaboration (NCD-RisC). Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: A pooled analysis of 2416 population-based measurement studies in 128.9 million children, adolescents, and adults. Lancet 2017, 390, 2627–2642. [Google Scholar] [CrossRef] [Green Version]
- Yun, Y.; Kim, H.N.; Lee, E.J.; Ryu, S.; Chang, Y.; Shin, H.; Kim, H.L.; Kim, T.H.; Yoo, K.; Kim, H.Y. Fecal and blood microbiota profiles and presence of nonalcoholic fatty liver disease in obese versus lean subjects. PLoS ONE 2019, 14, e0213692. [Google Scholar] [CrossRef] [Green Version]
- Vecchie, A.; Dallegri, F.; Carbone, F.; Bonaventura, A.; Liberale, L.; Portincasa, P.; Fruhbeck, G.; Montecucco, F. Obesity phenotypes and their paradoxical association with cardiovascular diseases. Eur. J. Intern. Med. 2018, 48, 6–17. [Google Scholar] [CrossRef]
- Molina-Molina, E.; Krawczyk, M.; Stachowska, E.; Lammert, F.; Portincasa, P. Non-alcoholic fatty liver disease in non-obese individuals: Prevalence, pathogenesis and treatment. Clin. Res. Hepatol. Gastroenterol. 2019, 43, 638–645. [Google Scholar] [CrossRef]
- Loomba, R.; Sanyal, A.J. The global NAFLD epidemic. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 686–690. [Google Scholar] [CrossRef]
- Wang, A.Y.; Dhaliwal, J.; Mouzaki, M. Lean non-alcoholic fatty liver disease. Clin. Nutr. 2019, 38, 975–981. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Stepanova, M.; Negro, F.; Hallaji, S.; Younossi, Y.; Lam, B.; Srishord, M. Nonalcoholic fatty liver disease in lean individuals in the United States. Medicine 2012, 91, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Ryu, S.; Sung, K.C.; Cho, Y.K.; Sung, E.; Kim, H.N.; Jung, H.S.; Yun, K.E.; Ahn, J.; Shin, H.; et al. Alcoholic and non-alcoholic fatty liver disease and associations with coronary artery calcification: Evidence from the Kangbuk Samsung Health Study. Gut 2019, 68, 1667–1675. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M. Non-alcoholic fatty liver disease—A global public health perspective. J. Hepatol. 2019, 70, 531–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.S.; Cho, H.J.; Kim, H.J.; Kang, D.R.; Berry, J.R.; Kim, J.H.; Yang, M.J.; Lim, S.G.; Kim, S.; Cheong, J.Y.; et al. Nonalcoholic fatty liver disease as a sentinel marker for the development of diabetes mellitus in non-obese subjects. Dig. Liver Dis. 2018, 50, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Golabi, P.; Otgonsuren, M.; de Avila, L.; Sayiner, M.; Rafiq, N.; Younossi, Z.M. Components of metabolic syndrome increase the risk of mortality in nonalcoholic fatty liver disease (NAFLD). Medicine 2018, 97, e0214. [Google Scholar] [CrossRef]
- Wong, V.W.; Wong, G.L.; Yip, G.W.; Lo, A.O.; Limquiaco, J.; Chu, W.C.; Chim, A.M.; Yu, C.M.; Yu, J.; Chan, F.K.; et al. Coronary artery disease and cardiovascular outcomes in patients with non-alcoholic fatty liver disease. Gut 2011, 60, 1721–1727. [Google Scholar] [CrossRef]
- Assimakopoulos, K.; Karaivazoglou, K.; Tsermpini, E.E.; Diamantopoulou, G.; Triantos, C. Quality of life in patients with nonalcoholic fatty liver disease: A systematic review. J. Psychosom. Res. 2018, 112, 73–80. [Google Scholar] [CrossRef]
- Faienza, M.F.; Chiarito, M.; Molina-Molina, E.; Shanmugam, H.; Lammert, F.; Krawczyk, M.; D’Amato, G.; Portincasa, P. Childhood obesity, cardiovascular and liver health: A growing epidemic with age. World J. Pediatr. WJP 2020. [Google Scholar] [CrossRef]
- Molina-Molina, E.; Lunardi Baccetto, R.; Wang, D.Q.; de Bari, O.; Krawczyk, M.; Portincasa, P. Exercising the hepatobiliary-gut axis. The impact of physical activity performance. Eur. J. Clin. Investig. 2018, 48, e12958. [Google Scholar] [CrossRef] [Green Version]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- Chen, Z.; Yu, Y.; Cai, J.; Li, H. Emerging Molecular Targets for Treatment of Nonalcoholic Fatty Liver Disease. Trends Endocrinol. Metab. 2019, 30, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Portincasa, P.; Krawczyk, M.; Smyk, W.; Lammert, F.; Di Ciaula, A. COVID-19 and nonalcoholic fatty liver disease: Two intersecting pandemics. Eur. J. Clin. Investig. 2020, e13338. [Google Scholar] [CrossRef] [PubMed]
- Martins, M.J.; Ascensao, A.; Magalhaes, J.; Collado, M.C.; Portincasa, P. Molecular mechanisms of NAFLD in metabolic syndrome. BioMed Res. Int. 2015, 2015, 621080. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Farrell, G.C.; Larter, C.Z. Nonalcoholic fatty liver disease: From steatosis to cirrhosis. Hepatology 2006, 43, S99–S112. [Google Scholar] [CrossRef]
- Bai, L.; Li, H. Innate immune regulatory networks in hepatic lipid metabolism. J. Mol. Med. 2019, 97, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Shulman, G.I. Nonalcoholic fatty liver disease as a nexus of metabolic and hepatic diseases. Cell Metab. 2018, 27, 22–41. [Google Scholar] [CrossRef] [Green Version]
- Arab, J.P.; Arrese, M.; Trauner, M. Recent insights into the pathogenesis of nonalcoholic fatty liver disease. Annu. Rev. Pathol. 2018, 13, 321–350. [Google Scholar] [CrossRef]
- Arab, J.P.; Karpen, S.J.; Dawson, P.A.; Arrese, M.; Trauner, M. Bile acids and nonalcoholic fatty liver disease: Molecular insights and therapeutic perspectives. Hepatology 2017, 65, 350–362. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef] [Green Version]
- Marra, F.; Svegliati-Baroni, G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J. Hepatol. 2018, 68, 280–295. [Google Scholar] [CrossRef] [PubMed]
- Sunny, N.E.; Parks, E.J.; Browning, J.D.; Burgess, S.C. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 2011, 14, 804–810. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.E.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, M.S.; Park, S.Y.; Shinzawa, K.; Kim, S.; Chung, K.W.; Lee, J.H.; Kwon, C.H.; Lee, K.W.; Lee, J.H.; Park, C.K.; et al. Lysophosphatidylcholine as a death effector in the lipoapoptosis of hepatocytes. J. Lipid Res. 2008, 49, 84–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mota, M.; Banini, B.A.; Cazanave, S.C.; Sanyal, A.J. Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metab. Clin. Exp. 2016, 65, 1049–1061. [Google Scholar] [CrossRef] [Green Version]
- Pagadala, M.; Kasumov, T.; McCullough, A.J.; Zein, N.N.; Kirwan, J.P. Role of ceramides in nonalcoholic fatty liver disease. Trends Endocrinol. Metab. 2012, 23, 365–371. [Google Scholar] [CrossRef] [Green Version]
- Bellanti, F.; Mitarotonda, D.; Tamborra, R.; Blonda, M.; Iannelli, G.; Petrella, A.; Sanginario, V.; Iuliano, L.; Vendemiale, G.; Serviddio, G. Oxysterols induce mitochondrial impairment and hepatocellular toxicity in non-alcoholic fatty liver disease. Free Radic. Biol. Med. 2014, 75 (Suppl. 1), S16–S17. [Google Scholar] [CrossRef]
- Ioannou, G.N. The role of cholesterol in the pathogenesis of NASH. Trends Endocrinol. Metab. 2016, 27, 84–95. [Google Scholar] [CrossRef]
- Tirosh, O. Hypoxic signaling and cholesterol lipotoxicity in fatty liver disease progression. Oxidative Med. Cell. Longev. 2018, 2018, 2548154. [Google Scholar] [CrossRef]
- Ho, C.M.; Ho, S.L.; Jeng, Y.M.; Lai, Y.S.; Chen, Y.H.; Lu, S.C.; Chen, H.L.; Chang, P.Y.; Hu, R.H.; Lee, P.H. Accumulation of free cholesterol and oxidized low-density lipoprotein is associated with portal inflammation and fibrosis in nonalcoholic fatty liver disease. J. Inflamm. 2019, 16, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahlang, B.; Jin, J.; Beier, J.I.; Hardesty, J.E.; Daly, E.F.; Schnegelberger, R.D.; Falkner, K.C.; Prough, R.A.; Kirpich, I.A.; Cave, M.C. Mechanisms of environmental contributions to fatty liver disease. Curr. Environ. Health Rep. 2019, 6, 80–94. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Watkins, S.M.; Hotamisligil, G.S. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab. 2012, 15, 623–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, R.J.; Samuel, V.T.; Petersen, K.F.; Shulman, G.I. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 2014, 510, 84–91. [Google Scholar] [CrossRef]
- Ertunc, M.E.; Hotamisligil, G.S. Lipid signaling and lipotoxicity in metaflammation: Indications for metabolic disease pathogenesis and treatment. J. Lipid Res. 2016, 57, 2099–2114. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Xu, M.; Zhang, X.; Li, H. Innate immune signaling in nonalcoholic fatty liver disease and cardiovascular diseases. Annu. Rev. Pathol. 2019, 14, 153–184. [Google Scholar] [CrossRef]
- Cai, J.; Zhang, X.J.; Li, H. The role of innate immune cells in nonalcoholic steatohepatitis. Hepatology 2019, 70, 1026–1037. [Google Scholar] [CrossRef]
- Wang, X.A.; Zhang, R.; She, Z.G.; Zhang, X.F.; Jiang, D.S.; Wang, T.; Gao, L.; Deng, W.; Zhang, S.M.; Zhu, L.H.; et al. Interferon regulatory factor 3 constrains IKKbeta/NF-kappaB signaling to alleviate hepatic steatosis and insulin resistance. Hepatology 2014, 59, 870–885. [Google Scholar] [CrossRef]
- Müller, F.A.; Sturla, S.J. Human in vitro models of nonalcoholic fatty liver disease. Curr. Opin. Toxicol. 2019, 16, 9–16. [Google Scholar] [CrossRef]
- Aung, H.H.; Altman, R.; Nyunt, T.; Kim, J.; Nuthikattu, S.; Budamagunta, M.; Voss, J.C.; Wilson, D.; Rutledge, J.C.; Villablanca, A.C. Lipotoxic brain microvascular injury is mediated by activating transcription factor 3-dependent inflammatory and oxidative stress pathways. J. Lipid Res. 2016, 57, 955–968. [Google Scholar] [CrossRef] [Green Version]
- Le Stunff, H.; Coant, N.; Migrenne, S.; Magnan, C. Targeting lipid sensing in the central nervous system: New therapy against the development of obesity and type 2 diabetes. Expert Opin. Ther. Targets 2013, 17, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Portincasa, P.; Wang, D.Q.H. Nonalcoholic fatty liver and gallstone disease. In Gallstones. Recent Advances in Epidemiology, Pathogenesis, Diagnosis and Management; Wang, D.Q.H., Portincasa, P., Eds.; Nova Science Publisher Inc.: New York, NY, USA, 2017; pp. 387–414. [Google Scholar]
- Grattagliano, I.; De Bari, O.; Di Palo, D.; Montecucco, F.; Carbone, F.; Oliveira, P.; Wang, D.Q.H.; Portincasa, P. Mitochondria in liver diseases. In Mitochondrial Biology and Experimental Therapeutics; Oliveira, P., Ed.; Springer Nature: Cham, Switzerland, 2018; pp. 91–126. [Google Scholar] [CrossRef]
- Karlsen, T.H.; Lammert, F.; Thompson, R.J. Genetics of liver disease: From pathophysiology to clinical practice. J. Hepatol. 2015, 62, S6–S14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krawczyk, M.; Portincasa, P.; Lammert, F. PNPLA3-associated steatohepatitis: Toward a gene-based classification of fatty liver disease. Semin. Liver Dis. 2013, 33, 369–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albillos, A.; Gottardi, A.; Rescigno, M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J. Hepatol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Yoo, E.R.; Li, A.A.; Cholankeril, G.; Tighe, S.P.; Kim, W.; Harrison, S.A.; Ahmed, A. Elevated urinary bisphenol a levels are associated with non-alcoholic fatty liver disease among adults in the United States. Liver Int. Off. J. Int. Assoc. Study Liver 2019, 39, 1335–1342. [Google Scholar] [CrossRef]
- Franco, M.E.; Fernandez-Luna, M.T.; Ramirez, A.J.; Lavado, R. Metabolomic-based assessment reveals dysregulation of lipid profiles in human liver cells exposed to environmental obesogens. Toxicol. Appl. Pharmacol. 2020, 398, 115009. [Google Scholar] [CrossRef]
- Wahlang, B.; Appana, S.; Falkner, K.C.; McClain, C.J.; Brock, G.; Cave, M.C. Insecticide and metal exposures are associated with a surrogate biomarker for non-alcoholic fatty liver disease in the National Health and Nutrition Examination Survey 2003–2004. Environ. Sci. Pollut. Res. Int. 2020, 27, 6476–6487. [Google Scholar] [CrossRef]
- Milosevic, N.; Milanovic, M.; Sudji, J.; Bosic Zivanovic, D.; Stojanoski, S.; Vukovic, B.; Milic, N.; Medic Stojanoska, M. Could phthalates exposure contribute to the development of metabolic syndrome and liver disease in humans? Environ. Sci. Pollut. Res. Int. 2020, 27, 772–784. [Google Scholar] [CrossRef]
- Wang, X.; Yang, Y.; Zhu, P.; Wu, Y.; Jin, Y.; Yu, S.; Wei, H.; Qian, M.; Cao, W.; Xu, S.; et al. Prenatal exposure to diesel exhaust PM2.5 programmed non-alcoholic fatty liver disease differently in adult male offspring of mice fed normal chow and a high-fat diet. Environ. Pollut. 2019, 255, 113366. [Google Scholar] [CrossRef]
- Chen, R.; Xu, Y.; Xu, C.; Shu, Y.; Ma, S.; Lu, C.; Mo, X. Associations between mercury exposure and the risk of nonalcoholic fatty liver disease (NAFLD) in US adolescents. Environ. Sci. Pollut. Res. Int. 2019, 26, 31384–31391. [Google Scholar] [CrossRef]
- Ding, S.; Yuan, C.; Si, B.; Wang, M.; Da, S.; Bai, L.; Wu, W. Combined effects of ambient particulate matter exposure and a high-fat diet on oxidative stress and steatohepatitis in mice. PLoS ONE 2019, 14, e0214680. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.X.; Ge, C.X.; Qin, Y.T.; Gu, T.T.; Lou, D.S.; Li, Q.; Hu, L.F.; Feng, J.; Huang, P.; Tan, J. Prolonged PM2.5 exposure elevates risk of oxidative stress-driven nonalcoholic fatty liver disease by triggering increase of dyslipidemia. Free Radic. Biol. Med. 2019, 130, 542–556. [Google Scholar] [CrossRef] [PubMed]
- Schnabl, B.; Brenner, D.A. Interactions between the intestinal microbiome and liver diseases. Gastroenterology 2014, 146, 1513–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liquori, G.E.; Mastrodonato, M.; Mentino, D.; Scillitani, G.; Desantis, S.; Portincasa, P.; Ferri, D. In situ characterization of O-linked glycans of Muc2 in mouse colon. Acta Histochem. 2012, 114, 723–732. [Google Scholar] [CrossRef]
- Mouries, J.; Brescia, P.; Silvestri, A.; Spadoni, I.; Sorribas, M.; Wiest, R.; Mileti, E.; Galbiati, M.; Invernizzi, P.; Adorini, L.; et al. Microbiota-driven gut vascular barrier disruption is a prerequisite for non-alcoholic steatohepatitis development. J. Hepatol. 2019, 71, 1216–1228. [Google Scholar] [CrossRef] [Green Version]
- Brandl, K.; Kumar, V.; Eckmann, L. Gut-liver axis at the frontier of host-microbial interactions. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G413–G419. [Google Scholar] [CrossRef]
- Garruti, G.; Di Ciaula, A.; Wang, H.H.; Wang, D.Q.; Portincasa, P. Cross-talk between bile acids and gastro-intestinal and thermogenic hormones: Clues from bariatric surgery. Ann. Hepatol. 2017, 16, S68–S82. [Google Scholar] [CrossRef]
- Garruti, G.; Wang, D.Q.; Di Ciaula, A.; Portincasa, P. Cholecystectomy: A way forward and back to metabolic syndrome? Lab. Investig. 2018, 98, 4–6. [Google Scholar] [CrossRef] [Green Version]
- Nicoletti, A.; Ponziani, F.R.; Biolato, M.; Valenza, V.; Marrone, G.; Sganga, G.; Gasbarrini, A.; Miele, L.; Grieco, A. Intestinal permeability in the pathogenesis of liver damage: From non-alcoholic fatty liver disease to liver transplantation. World J. Gastroenterol. WJG 2019, 25, 4814–4834. [Google Scholar] [CrossRef]
- Okumura, R.; Takeda, K. Maintenance of intestinal homeostasis by mucosal barriers. Inflamm. Regen. 2018, 38, 5. [Google Scholar] [CrossRef]
- Meyer-Hoffert, U.; Hornef, M.W.; Henriques-Normark, B.; Axelsson, L.G.; Midtvedt, T.; Putsep, K.; Andersson, M. Secreted enteric antimicrobial activity localises to the mucus surface layer. Gut 2008, 57, 764–771. [Google Scholar] [CrossRef] [PubMed]
- Savage, D.C. Microbial ecology of the gastrointestinal tract. Annu. Rev. Microbiol. 1977, 31, 107–133. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.V.; Pedersen, O. The human intestinal microbiome in health and disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jandhyala, S.M.; Talukdar, R.; Subramanyam, C.; Vuyyuru, H.; Sasikala, M.; Nageshwar Reddy, D. Role of the normal gut microbiota. World J. Gastroenterol. WJG 2015, 21, 8787–8803. [Google Scholar] [CrossRef]
- Kim, Y.S.; Ho, S.B. Intestinal goblet cells and mucins in health and disease: Recent insights and progress. Curr. Gastroenterol. Rep. 2010, 12, 319–330. [Google Scholar] [CrossRef] [Green Version]
- Johansson, M.E.; Phillipson, M.; Petersson, J.; Velcich, A.; Holm, L.; Hansson, G.C. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc. Natl. Acad. Sci. USA 2008, 105, 15064–15069. [Google Scholar] [CrossRef] [Green Version]
- Vereecke, L.; Beyaert, R.; van Loo, G. Enterocyte death and intestinal barrier maintenance in homeostasis and disease. Trends Mol. Med. 2011, 17, 584–593. [Google Scholar] [CrossRef]
- Tsilingiri, K.; Barbosa, T.; Penna, G.; Caprioli, F.; Sonzogni, A.; Viale, G.; Rescigno, M. Probiotic and postbiotic activity in health and disease: Comparison on a novel polarised ex-vivo organ culture model. Gut 2012, 61, 1007–1015. [Google Scholar] [CrossRef] [Green Version]
- Tsilingiri, K.; Rescigno, M. Postbiotics: What else? In Beneficial Microbes 4; Wageningen Academic Publishers: Wageningen, The Netherlands, 2013; pp. 101–107. [Google Scholar]
- Levy, M.; Blacher, E.; Elinav, E. Microbiome, metabolites and host immunity. Curr. Opin. Microbiol. 2017, 35, 8–15. [Google Scholar] [CrossRef]
- Blacher, E.; Levy, M.; Tatirovsky, E.; Elinav, E. Microbiome-modulated metabolites at the interface of host immunity. J. Immunol. 2017, 198, 572–580. [Google Scholar] [CrossRef] [Green Version]
- Mosca, F.; Gianni, M.L.; Rescigno, M. Can Postbiotics represent a new strategy for NEC? In Probiotics and Child Gastrointestinal Health; Springer: Cham, Switzerland, 2019. [Google Scholar]
- Gibbins, H.L.; Proctor, G.B.; Yakubov, G.E.; Wilson, S.; Carpenter, G.H. SIgA binding to mucosal surfaces is mediated by mucin-mucin interactions. PLoS ONE 2015, 10, e0119677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakobsson, H.E.; Rodriguez-Pineiro, A.M.; Schutte, A.; Ermund, A.; Boysen, P.; Bemark, M.; Sommer, F.; Backhed, F.; Hansson, G.C.; Johansson, M.E. The composition of the gut microbiota shapes the colon mucus barrier. EMBO Rep. 2015, 16, 164–177. [Google Scholar] [CrossRef] [PubMed]
- Birchenough, G.M.; Nystrom, E.E.; Johansson, M.E.; Hansson, G.C. A sentinel goblet cell guards the colonic crypt by triggering Nlrp6-dependent Muc2 secretion. Science 2016, 352, 1535–1542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abreu, M.T. Toll-like receptor signalling in the intestinal epithelium: How bacterial recognition shapes intestinal function. Nat. Rev. Immunol. 2010, 10, 131. [Google Scholar] [CrossRef] [PubMed]
- Ouwerkerk, J.P.; de Vos, W.M.; Belzer, C. Glycobiome: Bacteria and mucus at the epithelial interface. Best Pract. Res. Clin. Gastroenterol. 2013, 27, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Derrien, M.; Van Baarlen, P.; Hooiveld, G.; Norin, E.; Muller, M.; de Vos, W.M. Modulation of mucosal immune response, tolerance, and proliferation in mice colonized by the mucin-degrader akkermansia muciniphila. Front. Microbiol. 2011, 2, 166. [Google Scholar] [CrossRef] [Green Version]
- Dao, M.C.; Everard, A.; Aron-Wisnewsky, J.; Sokolovska, N.; Prifti, E.; Verger, E.O.; Kayser, B.D.; Levenez, F.; Chilloux, J.; Hoyles, L.; et al. Akkermansia muciniphila and improved metabolic health during a dietary intervention in obesity: Relationship with gut microbiome richness and ecology. Gut 2016, 65, 426–436. [Google Scholar] [CrossRef] [Green Version]
- Grander, C.; Adolph, T.E.; Wieser, V.; Lowe, P.; Wrzosek, L.; Gyongyosi, B.; Ward, D.V.; Grabherr, F.; Gerner, R.R.; Pfister, A.; et al. Recovery of ethanol-induced Akkermansia muciniphila depletion ameliorates alcoholic liver disease. Gut 2018, 67, 891–901. [Google Scholar] [CrossRef]
- Everard, A.; Belzer, C.; Geurts, L.; Ouwerkerk, J.P.; Druart, C.; Bindels, L.B.; Guiot, Y.; Derrien, M.; Muccioli, G.G.; Delzenne, N.M. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. USA 2013, 110, 9066–9071. [Google Scholar] [CrossRef] [Green Version]
- Desai, M.S.; Seekatz, A.M.; Koropatkin, N.M.; Kamada, N.; Hickey, C.A.; Wolter, M.; Pudlo, N.A.; Kitamoto, S.; Terrapon, N.; Muller, A.; et al. A dietary fiber-deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell 2016, 167, 1339–1353.e21. [Google Scholar] [CrossRef] [Green Version]
- Wrzosek, L.; Miquel, S.; Noordine, M.L.; Bouet, S.; Chevalier-Curt, M.J.; Robert, V.; Philippe, C.; Bridonneau, C.; Cherbuy, C.; Robbe-Masselot, C.; et al. Bacteroides thetaiotaomicron and Faecalibacterium prausnitzii influence the production of mucus glycans and the development of goblet cells in the colonic epithelium of a gnotobiotic model rodent. BMC Biol. 2013, 11, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.-H.; Eberl, G. Type 3 regulatory T cells at the interface of symbiosis. J. Microbiol. 2018, 56, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y.; Hand, T.W. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynes, B.; Palou, M.; Rodriguez, A.M.; Palou, A. Regulation of adaptive thermogenesis and browning by prebiotics and postbiotics. Front. Physiol. 2018, 9, 1908. [Google Scholar] [CrossRef]
- Bergström, J.H.; Birchenough, G.M.H.; Katona, G.; Schroeder, B.O.; Schütte, A.; Ermund, A.; Johansson, M.E.V.; Hansson, G.C. Gram-positive bacteria are held at a distance in the colon mucus by the lectin-like protein ZG16. Proc. Natl. Acad. Sci. USA 2016, 113, 13833–13838. [Google Scholar] [CrossRef] [Green Version]
- Ismail, A.S.; Severson, K.M.; Vaishnava, S.; Behrendt, C.L.; Yu, X.; Benjamin, J.L.; Ruhn, K.A.; Hou, B.; DeFranco, A.L.; Yarovinsky, F. γδ intraepithelial lymphocytes are essential mediators of host–microbial homeostasis at the intestinal mucosal surface. Proc. Natl. Acad. Sci. USA 2011, 108, 8743–8748. [Google Scholar] [CrossRef] [Green Version]
- Johansson, M.E.V. Fast renewal of the distal colonic mucus layers by the surface goblet cells as measured by in vivo labeling of mucin glycoproteins. PLoS ONE 2012, 7, e41009. [Google Scholar] [CrossRef]
- Mastrodonato, M.; Mentino, D.; Portincasa, P.; Calamita, G.; Liquori, G.E.; Ferri, D. High-fat diet alters the oligosaccharide chains of colon mucins in mice. Histochem. Cell Biol. 2014, 142, 449–459. [Google Scholar] [CrossRef]
- Ponziani, F.R.; Gerardi, V.; Gasbarrini, A. Diagnosis and treatment of small intestinal bacterial overgrowth. Expert Rev. Gastroenterol. Hepatol. 2016, 10, 215–227. [Google Scholar] [CrossRef]
- Inagaki, T.; Choi, M.; Moschetta, A.; Peng, L.; Cummins, C.L.; McDonald, J.G.; Luo, G.; Jones, S.A.; Goodwin, B.; Richardson, J.A.; et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005, 2, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Garruti, G.; Wang, H.H.; Bonfrate, L.; de Bari, O.; Wang, D.Q.; Portincasa, P. A pleiotropic role for the orphan nuclear receptor small heterodimer partner in lipid homeostasis and metabolic pathways. J. Lipids 2012, 2012, 304292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Hu, C.; Zhang, X.; Jia, W. Role of gut microbiota, bile acids and their cross-talk in the effects of bariatric surgery on obesity and type 2 diabetes. J. Diabetes Investig. 2018, 9, 13–20. [Google Scholar] [CrossRef]
- Wahlström, A.; Sayin, S.I.; Marschall, H.-U.; Bäckhed, F. Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab. 2016, 24, 41–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ory, D.S. Nuclear receptor signaling in the control of cholesterol homeostasis: Have the orphans found a home? Circ. Res. 2004, 95, 660–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Aguiar Vallim, T.Q.; Tarling, E.J.; Edwards, P.A. Pleiotropic roles of bile acids in metabolism. Cell Metab. 2013, 17, 657–669. [Google Scholar] [CrossRef] [Green Version]
- Luiking, Y.C.; Peeters, T.L.; Stolk, M.F.; Nieuwenhuijs, V.B.; Portincasa, P.; Depoortere, I.; van Berge Henegouwen, G.P.; Akkermans, L.M. Motilin induces gall bladder emptying and antral contractions in the fasted state in humans. Gut 1998, 42, 830–835. [Google Scholar] [CrossRef] [PubMed]
- Portincasa, P.; Peeters, T.L.; van Berge-Henegouwen, G.P.; van Solinge, W.W.; Palasciano, G.; van Erpecum, K.J. Acute intraduodenal bile salt depletion leads to strong gallbladder contraction, altered antroduodenal motility and high plasma motilin levels in humans. Neurogastroenterol. Motil. 2000, 12, 421–430. [Google Scholar] [CrossRef]
- Portincasa, P.; Di Ciaula, A.; Wang, H.H.; Palasciano, G.; van Erpecum, K.J.; Moschetta, A.; Wang, D.Q. Coordinate regulation of gallbladder motor function in the gut-liver axis. Hepatology 2008, 47, 2112–2126. [Google Scholar] [CrossRef]
- Jansson, R.; Steen, G.; Svanvik, J. Effects of intravenous vasoactive intestinal peptide (VIP) on gallbladder function in the cat. Gastroenterology 1978, 75, 47–50. [Google Scholar] [CrossRef]
- Housset, C.; Chretien, Y.; Debray, D.; Chignard, N. Functions of the Gallbladder. Compr. Physiol. 2016, 6, 1549–1577. [Google Scholar] [CrossRef]
- Choi, M.; Moschetta, A.; Bookout, A.L.; Peng, L.; Umetani, M.; Holmstrom, S.R.; Suino-Powell, K.; Xu, H.E.; Richardson, J.A.; Gerard, R.D.; et al. Identification of a hormonal basis for gallbladder filling. Nat. Med. 2006, 12, 1253–1255. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y. Bile acid metabolism and signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar] [CrossRef] [Green Version]
- Dawson, P.A.; Lan, T.; Rao, A. Bile acid transporters. J. Lipid Res. 2009, 50, 2340–2357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjorkhem, I.; Blomstrand, R.; Lewenhaupt, A.; Svensson, L. Effect of lymphatic drainage on 7alpha-hydroxylation of cholesterol in rat liver. Biochem. Biophys. Res. Commun. 1978, 85, 532–540. [Google Scholar] [CrossRef]
- Kemper, J.K. Regulation of FXR transcriptional activity in health and disease: Emerging roles of FXR cofactors and post-translational modifications. Biochim. Biophys. Acta 2011, 1812, 842–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodwin, B.; Jones, S.A.; Price, R.R.; Watson, M.A.; McKee, D.D.; Moore, L.B.; Galardi, C.; Wilson, J.G.; Lewis, M.C.; Roth, M.E.; et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol. Cell 2000, 6, 517–526. [Google Scholar] [CrossRef]
- Lu, T.T.; Makishima, M.; Repa, J.J.; Schoonjans, K.; Kerr, T.A.; Auwerx, J.; Mangelsdorf, D.J. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol. Cell 2000, 6, 507–515. [Google Scholar] [CrossRef]
- Nishimaki-Mogami, T.; Une, M.; Fujino, T.; Sato, Y.; Tamehiro, N.; Kawahara, Y.; Shudo, K.; Inoue, K. Identification of intermediates in the bile acid synthetic pathway as ligands for the farnesoid X receptor. J. Lipid Res. 2004, 45, 1538–1545. [Google Scholar] [CrossRef] [Green Version]
- Modica, S.; Bellafante, E.; Moschetta, A. Master regulation of bile acid and xenobiotic metabolism via the FXR, PXR and CAR trio. Front. Biosci. (Landmark Ed.) 2008, 14, 4719–4745. [Google Scholar] [CrossRef] [Green Version]
- Brighton, C.A.; Rievaj, J.; Kuhre, R.E.; Glass, L.L.; Schoonjans, K.; Holst, J.J.; Gribble, F.M.; Reimann, F. Bile acids trigger GLP-1 release predominantly by accessing basolaterally located G Protein-coupled bile acid receptors. Endocrinology 2015, 156, 3961–3970. [Google Scholar] [CrossRef] [Green Version]
- Sinal, C.J.; Tohkin, M.; Miyata, M.; Ward, J.M.; Lambert, G.; Gonzalez, F.J. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 2000, 102, 731–744. [Google Scholar] [CrossRef] [Green Version]
- Pols, T.W.; Noriega, L.G.; Nomura, M.; Auwerx, J.; Schoonjans, K. The bile acid membrane receptor TGR5: A valuable metabolic target. Dig. Dis. 2011, 29, 37–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perino, A.; Schoonjans, K. TGR5 and immunometabolism: Insights from physiology and pharmacology. Trends Pharm. Sci. 2015, 36, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chiang, J.Y. Bile acid signaling in metabolic disease and drug therapy. Pharm. Rev. 2014, 66, 948–983. [Google Scholar] [CrossRef] [Green Version]
- Wagner, M.; Zollner, G.; Trauner, M. New molecular insights into the mechanisms of cholestasis. J. Hepatol. 2009, 51, 565–580. [Google Scholar] [CrossRef] [Green Version]
- Kurashima, Y.; Kiyono, H. Mucosal ecological network of epithelium and immune cells for gut homeostasis and tissue healing. Annu. Rev. Immunol. 2017, 35, 119–147. [Google Scholar] [CrossRef]
- Nevo, S.; Kadouri, N.; Abramson, J. Tuft cells: From the mucosa to the thymus. Immunol. Lett. 2019, 210, 1–9. [Google Scholar] [CrossRef]
- Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 2009, 9, 799–809. [Google Scholar] [CrossRef]
- Odenwald, M.A.; Turner, J.R. The intestinal epithelial barrier: A therapeutic target? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 9–21. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Okawa, K.; Yano, T.; Tsukita, S.; Tsukita, S. Optimized proteomic analysis on gels of cell-cell adhering junctional membrane proteins. Biochemistry 2008, 47, 5378–5386. [Google Scholar] [CrossRef]
- Schneeberger, E.E.; Lynch, R.D. The tight junction: A multifunctional complex. Am. J. Physiol. Cell Physiol. 2004, 286, C1213–C1228. [Google Scholar] [CrossRef] [PubMed]
- Van Itallie, C.M.; Anderson, J.M. Architecture of tight junctions and principles of molecular composition. Semin. Cell Dev. Biol. 2014, 36, 157–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, J.M.; Van Itallie, C.M. Physiology and function of the tight junction. Cold Spring Harb. Perspect. Biol. 2009, 1, a002584. [Google Scholar] [CrossRef] [PubMed]
- Van Itallie, C.M.; Holmes, J.; Bridges, A.; Gookin, J.L.; Coccaro, M.R.; Proctor, W.; Colegio, O.R.; Anderson, J.M. The density of small tight junction pores varies among cell types and is increased by expression of claudin-2. J. Cell Sci. 2008, 121, 298–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, C.T.; Dzus, A.L.; Colgan, S.P. Autocrine regulation of epithelial permeability by hypoxia: Role for polarized release of tumor necrosis factor alpha. Gastroenterology 1998, 114, 657–668. [Google Scholar] [CrossRef]
- Madara, J.L.; Stafford, J. Interferon-gamma directly affects barrier function of cultured intestinal epithelial monolayers. J. Clin. Investig. 1989, 83, 724–727. [Google Scholar] [CrossRef]
- Turner, J.R.; Rill, B.K.; Carlson, S.L.; Carnes, D.; Kerner, R.; Mrsny, R.J.; Madara, J.L. Physiological regulation of epithelial tight junctions is associated with myosin light-chain phosphorylation. Am. J. Physiol. 1997, 273, C1378–C1385. [Google Scholar] [CrossRef]
- Hartmann, P.; Haimerl, M.; Mazagova, M.; Brenner, D.A.; Schnabl, B. Toll-like receptor 2-mediated intestinal injury and enteric tumor necrosis factor receptor I contribute to liver fibrosis in mice. Gastroenterology 2012, 143, 1330–1340.e1. [Google Scholar] [CrossRef] [Green Version]
- Cariello, R.; Federico, A.; Sapone, A.; Tuccillo, C.; Scialdone, V.R.; Tiso, A.; Miranda, A.; Portincasa, P.; Carbonara, V.; Palasciano, G.; et al. Intestinal permeability in patients with chronic liver diseases: Its relationship with the aetiology and the entity of liver damage. Dig. Liver Dis. 2010, 42, 200–204. [Google Scholar] [CrossRef]
- Assimakopoulos, S.F.; Tsamandas, A.C.; Tsiaoussis, G.I.; Karatza, E.; Triantos, C.; Vagianos, C.E.; Spiliopoulou, I.; Kaltezioti, V.; Charonis, A.; Nikolopoulou, V.N.; et al. Altered intestinal tight junctions’ expression in patients with liver cirrhosis: A pathogenetic mechanism of intestinal hyperpermeability. Eur. J. Clin. Investig. 2012, 42, 439–446. [Google Scholar] [CrossRef]
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Masciana, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Bennett, K.M.; Walker, S.L.; Lo, D.D. Epithelial microvilli establish an electrostatic barrier to microbial adhesion. Infect. Immun. 2014, 82, 2860–2871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Palo, D.M.; Garruti, G.; Di Ciaula, A.; Molina-Molina, E.; Shanmugam, H.; De Angelis, M.; Portincasa, P. Increased colonic permeability and lifestyles as contributing factors to obesity and liver steatosis. Nutrients 2020, 12, 564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiest, R.; Lawson, M.; Geuking, M. Pathological bacterial translocation in liver cirrhosis. J. Hepatol. 2014, 60, 197–209. [Google Scholar] [CrossRef] [Green Version]
- McDonald, B.D.; Jabri, B.; Bendelac, A. Diverse developmental pathways of intestinal intraepithelial lymphocytes. Nat. Rev. Immunol. 2018, 18, 514–525. [Google Scholar] [CrossRef]
- Chieppa, M.; Rescigno, M.; Huang, A.Y.C.; Germain, R.N. Dynamic imaging of dendritic cell extension into the small bowel lumen in response to epithelial cell TLR engagement. J. Exp. Med. 2006, 203, 2841–2852. [Google Scholar] [CrossRef] [Green Version]
- Niess, J.H.; Brand, S.; Gu, X.; Landsman, L.; Jung, S.; McCormick, B.A.; Vyas, J.M.; Boes, M.; Ploegh, H.L.; Fox, J.G.; et al. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science 2005, 307, 254–258. [Google Scholar] [CrossRef] [Green Version]
- Mazzini, E.; Massimiliano, L.; Penna, G.; Rescigno, M. Oral tolerance can be established via gap junction transfer of fed antigens from CX3CR1+ macrophages to CD103+ dendritic cells. Immunity 2014, 40, 248–261. [Google Scholar] [CrossRef] [Green Version]
- Brennan, P.J.; Brigl, M.; Brenner, M.B. Invariant natural killer T cells: An innate activation scheme linked to diverse effector functions. Nat. Rev. Immunol. 2013, 13, 101–117. [Google Scholar] [CrossRef]
- Dias, J.; Leeansyah, E.; Sandberg, J.K. Multiple layers of heterogeneity and subset diversity in human MAIT cell responses to distinct microorganisms and to innate cytokines. Proc. Natl. Acad. Sci. USA 2017, 114, E5434–E5443. [Google Scholar] [CrossRef] [Green Version]
- Corbett, A.J.; Eckle, S.B.; Birkinshaw, R.W.; Liu, L.; Patel, O.; Mahony, J.; Chen, Z.; Reantragoon, R.; Meehan, B.; Cao, H.; et al. T-cell activation by transitory neo-antigens derived from distinct microbial pathways. Nature 2014, 509, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Sandquist, I.; Kolls, J. Update on regulation and effector functions of Th17 cells. F1000Res 2018, 7, 205. [Google Scholar] [CrossRef]
- Hirota, K.; Turner, J.-E.; Villa, M.; Duarte, J.H.; Demengeot, J.; Steinmetz, O.M.; Stockinger, B. Plasticity of T H 17 cells in Peyer’s patches is responsible for the induction of T cell–dependent IgA responses. Nat. Immunol. 2013, 14, 372. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Tanoue, T.; Ando, M.; Kamada, N.; Nagano, Y.; Narushima, S.; Suda, W.; Imaoka, A.; Setoyama, H.; Nagamori, T.; et al. Th17 Cell Induction by Adhesion of Microbes to Intestinal Epithelial Cells. Cell 2015, 163, 367–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaboriau-Routhiau, V.; Rakotobe, S.; Lecuyer, E.; Mulder, I.; Lan, A.; Bridonneau, C.; Rochet, V.; Pisi, A.; De Paepe, M.; Brandi, G.; et al. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity 2009, 31, 677–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Rudra, D. Emerging functions of regulatory T cells in tissue homeostasis. Front. Immunol. 2018, 9, 883. [Google Scholar] [CrossRef]
- Wojno, E.D.T.; Artis, D. Emerging concepts and future challenges in innate lymphoid cell biology. J. Exp. Med. 2016, 213, 2229–2248. [Google Scholar] [CrossRef] [Green Version]
- Gautreaux, M.D.; Gelder, F.B.; Deitch, E.A.; Berg, R.D. Adoptive transfer of T lymphocytes to T-cell-depleted mice inhibits Escherichia coli translocation from the gastrointestinal tract. Infect. Immun. 1995, 63, 3827–3834. [Google Scholar] [CrossRef] [Green Version]
- Gautreaux, M.D.; Deitch, E.A.; Berg, R.D. T lymphocytes in host defense against bacterial translocation from the gastrointestinal tract. Infect. Immun. 1994, 62, 2874–2884. [Google Scholar] [CrossRef] [Green Version]
- Spadoni, I.; Zagato, E.; Bertocchi, A.; Paolinelli, R.; Hot, E.; Di Sabatino, A.; Caprioli, F.; Bottiglieri, L.; Oldani, A.; Viale, G.; et al. A gut-vascular barrier controls the systemic dissemination of bacteria. Science 2015, 350, 830–834. [Google Scholar] [CrossRef] [PubMed]
- Spadoni, I.; Fornasa, G.; Rescigno, M. Organ-specific protection mediated by cooperation between vascular and epithelial barriers. Nat. Rev. Immunol. 2017, 17, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Cornet, A.; Savidge, T.C.; Cabarrocas, J.; Deng, W.L.; Colombel, J.F.; Lassmann, H.; Desreumaux, P.; Liblau, R.S. Enterocolitis induced by autoimmune targeting of enteric glial cells: A possible mechanism in Crohn’s disease? Proc. Natl. Acad. Sci. USA 2001, 98, 13306–13311. [Google Scholar] [CrossRef] [Green Version]
- Ciccia, F.; Guggino, G.; Rizzo, A.; Alessandro, R.; Luchetti, M.M.; Milling, S.; Saieva, L.; Cypers, H.; Stampone, T.; Di Benedetto, P.; et al. Dysbiosis and zonulin upregulation alter gut epithelial and vascular barriers in patients with ankylosing spondylitis. Ann. Rheum. Dis. 2017, 76, 1123–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macpherson, A.J.; Harris, N.L. Interactions between commensal intestinal bacteria and the immune system. Nat. Rev. Immunol. 2004, 4, 478–485. [Google Scholar] [CrossRef]
- Maynard, C.L.; Elson, C.O.; Hatton, R.D.; Weaver, C.T. Reciprocal interactions of the intestinal microbiota and immune system. Nature 2012, 489, 231–241. [Google Scholar] [CrossRef] [Green Version]
- Macpherson, A.J.; Uhr, T. Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science 2004, 303, 1662–1665. [Google Scholar] [CrossRef] [Green Version]
- Macpherson, A.J.; Gatto, D.; Sainsbury, E.; Harriman, G.R.; Hengartner, H.; Zinkernagel, R.M. A primitive T cell-independent mechanism of intestinal mucosal IgA responses to commensal bacteria. Science 2000, 288, 2222–2226. [Google Scholar] [CrossRef]
- Brun, P.; Castagliuolo, I.; Di Leo, V.; Buda, A.; Pinzani, M.; Palu, G.; Martines, D. Increased intestinal permeability in obese mice: New evidence in the pathogenesis of nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G518–G525. [Google Scholar] [CrossRef] [Green Version]
- Etienne-Mesmin, L.; Vijay-Kumar, M.; Gewirtz, A.T.; Chassaing, B. Hepatocyte Toll-Like Receptor 5 Promotes Bacterial Clearance and Protects Mice Against High-Fat Diet-Induced Liver Disease. Cell Mol. Gastroenterol. Hepatol. 2016, 2, 584–604. [Google Scholar] [CrossRef] [Green Version]
- Balmer, M.L.; Slack, E.; de Gottardi, A.; Lawson, M.A.; Hapfelmeier, S.; Miele, L.; Grieco, A.; Van Vlierberghe, H.; Fahrner, R.; Patuto, N.; et al. The liver may act as a firewall mediating mutualism between the host and its gut commensal microbiota. Sci. Transl. Med. 2014, 6, 237ra266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, N.J. Liver: The liver as a firewall—Clearance of commensal bacteria that have escaped from the gut. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 391. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.Y.; Moriarty, T.J.; Wong, C.H.; Zhou, H.; Strieter, R.M.; van Rooijen, N.; Chaconas, G.; Kubes, P. An intravascular immune response to Borrelia burgdorferi involves Kupffer cells and iNKT cells. Nat. Immunol. 2010, 11, 295–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knook, D.L.; Barkway, C.; Sleyster, E.C. Lysosomal enzyme content of Kupffer and endothelial liver cells isolated from germfree and clean conventional rats. Infect. Immun. 1981, 33, 620–622. [Google Scholar] [CrossRef] [Green Version]
- Schwabe, R.F.; Seki, E.; Brenner, D.A. Toll-like receptor signaling in the liver. Gastroenterology 2006, 130, 1886–1900. [Google Scholar] [CrossRef] [Green Version]
- Fox, E.S.; Thomas, P.; Broitman, S.A. Clearance of gut-derived endotoxins by the liver. Release and modification of 3H, 14C-lipopolysaccharide by isolated rat Kupffer cells. Gastroenterology 1989, 96, 456–461. [Google Scholar] [CrossRef]
- Su, G.L.; Klein, R.D.; Aminlari, A.; Zhang, H.Y.; Steinstraesser, L.; Alarcon, W.H.; Remick, D.G.; Wang, S.C. Kupffer cell activation by lipopolysaccharide in rats: Role for lipopolysaccharide binding protein and toll-like receptor 4. Hepatology 2000, 31, 932–936. [Google Scholar] [CrossRef]
- Schumann, R.R.; Kirschning, C.J.; Unbehaun, A.; Aberle, H.P.; Knope, H.P.; Lamping, N.; Ulevitch, R.J.; Herrmann, F. The lipopolysaccharide-binding protein is a secretory class 1 acute-phase protein whose gene is transcriptionally activated by APRF/STAT/3 and other cytokine-inducible nuclear proteins. Mol. Cell. Biol. 1996, 16, 3490–3503. [Google Scholar] [CrossRef] [Green Version]
- Pugin, J.; Schurer-Maly, C.C.; Leturcq, D.; Moriarty, A.; Ulevitch, R.J.; Tobias, P.S. Lipopolysaccharide activation of human endothelial and epithelial cells is mediated by lipopolysaccharide-binding protein and soluble CD14. Proc. Natl. Acad. Sci. USA 1993, 90, 2744–2748. [Google Scholar] [CrossRef] [Green Version]
- Landmann, R.; Knopf, H.P.; Link, S.; Sansano, S.; Schumann, R.; Zimmerli, W. Human monocyte CD14 is upregulated by lipopolysaccharide. Infect. Immun. 1996, 64, 1762–1769. [Google Scholar] [CrossRef] [Green Version]
- Frey, E.A.; Miller, D.S.; Jahr, T.G.; Sundan, A.; Bazil, V.; Espevik, T.; Finlay, B.B.; Wright, S.D. Soluble CD14 participates in the response of cells to lipopolysaccharide. J. Exp. Med. 1992, 176, 1665–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, R.S.; Lee, F.Y.; Lee, S.D.; Tsai, Y.T.; Lin, H.C.; Lu, R.H.; Hsu, W.C.; Huang, C.C.; Wang, S.S.; Lo, K.J. Endotoxemia in patients with chronic liver diseases: Relationship to severity of liver diseases, presence of esophageal varices, and hyperdynamic circulation. J. Hepatol. 1995, 22, 165–172. [Google Scholar] [CrossRef]
- Garcia-Tsao, G.; Lee, F.Y.; Barden, G.E.; Cartun, R.; West, A.B. Bacterial translocation to mesenteric lymph nodes is increased in cirrhotic rats with ascites. Gastroenterology 1995, 108, 1835–1841. [Google Scholar] [CrossRef]
- Cirera, I.; Bauer, T.M.; Navasa, M.; Vila, J.; Grande, L.; Taura, P.; Fuster, J.; Garcia-Valdecasas, J.C.; Lacy, A.; Suarez, M.J.; et al. Bacterial translocation of enteric organisms in patients with cirrhosis. J. Hepatol. 2001, 34, 32–37. [Google Scholar] [CrossRef]
- Bellot, P.; Garcia-Pagan, J.C.; Frances, R.; Abraldes, J.G.; Navasa, M.; Perez-Mateo, M.; Such, J.; Bosch, J. Bacterial DNA translocation is associated with systemic circulatory abnormalities and intrahepatic endothelial dysfunction in patients with cirrhosis. Hepatology 2010, 52, 2044–2052. [Google Scholar] [CrossRef]
- Ramadori, G.; Moriconi, F.; Malik, I.; Dudas, J. Physiology and pathophysiology of liver inflammation, damage and repair. J. Physiol. Pharm. 2008, 59 (Suppl. 1), 107–117. [Google Scholar]
- Kudo, H.; Takahara, T.; Yata, Y.; Kawai, K.; Zhang, W.; Sugiyama, T. Lipopolysaccharide triggered TNF-alpha-induced hepatocyte apoptosis in a murine non-alcoholic steatohepatitis model. J. Hepatol. 2009, 51, 168–175. [Google Scholar] [CrossRef]
- Heymann, F.; Tacke, F. Immunology in the liver--from homeostasis to disease. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 88–110. [Google Scholar] [CrossRef]
- Brenner, C.; Galluzzi, L.; Kepp, O.; Kroemer, G. Decoding cell death signals in liver inflammation. J. Hepatol. 2013, 59, 583–594. [Google Scholar] [CrossRef] [Green Version]
- Tilg, H.; Moschen, A.R.; Szabo, G. Interleukin-1 and inflammasomes in alcoholic liver disease/acute alcoholic hepatitis and nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2016, 64, 955–965. [Google Scholar] [CrossRef]
- Wenfeng, Z.; Yakun, W.; Di, M.; Jianping, G.; Chuanxin, W.; Chun, H. Kupffer cells: Increasingly significant role in nonalcoholic fatty liver disease. Ann. Hepatol. 2014, 13, 489–495. [Google Scholar] [CrossRef]
- Duffield, J.S.; Forbes, S.J.; Constandinou, C.M.; Clay, S.; Partolina, M.; Vuthoori, S.; Wu, S.; Lang, R.; Iredale, J.P. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J. Clin. Investig. 2005, 115, 56–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seki, E.; De Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; Schnabl, B. Role of innate immunity and the microbiota in liver fibrosis: Crosstalk between the liver and gut. J. Physiol. 2012, 590, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Wree, A.; Broderick, L.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. From NAFLD to NASH to cirrhosis-new insights into disease mechanisms. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Roderfeld, M. Matrix metalloproteinase functions in hepatic injury and fibrosis. Matrix Biol. J. Int. Soc. Matrix Biol. 2018, 68–69, 452–462. [Google Scholar] [CrossRef]
- Benyon, R.C.; Arthur, M.J. Extracellular matrix degradation and the role of hepatic stellate cells. Semin. Liver Dis. 2001, 21, 373–384. [Google Scholar] [CrossRef]
- Schuppan, D.; Ruehl, M.; Somasundaram, R.; Hahn, E.G. Matrix as a modulator of hepatic fibrogenesis. Semin. Liver Dis. 2001, 21, 351–372. [Google Scholar] [CrossRef]
- Knittel, T.; Mehde, M.; Kobold, D.; Saile, B.; Dinter, C.; Ramadori, G. Expression patterns of matrix metalloproteinases and their inhibitors in parenchymal and non-parenchymal cells of rat liver: Regulation by TNF-alpha and TGF-beta1. J. Hepatol. 1999, 30, 48–60. [Google Scholar] [CrossRef]
- Miele, L.; Forgione, A.; La Torre, G.; Vero, V.; Cefalo, C.; Racco, S.; Vellone, V.G.; Vecchio, F.M.; Gasbarrini, G.; Rapaccini, G.L.; et al. Serum levels of hyaluronic acid and tissue metalloproteinase inhibitor-1 combined with age predict the presence of nonalcoholic steatohepatitis in a pilot cohort of subjects with nonalcoholic fatty liver disease. Transl. Res. J. Lab. Clin. Med. 2009, 154, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Cichoz-Lach, H.; Michalak, A. Oxidative stress as a crucial factor in liver diseases. World J. Gastroenterol. WJG 2014, 20, 8082–8091. [Google Scholar] [CrossRef] [PubMed]
- Grattagliano, I.; Caraceni, P.; Calamita, G.; Ferri, D.; Gargano, I.; Palasciano, G.; Portincasa, P. Severe liver steatosis correlates with nitrosative and oxidative stress in rats. Eur. J. Clin. Investig. 2008, 38, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Luangmonkong, T.; Suriguga, S.; Mutsaers, H.A.M.; Groothuis, G.M.M.; Olinga, P.; Boersema, M. Targeting oxidative stress for the treatment of liver fibrosis. Rev. Physiol. Biochem. Pharmacol. 2018, 175, 71–102. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Tan, H.Y.; Wang, N.; Zhang, Z.J.; Lao, L.; Wong, C.W.; Feng, Y. The role of oxidative stress and antioxidants in liver diseases. Int. J. Mol. Sci. 2015, 16, 26087–26124. [Google Scholar] [CrossRef] [Green Version]
- Gil-Cardoso, K.; Gines, I.; Pinent, M.; Ardevol, A.; Terra, X.; Blay, M. A cafeteria diet triggers intestinal inflammation and oxidative stress in obese rats. Br. J. Nutr. 2017, 117, 218–229. [Google Scholar] [CrossRef] [Green Version]
- Keshavarzian, A.; Farhadi, A.; Forsyth, C.B.; Rangan, J.; Jakate, S.; Shaikh, M.; Banan, A.; Fields, J.Z. Evidence that chronic alcohol exposure promotes intestinal oxidative stress, intestinal hyperpermeability and endotoxemia prior to development of alcoholic steatohepatitis in rats. J. Hepatol. 2009, 50, 538–547. [Google Scholar] [CrossRef] [Green Version]
- Van Ampting, M.T.; Schonewille, A.J.; Vink, C.; Brummer, R.J.; van der Meer, R.; Bovee-Oudenhoven, I.M. Intestinal barrier function in response to abundant or depleted mucosal glutathione in Salmonella-infected rats. BMC Physiol. 2009, 9, 6. [Google Scholar] [CrossRef] [Green Version]
- Novak, E.A.; Mollen, K.P. Mitochondrial dysfunction in inflammatory bowel disease. Front. Cell Dev. Biol. 2015, 3, 62. [Google Scholar] [CrossRef] [Green Version]
- Utzeri, E.; Usai, P. Role of non-steroidal anti-inflammatory drugs on intestinal permeability and nonalcoholic fatty liver disease. World J. Gastroenterol. WJG 2017, 23, 3954–3963. [Google Scholar] [CrossRef]
- Ramachandran, A.; Prabhu, R.; Thomas, S.; Reddy, J.B.; Pulimood, A.; Balasubramanian, K.A. Intestinal mucosal alterations in experimental cirrhosis in the rat: Role of oxygen free radicals. Hepatology 2002, 35, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Kisseleva, T.; Brenner, D.A. The Role of NADPH Oxidases (NOXs) in Liver Fibrosis and the Activation of Myofibroblasts. Front. Physiol. 2016, 7, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieto, N. Oxidative-stress and IL-6 mediate the fibrogenic effects of [corrected] Kupffer cells on stellate cells. Hepatology 2006, 44, 1487–1501. [Google Scholar] [CrossRef] [PubMed]
- Krause, P.; Morris, V.; Greenbaum, J.A.; Park, Y.; Bjoerheden, U.; Mikulski, Z.; Muffley, T.; Shui, J.W.; Kim, G.; Cheroutre, H.; et al. IL-10-producing intestinal macrophages prevent excessive antibacterial innate immunity by limiting IL-23 synthesis. Nat. Commun. 2015, 6, 7055. [Google Scholar] [CrossRef]
- Gomez-Hurtado, I.; Moratalla, A.; Moya-Perez, A.; Peiro, G.; Zapater, P.; Gonzalez-Navajas, J.M.; Gimenez, P.; Such, J.; Sanz, Y.; Frances, R. Role of interleukin 10 in norfloxacin prevention of luminal free endotoxin translocation in mice with cirrhosis. J. Hepatol. 2014, 61, 799–808. [Google Scholar] [CrossRef]
- Thompson, K.; Maltby, J.; Fallowfield, J.; McAulay, M.; Millward-Sadler, H.; Sheron, N. Interleukin-10 expression and function in experimental murine liver inflammation and fibrosis. Hepatology 1998, 28, 1597–1606. [Google Scholar] [CrossRef]
- De Souza-Cruz, S.; Victoria, M.B.; Tarrago, A.M.; da Costa, A.G.; Pimentel, J.P.; Pires, E.F.; Araujo Lde, P.; Coelho-dos-Reis, J.G.; Gomes Mde, S.; Amaral, L.R.; et al. Liver and blood cytokine microenvironment in HCV patients is associated to liver fibrosis score: A proinflammatory cytokine ensemble orchestrated by TNF and tuned by IL-10. BMC Microbiol. 2016, 16, 3. [Google Scholar] [CrossRef] [Green Version]
- Melhem, A.; Muhanna, N.; Bishara, A.; Alvarez, C.E.; Ilan, Y.; Bishara, T.; Horani, A.; Nassar, M.; Friedman, S.L.; Safadi, R. Anti-fibrotic activity of NK cells in experimental liver injury through killing of activated HSC. J. Hepatol. 2006, 45, 60–71. [Google Scholar] [CrossRef]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of activated stellate cells limits liver fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef] [Green Version]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Serino, M.; Luche, E.; Chabo, C.; Amar, J.; Burcelin, R. Intestinal microflora and metabolic diseases. Diabetes Metab. 2009, 35, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Serino, M.; Luche, E.; Gres, S.; Baylac, A.; Berge, M.; Cenac, C.; Waget, A.; Klopp, P.; Iacovoni, J.; Klopp, C.; et al. Metabolic adaptation to a high-fat diet is associated with a change in the gut microbiota. Gut 2012, 61, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Boursier, J.; Mueller, O.; Barret, M.; Machado, M.; Fizanne, L.; Araujo-Perez, F.; Guy, C.D.; Seed, P.C.; Rawls, J.F.; David, L.A.; et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016, 63, 764–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, C.; Rivera, L.; Furness, J.B.; Angus, P.W. The role of the gut microbiota in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Minemura, M.; Shimizu, Y. Gut microbiota and liver diseases. World J. Gastroenterol. WJG 2015, 21, 1691–1702. [Google Scholar] [CrossRef]
- Rahman, K.; Desai, C.; Iyer, S.S.; Thorn, N.E.; Kumar, P.; Liu, Y.; Smith, T.; Neish, A.S.; Li, H.; Tan, S.; et al. Loss of junctional adhesion molecule a promotes severe steatohepatitis in mice on a diet high in saturated fat, fructose, and cholesterol. Gastroenterology 2016, 151, 733–746.e12. [Google Scholar] [CrossRef] [Green Version]
- Pappo, I.; Bercovier, H.; Berry, E.; Gallilly, R.; Feigin, E.; Freund, H.R. Antitumor necrosis factor antibodies reduce hepatic steatosis during total parenteral nutrition and bowel rest in the rat. Jpn. J. Parenter. Enter. Nutr. 1995, 19, 80–82. [Google Scholar] [CrossRef]
- Kirsch, R.; Clarkson, V.; Verdonk, R.C.; Marais, A.D.; Shephard, E.G.; Ryffel, B.; de la M Hall, P. Rodent nutritional model of steatohepatitis: Effects of endotoxin (lipopolysaccharide) and tumor necrosis factor alpha deficiency. J. Gastroenterol. Hepatol. 2006, 21, 174–182. [Google Scholar] [CrossRef]
- Jin, X.; Yu, C.H.; Lv, G.C.; Li, Y.M. Increased intestinal permeability in pathogenesis and progress of nonalcoholic steatohepatitis in rats. World J. Gastroenterol. WJG 2007, 13, 1732–1736. [Google Scholar] [CrossRef]
- Imajo, K.; Fujita, K.; Yoneda, M.; Nozaki, Y.; Ogawa, Y.; Shinohara, Y.; Kato, S.; Mawatari, H.; Shibata, W.; Kitani, H.; et al. Hyperresponsivity to low-dose endotoxin during progression to nonalcoholic steatohepatitis is regulated by leptin-mediated signaling. Cell Metab. 2012, 16, 44–54. [Google Scholar] [CrossRef] [Green Version]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgio, V.; Miele, L.; Principessa, L.; Ferretti, F.; Villa, M.P.; Negro, V.; Grieco, A.; Alisi, A.; Nobili, V. Intestinal permeability is increased in children with non-alcoholic fatty liver disease, and correlates with liver disease severity. Dig. Liver Dis. 2014, 46, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Wigg, A.J.; Roberts-Thomson, I.C.; Dymock, R.B.; McCarthy, P.J.; Grose, R.H.; Cummins, A.G. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut 2001, 48, 206–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasbarrini, A.; Corazza, G.R.; Gasbarrini, G.; Montalto, M.; Di Stefano, M.; Basilisco, G.; Parodi, A.; Usai-Satta, P.; Vernia, P.; Anania, C.; et al. Methodology and indications of H2-breath testing in gastrointestinal diseases: The Rome Consensus Conference. Aliment. Pharmacol. Ther. 2009, 29 (Suppl. 1), 1–49. [Google Scholar] [CrossRef]
- Gasbarrini, A.; Lauritano, E.C.; Gabrielli, M.; Scarpellini, E.; Lupascu, A.; Ojetti, V.; Gasbarrini, G. Small intestinal bacterial overgrowth: Diagnosis and treatment. Dig. Dis. 2007, 25, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Seguritan, V.; Li, W.; Long, T.; Klitgord, N.; Bhatt, A.; Dulai, P.S.; Caussy, C.; Bettencourt, R.; Highlander, S.K.; et al. Gut Microbiome-Based Metagenomic Signature for Non-invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab. 2017, 25, 1054–1062.e5. [Google Scholar] [CrossRef]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef] [PubMed]
- De Wit, N.J.; Afman, L.A.; Mensink, M.; Muller, M. Phenotyping the effect of diet on non-alcoholic fatty liver disease. J. Hepatol. 2012, 57, 1370–1373. [Google Scholar] [CrossRef]
- O’Sullivan, A.; He, X.; McNiven, E.M.; Haggarty, N.W.; Lonnerdal, B.; Slupsky, C.M. Early diet impacts infant rhesus gut microbiome, immunity, and metabolism. J. Proteome Res. 2013, 12, 2833–2845. [Google Scholar] [CrossRef]
- Lelouvier, B.; Servant, F.; Paisse, S.; Brunet, A.C.; Benyahya, S.; Serino, M.; Valle, C.; Ortiz, M.R.; Puig, J.; Courtney, M.; et al. Changes in blood microbiota profiles associated with liver fibrosis in obese patients: A pilot analysis. Hepatology 2016, 64, 2015–2027. [Google Scholar] [CrossRef] [PubMed]
- Amar, J.; Lange, C.; Payros, G.; Garret, C.; Chabo, C.; Lantieri, O.; Courtney, M.; Marre, M.; Charles, M.A.; Balkau, B.; et al. Blood microbiota dysbiosis is associated with the onset of cardiovascular events in a large general population: The D.E.S.I.R. study. PLoS ONE 2013, 8, e54461. [Google Scholar] [CrossRef] [PubMed]
- Amar, J.; Serino, M.; Lange, C.; Chabo, C.; Iacovoni, J.; Mondot, S.; Lepage, P.; Klopp, C.; Mariette, J.; Bouchez, O.; et al. Involvement of tissue bacteria in the onset of diabetes in humans: Evidence for a concept. Diabetologia 2011, 54, 3055–3061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raman, M.; Ahmed, I.; Gillevet, P.M.; Probert, C.S.; Ratcliffe, N.M.; Smith, S.; Greenwood, R.; Sikaroodi, M.; Lam, V.; Crotty, P.; et al. Fecal microbiome and volatile organic compound metabolome in obese humans with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2013, 11, 868–875.e3. [Google Scholar] [CrossRef] [PubMed]
- Caussy, C.; Hsu, C.; Lo, M.T.; Liu, A.; Bettencourt, R.; Ajmera, V.H.; Bassirian, S.; Hooker, J.; Sy, E.; Richards, L.; et al. Link between gut-microbiome derived metabolite and shared gene-effects with hepatic steatosis and fibrosis in NAFLD. Hepatology 2018. [Google Scholar] [CrossRef] [Green Version]
- Di Ciaula, A.; Wang, D.Q.; Molina-Molina, E.; Lunardi Baccetto, R.; Calamita, G.; Palmieri, V.O.; Portincasa, P. Bile acids and cancer: Direct and environmental-dependent effects. Ann. Hepatol. 2017, 16, S87–S105. [Google Scholar] [CrossRef]
- Grattagliano, I.; Diogo, C.V.; Mastrodonato, M.; de Bari, O.; Persichella, M.; Wang, D.Q.; Liquori, A.; Ferri, D.; Carratu, M.R.; Oliveira, P.J.; et al. A silybin-phospholipids complex counteracts rat fatty liver degeneration and mitochondrial oxidative changes. World J. Gastroenterol. WJG 2013, 19, 3007–3017. [Google Scholar] [CrossRef]
- Mastrodonato, M.; Calamita, G.; Rossi, R.; Mentino, D.; Bonfrate, L.; Portincasa, P.; Ferri, D.; Liquori, G.E. Altered distribution of caveolin-1 in early liver steatosis. Eur. J. Clin. Investig. 2011, 41, 642–651. [Google Scholar] [CrossRef]
- Pacelli, C.; Coluccia, A.; Grattagliano, I.; Cocco, T.; Petrosillo, G.; Paradies, G.; De Nitto, E.; Massaro, A.; Persichella, M.; Borracci, P.; et al. Dietary choline deprivation impairs rat brain mitochondrial function and behavioral phenotype. J. Nutr. 2010, 140, 1072–1079. [Google Scholar] [CrossRef] [Green Version]
- Petrosillo, G.; Portincasa, P.; Grattagliano, I.; Casanova, G.; Matera, M.; Ruggiero, F.M.; Ferri, D.; Paradies, G. Mitochondrial dysfunction in rat with nonalcoholic fatty liver Involvement of complex I, reactive oxygen species and cardiolipin. Biochim. Biophys. Acta 2007, 1767, 1260–1267. [Google Scholar] [CrossRef] [Green Version]
- Ponziani, F.R.; Bhoori, S.; Castelli, C.; Putignani, L.; Rivoltini, L.; Del Chierico, F.; Sanguinetti, M.; Morelli, D.; Sterbini, F.P.; Petito, V.; et al. Hepatocellular carcinoma is associated with gut microbiota profile and inflammation in nonalcoholic fatty liver disease. Hepatology 2019, 69, 107–120. [Google Scholar] [CrossRef]
- Philips, C.A.; Pande, A.; Shasthry, S.M.; Jamwal, K.D.; Khillan, V.; Chandel, S.S.; Kumar, G.; Sharma, M.K.; Maiwall, R.; Jindal, A. A pilot study. Clin. Gastroenterol. Hepatol. 2017, 15, 600–602. [Google Scholar] [CrossRef] [PubMed]
- Luther, J.; Garber, J.J.; Khalili, H.; Dave, M.; Bale, S.S.; Jindal, R.; Motola, D.L.; Luther, S.; Bohr, S.; Jeoung, S.W.; et al. Hepatic injury in nonalcoholic steatohepatitis contributes to altered intestinal permeability. Cell Mol. Gastroenterol. Hepatol. 2015, 1, 222–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazim, M.; Stamp, G.; Hodgson, H.J. Non-alcoholic steatohepatitis associated with small intestinal diverticulosis and bacterial overgrowth. Hepatogastroenterology 1989, 36, 349–351. [Google Scholar]
- Lichtman, S.N.; Sartor, R.B.; Keku, J.; Schwab, J.H. Hepatic inflammation in rats with experimental small intestinal bacterial overgrowth. Gastroenterology 1990, 98, 414–423. [Google Scholar] [CrossRef]
- Lichtman, S.N.; Keku, J.; Schwab, J.H.; Sartor, R.B. Hepatic injury associated with small bowel bacterial overgrowth in rats is prevented by metronidazole and tetracycline. Gastroenterology 1991, 100, 513–519. [Google Scholar] [CrossRef]
- Kapil, S.; Duseja, A.; Sharma, B.K.; Singla, B.; Chakraborti, A.; Das, A.; Ray, P.; Dhiman, R.K.; Chawla, Y. Small intestinal bacterial overgrowth and toll-like receptor signaling in patients with non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2016, 31, 213–221. [Google Scholar] [CrossRef]
- Diehl, A.M.; Li, Z.P.; Lin, H.Z.; Yang, S.Q. Cytokines and the pathogenesis of non-alcoholic steatohepatitis. Gut 2005, 54, 303–306. [Google Scholar] [CrossRef] [Green Version]
- Vetrano, S.; Rescigno, M.; Cera, M.R.; Correale, C.; Rumio, C.; Doni, A.; Fantini, M.; Sturm, A.; Borroni, E.; Repici, A.; et al. Unique role of junctional adhesion molecule-a in maintaining mucosal homeostasis in inflammatory bowel disease. Gastroenterology 2008, 135, 173–184. [Google Scholar] [CrossRef] [Green Version]
- Monteiro, A.C.; Sumagin, R.; Rankin, C.R.; Leoni, G.; Mina, M.J.; Reiter, D.M.; Stehle, T.; Dermody, T.S.; Schaefer, S.A.; Hall, R.A.; et al. JAM-A associates with ZO-2, afadin, and PDZ-GEF1 to activate Rap2c and regulate epithelial barrier function. Mol. Biol. Cell 2013, 24, 2849–2860. [Google Scholar] [CrossRef]
- Menard, S.; Cerf-Bensussan, N.; Heyman, M. Multiple facets of intestinal permeability and epithelial handling of dietary antigens. Mucosal. Immunol. 2010, 3, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Laukoetter, M.G.; Nava, P.; Lee, W.Y.; Severson, E.A.; Capaldo, C.T.; Babbin, B.A.; Williams, I.R.; Koval, M.; Peatman, E.; Campbell, J.A.; et al. JAM-A regulates permeability and inflammation in the intestine in vivo. J. Exp. Med. 2007, 204, 3067–3076. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Tan, J.; Qian, W.; Zhang, L.; Hou, X. Gut inflammation exacerbates hepatic injury in the high-fat diet induced NAFLD mouse: Attention to the gut-vascular barrier dysfunction. Life Sci. 2018, 209, 157–166. [Google Scholar] [CrossRef] [PubMed]
- DeMeo, M.T.; Mutlu, E.A.; Keshavarzian, A.; Tobin, M.C. Intestinal permeation and gastrointestinal disease. J. Clin. Gastroenterol. 2002, 34, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Arslan, G.; Atasever, T.; Cindoruk, M.; Yildirim, I.S. (51)CrEDTA colonic permeability and therapy response in patients with ulcerative colitis. Nucl. Med. Commun. 2001, 22, 997–1001. [Google Scholar] [CrossRef]
- Ponziani, F.R.; Zocco, M.A.; Cerrito, L.; Gasbarrini, A.; Pompili, M. Bacterial translocation in patients with liver cirrhosis: Physiology, clinical consequences, and practical implications. Expert Rev. Gastroenterol. Hepatol. 2018, 12, 641–656. [Google Scholar] [CrossRef]
- Gabele, E.; Muhlbauer, M.; Dorn, C.; Weiss, T.S.; Froh, M.; Schnabl, B.; Wiest, R.; Scholmerich, J.; Obermeier, F.; Hellerbrand, C. Role of TLR9 in hepatic stellate cells and experimental liver fibrosis. Biochem. Biophys. Res. Commun. 2008, 376, 271–276. [Google Scholar] [CrossRef]
- Lebeaupin, C.; Proics, E.; de Bieville, C.H.; Rousseau, D.; Bonnafous, S.; Patouraux, S.; Adam, G.; Lavallard, V.J.; Rovere, C.; Le Thuc, O.; et al. ER stress induces NLRP3 inflammasome activation and hepatocyte death. Cell Death Dis. 2015, 6, e1879. [Google Scholar] [CrossRef] [Green Version]
- Miura, K.; Kodama, Y.; Inokuchi, S.; Schnabl, B.; Aoyama, T.; Ohnishi, H.; Olefsky, J.M.; Brenner, D.A.; Seki, E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology 2010, 139, 323–334.e7. [Google Scholar] [CrossRef] [Green Version]
- Saberi, M.; Woods, N.B.; de Luca, C.; Schenk, S.; Lu, J.C.; Bandyopadhyay, G.; Verma, I.M.; Olefsky, J.M. Hematopoietic cell-specific deletion of toll-like receptor 4 ameliorates hepatic and adipose tissue insulin resistance in high-fat-fed mice. Cell Metab. 2009, 10, 419–429. [Google Scholar] [CrossRef] [Green Version]
- Rivera, C.A.; Adegboyega, P.; van Rooijen, N.; Tagalicud, A.; Allman, M.; Wallace, M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J. Hepatol. 2007, 47, 571–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levitt, M.D.; Li, R.; Demaster, E.G.; Elson, M.; Furne, J.; Levitt, D.G. Use of measurements of ethanol absorption from stomach and intestine to assess human ethanol metabolism. Am. J. Physiol. Gastrointest. Liver Physiol. 1997, 273, G951–G957. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Miyamoto, Y.; Mazagova, M.; Lee, K.C.; Eckmann, L.; Schnabl, B. Microbiota Protects Mice Against Acute Alcohol-Induced Liver Injury. Alcohol. Clin. Exp. Res. 2015, 39, 2313–2323. [Google Scholar] [CrossRef] [Green Version]
- Ansari, R.A.; Husain, K.; Rizvi, S.A. Role of Transcription Factors in Steatohepatitis and Hypertension after Ethanol: The Epicenter of Metabolism. Biomolecules 2016, 6, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamarneh, S.R.; Kim, B.M.; Kaliannan, K.; Morrison, S.A.; Tantillo, T.J.; Tao, Q.; Mohamed, M.M.R.; Ramirez, J.M.; Karas, A.; Liu, W.; et al. Intestinal alkaline phosphatase attenuates alcohol-induced hepatosteatosis in mice. Dig. Dis. Sci. 2017, 62, 2021–2034. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Jeong, D.; Kang, I.B.; Kim, H.; Song, K.Y.; Seo, K.H. Dual function of Lactobacillus kefiri DH5 in preventing high-fat-diet-induced obesity: Direct reduction of cholesterol and upregulation of PPAR-alpha in adipose tissue. Mol. Nutr. Food Res. 2017, 61, 1700252. [Google Scholar] [CrossRef]
- Elamin, E.; Jonkers, D.; Juuti-Uusitalo, K.; van Ijzendoorn, S.; Troost, F.; Duimel, H.; Broers, J.; Verheyen, F.; Dekker, J.; Masclee, A. Effects of ethanol and acetaldehyde on tight junction integrity: In vitro study in a three dimensional intestinal epithelial cell culture model. PLoS ONE 2012, 7, e35008. [Google Scholar] [CrossRef] [Green Version]
- Samak, G.; Aggarwal, S.; Rao, R.K. ERK is involved in EGF-mediated protection of tight junctions, but not adherens junctions, in acetaldehyde-treated Caco-2 cell monolayers. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G50–G59. [Google Scholar] [CrossRef] [Green Version]
- Basuroy, S.; Sheth, P.; Mansbach, C.M.; Rao, R.K. Acetaldehyde disrupts tight junctions and adherens junctions in human colonic mucosa: Protection by EGF and L-glutamine. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G367–G375. [Google Scholar] [CrossRef]
- Yan, A.W.; Fouts, D.E.; Brandl, J.; Starkel, P.; Torralba, M.; Schott, E.; Tsukamoto, H.; Nelson, K.E.; Brenner, D.A.; Schnabl, B. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology 2011, 53, 96–105. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, P.; Chen, P.; Wang, H.J.; Wang, L.; McCole, D.F.; Brandl, K.; Starkel, P.; Belzer, C.; Hellerbrand, C.; Tsukamoto, H.; et al. Deficiency of intestinal mucin-2 ameliorates experimental alcoholic liver disease in mice. Hepatology 2013, 58, 108–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, B.; Lee, H.R.; Lee, Y.J. Alcoholic liver disease: Focus on prodromal gut health. J. Dig. Dis. 2016, 17, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lafdil, F.; Kong, X.; Gao, B. Signal transducer and activator of transcription 3 in liver diseases: A novel therapeutic target. Int. J. Biol. Sci. 2011, 7, 536–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mottaran, E.; Stewart, S.F.; Rolla, R.; Vay, D.; Cipriani, V.; Moretti, M.; Vidali, M.; Sartori, M.; Rigamonti, C.; Day, C.P.; et al. Lipid peroxidation contributes to immune reactions associated with alcoholic liver disease. Free Radic. Biol. Med. 2002, 32, 38–45. [Google Scholar] [CrossRef]
- Chen, P.; Torralba, M.; Tan, J.; Embree, M.; Zengler, K.; Starkel, P.; van Pijkeren, J.P.; DePew, J.; Loomba, R.; Ho, S.B.; et al. Supplementation of saturated long-chain fatty acids maintains intestinal eubiosis and reduces ethanol-induced liver injury in mice. Gastroenterology 2015, 148, 203–214.e16. [Google Scholar] [CrossRef] [Green Version]
- Xie, G.; Zhong, W.; Zheng, X.; Li, Q.; Qiu, Y.; Li, H.; Chen, H.; Zhou, Z.; Jia, W. Chronic ethanol consumption alters mammalian gastrointestinal content metabolites. J. Proteome Res. 2013, 12, 3297–3306. [Google Scholar] [CrossRef] [Green Version]
- Couch, R.D.; Dailey, A.; Zaidi, F.; Navarro, K.; Forsyth, C.B.; Mutlu, E.; Engen, P.A.; Keshavarzian, A. Alcohol induced alterations to the human fecal VOC metabolome. PLoS ONE 2015, 10, e0119362. [Google Scholar] [CrossRef] [Green Version]
- Cresci, G.A.; Glueck, B.; McMullen, M.R.; Xin, W.; Allende, D.; Nagy, L.E. Prophylactic tributyrin treatment mitigates chronic-binge ethanol-induced intestinal barrier and liver injury. J. Gastroenterol. Hepatol. 2017, 32, 1587–1597. [Google Scholar] [CrossRef]
- Leclercq, S.; Matamoros, S.; Cani, P.D.; Neyrinck, A.M.; Jamar, F.; Starkel, P.; Windey, K.; Tremaroli, V.; Backhed, F.; Verbeke, K.; et al. Intestinal permeability, gut-bacterial dysbiosis, and behavioral markers of alcohol-dependence severity. Proc. Natl. Acad. Sci. USA 2014, 111, E4485–E4493. [Google Scholar] [CrossRef] [Green Version]
- Arroyo, V.; Moreau, R.; Kamath, P.S.; Jalan, R.; Ginès, P.; Nevens, F.; Fernández, J.; To, U.; García-Tsao, G.; Schnabl, B. Acute-on-chronic liver failure in cirrhosis. Nat. Rev. Dis. Primers 2016, 2, 16041. [Google Scholar] [CrossRef] [Green Version]
- Cresci, G.A.; Bush, K.; Nagy, L.E. Tributyrin supplementation protects mice from acute ethanol-induced gut injury. Alcohol. Clin. Exp. Res. 2014, 38, 1489–1501. [Google Scholar] [CrossRef] [PubMed]
- Mezey, E.; Imbembo, A.L.; Potter, J.J.; Rent, K.C.; Lombardo, R.; Holt, P.R. Endogenous ethanol production and hepatic disease following jejunoileal bypass for morbid obesity. Am. J. Clin. Nutr. 1975, 28, 1277–1283. [Google Scholar] [CrossRef] [PubMed]
- Baraona, E.; Julkunen, R.; Tannenbaum, L.; Lieber, C.S. Role of intestinal bacterial overgrowth in ethanol production and metabolism in rats. Gastroenterology 1986, 90, 103–110. [Google Scholar] [CrossRef]
- Kaji, H.; Asanuma, Y.; Yahara, O.; Shibue, H.; Hisamura, M.; Saito, N.; Kawakami, Y.; Murao, M. Intragastrointestinal alcohol fermentation syndrome: Report of two cases and review of the literature. J. Forensic Sci. Soc. 1984, 24, 461–471. [Google Scholar] [CrossRef]
- Nair, S.; Cope, K.; Risby, T.H.; Diehl, A.M. Obesity and female gender increase breath ethanol concentration: Potential implications for the pathogenesis of nonalcoholic steatohepatitis. Am. J. Gastroenterol. 2001, 96, 1200–1204. [Google Scholar] [CrossRef]
- Cope, K.; Risby, T.; Diehl, A.M. Increased gastrointestinal ethanol production in obese mice: Implications for fatty liver disease pathogenesis. Gastroenterology 2000, 119, 1340–1347. [Google Scholar] [CrossRef]
- Salaspuro, M. Bacteriocolonic pathway for ethanol oxidation: Characteristics and implications. Ann. Med. 1996, 28, 195–200. [Google Scholar] [CrossRef]
- Dawes, E.A.; Foster, S.M. The formation of ethanol in Escherichia coli. Biochim. Biophys. Acta 1956, 22, 253–265. [Google Scholar] [CrossRef]
- Engstler, A.J.; Aumiller, T.; Degen, C.; Durr, M.; Weiss, E.; Maier, I.B.; Schattenberg, J.M.; Jin, C.J.; Sellmann, C.; Bergheim, I. Insulin resistance alters hepatic ethanol metabolism: Studies in mice and children with non-alcoholic fatty liver disease. Gut 2016, 65, 1564–1571. [Google Scholar] [CrossRef]
- Christopherson, M.R.; Dawson, J.A.; Stevenson, D.M.; Cunningham, A.C.; Bramhacharya, S.; Weimer, P.J.; Kendziorski, C.; Suen, G. Unique aspects of fiber degradation by the ruminal ethanologen Ruminococcus albus 7 revealed by physiological and transcriptomic analysis. BMC Genom. 2014, 15, 1066. [Google Scholar] [CrossRef] [Green Version]
- Setshedi, M.; Wands, J.R.; Monte, S.M. Acetaldehyde adducts in alcoholic liver disease. Oxidative Med. Cell. Longev. 2010, 3, 178–185. [Google Scholar] [CrossRef] [Green Version]
- Ni, Y.H.; Huo, L.J.; Li, T.T. Effect of interleukin-22 on proliferation and activation of hepatic stellate cells induced by acetaldehyde and related mechanism. Chin. J. Hepatol. 2017, 25, 9–14. [Google Scholar] [CrossRef]
- Wu, X.; Wang, Y.; Wang, S.; Xu, R.; Lv, X. Purinergic P2X7 receptor mediates acetaldehyde-induced hepatic stellate cells activation via PKC-dependent GSK3beta pathway. Int. Immunopharmacol. 2017, 43, 164–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Lázaro, M. A local mechanism by which alcohol consumption causes cancer. Oral Oncol. 2016, 62, 149–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, S.S.; Baker, R.D.; Liu, W.; Nowak, N.J.; Zhu, L. Role of alcohol metabolism in non-alcoholic steatohepatitis. PLoS ONE 2010, 5, e9570. [Google Scholar] [CrossRef] [Green Version]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef] [Green Version]
- Sayin, S.I.; Wahlstrom, A.; Felin, J.; Jantti, S.; Marschall, H.U.; Bamberg, K.; Angelin, B.; Hyotylainen, T.; Oresic, M.; Backhed, F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013, 17, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Yokota, A.; Fukiya, S.; Islam, K.B.; Ooka, T.; Ogura, Y.; Hayashi, T.; Hagio, M.; Ishizuka, S. Is bile acid a determinant of the gut microbiota on a high-fat diet? Gut Microbes 2012, 3, 455–459. [Google Scholar] [CrossRef] [Green Version]
- Inagaki, T.; Moschetta, A.; Lee, Y.K.; Peng, L.; Zhao, G.; Downes, M.; Yu, R.T.; Shelton, J.M.; Richardson, J.A.; Repa, J.J.; et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 3920–3925. [Google Scholar] [CrossRef] [Green Version]
- Parseus, A.; Sommer, N.; Sommer, F.; Caesar, R.; Molinaro, A.; Stahlman, M.; Greiner, T.U.; Perkins, R.; Backhed, F. Microbiota-induced obesity requires farnesoid X receptor. Gut 2017, 66, 429–437. [Google Scholar] [CrossRef] [Green Version]
- Gadaleta, R.M.; van Erpecum, K.J.; Oldenburg, B.; Willemsen, E.C.; Renooij, W.; Murzilli, S.; Klomp, L.W.; Siersema, P.D.; Schipper, M.E.; Danese, S.; et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut 2011, 60, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Jiang, C.; Krausz, K.W.; Li, Y.; Albert, I.; Hao, H.; Fabre, K.M.; Mitchell, J.B.; Patterson, A.D.; Gonzalez, F.J. Microbiome remodelling leads to inhibition of intestinal farnesoid X receptor signalling and decreased obesity. Nat. Commun. 2013, 4, 2384. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Xu, M.; Dong, W.; Deng, B.; Wang, S.; Zhang, Y.; Wang, S.; Luo, S.; Wang, W.; Qi, Y.; et al. Secondary bile acid-induced dysbiosis promotes intestinal carcinogenesis. Int. J. Cancer 2017, 140, 2545–2556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, C.; Xie, C.; Li, F.; Zhang, L.; Nichols, R.G.; Krausz, K.W.; Cai, J.; Qi, Y.; Fang, Z.Z.; Takahashi, S.; et al. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J. Clin. Investig. 2015, 125, 386–402. [Google Scholar] [CrossRef] [PubMed]
- Jiao, N.; Baker, S.S.; Chapa-Rodriguez, A.; Liu, W.; Nugent, C.A.; Tsompana, M.; Mastrandrea, L.; Buck, M.J.; Baker, R.D.; Genco, R.J.; et al. Suppressed hepatic bile acid signalling despite elevated production of primary and secondary bile acids in NAFLD. Gut 2018, 67, 1881–1891. [Google Scholar] [CrossRef] [PubMed]
- Ferslew, B.C.; Xie, G.; Johnston, C.K.; Su, M.; Stewart, P.W.; Jia, W.; Brouwer, K.L.; Barritt, A.S.T. Altered bile acid metabolome in patients with nonalcoholic steatohepatitis. Dig. Dis. Sci. 2015, 60, 3318–3328. [Google Scholar] [CrossRef] [Green Version]
- Mouzaki, M.; Wang, A.Y.; Bandsma, R.; Comelli, E.M.; Arendt, B.M.; Zhang, L.; Fung, S.; Fischer, S.E.; McGilvray, I.G.; Allard, J.P. Bile acids and dysbiosis in non-alcoholic fatty liver disease. PLoS ONE 2016, 11, e0151829. [Google Scholar] [CrossRef] [Green Version]
- Duncan, S.H.; Louis, P.; Thomson, J.M.; Flint, H.J. The role of pH in determining the species composition of the human colonic microbiota. Environ. Microbiol. 2009, 11, 2112–2122. [Google Scholar] [CrossRef]
- Sawicki, C.M.; Livingston, K.A.; Obin, M.; Roberts, S.B.; Chung, M.; McKeown, N.M. Dietary Fiber and the Human Gut Microbiota: Application of Evidence Mapping Methodology. Nutrients 2017, 9, 125. [Google Scholar] [CrossRef] [Green Version]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From dietary fiber to host physiology: Short-chain fatty acids as key bacterial metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [Green Version]
- Brussow, H.; Parkinson, S.J. You are what you eat. Nat. Biotechnol. 2014, 32, 243–245. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Goodspeed, L.; Wang, S.; Kim, J.; Zeng, L.; Ioannou, G.N.; Haigh, W.G.; Yeh, M.M.; Kowdley, K.V.; O’Brien, K.D.; et al. Dietary cholesterol exacerbates hepatic steatosis and inflammation in obese LDL receptor-deficient mice. J. Lipid Res. 2011, 52, 1626–1635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musso, G.; Gambino, R.; Cassader, M. Obesity, diabetes, and gut microbiota: The hygiene hypothesis expanded? Diabetes Care 2010, 33, 2277–2284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svegliati-Baroni, G.; Saccomanno, S.; Rychlicki, C.; Agostinelli, L.; De Minicis, S.; Candelaresi, C.; Faraci, G.; Pacetti, D.; Vivarelli, M.; Nicolini, D.; et al. Glucagon-like peptide-1 receptor activation stimulates hepatic lipid oxidation and restores hepatic signalling alteration induced by a high-fat diet in nonalcoholic steatohepatitis. Liver Int. Off. J. Int. Assoc. Study Liver 2011, 31, 1285–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, F.; Ding, X.; Wu, G.; Lam, Y.Y.; Wang, X.; Fu, H.; Xue, X.; Lu, C.; Ma, J.; et al. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science 2018, 359, 1151–1156. [Google Scholar] [CrossRef] [Green Version]
- Backhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [Green Version]
- Alex, S.; Lange, K.; Amolo, T.; Grinstead, J.S.; Haakonsson, A.K.; Szalowska, E.; Koppen, A.; Mudde, K.; Haenen, D.; Al-Lahham, S.; et al. Short-chain fatty acids stimulate angiopoietin-like 4 synthesis in human colon adenocarcinoma cells by activating peroxisome proliferator-activated receptor gamma. Mol. Cell. Biol. 2013, 33, 1303–1316. [Google Scholar] [CrossRef] [Green Version]
- Mehedint, M.G.; Zeisel, S.H. Choline’s role in maintaining liver function: New evidence for epigenetic mechanisms. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 339–345. [Google Scholar] [CrossRef] [Green Version]
- Zeisel, S.H.; da Costa, K.A. Choline: An essential nutrient for public health. Nutr. Rev. 2009, 67, 615–623. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grattagliano, I.; Caraceni, P.; Portincasa, P.; Domenicali, M.; Palmieri, V.O.; Trevisani, F.; Bernardi, M.; Palasciano, G. Adaptation of subcellular glutathione detoxification system to stress conditions in choline-deficient diet induced rat fatty liver. Cell Biol. Toxicol. 2003, 19, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Dumas, M.E.; Barton, R.H.; Toye, A.; Cloarec, O.; Blancher, C.; Rothwell, A.; Fearnside, J.; Tatoud, R.; Blanc, V.; Lindon, J.C.; et al. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc. Natl. Acad. Sci. USA 2006, 103, 12511–12516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, M.D.; Hamp, T.J.; Reid, R.W.; Fischer, L.M.; Zeisel, S.H.; Fodor, A.A. Association between composition of the human gastrointestinal microbiome and development of fatty liver with choline deficiency. Gastroenterology 2011, 140, 976–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velasquez, M.T.; Ramezani, A.; Manal, A.; Raj, D.S. Trimethylamine N-Oxide: The good, the bad and the unknown. Toxins 2016, 8, 326. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.M.; Liu, Y.; Zhou, R.F.; Chen, X.L.; Wang, C.; Tan, X.Y.; Wang, L.J.; Zheng, R.D.; Zhang, H.W.; Ling, W.H.; et al. Associations of gut-flora-dependent metabolite trimethylamine-N-oxide, betaine and choline with non-alcoholic fatty liver disease in adults. Sci. Rep. 2016, 6, 19076. [Google Scholar] [CrossRef]
- Hoyles, L.; Fernández-Real, J.-M.; Federici, M.; Serino, M.; Abbott, J.; Charpentier, J.; Heymes, C.; Luque, J.L.; Anthony, E.; Barton, R.H. Molecular phenomics and metagenomics of hepatic steatosis in non-diabetic obese women. Nat. Med. 2018, 24, 1070. [Google Scholar] [CrossRef]
- Machado, M.V.; Cortez-Pinto, H. Diet, Microbiota, obesity, and NAFLD: A dangerous quartet. Int. J. Mol. Sci. 2016, 17, 481. [Google Scholar] [CrossRef] [Green Version]
- Biedermann, L.; Zeitz, J.; Mwinyi, J.; Sutter-Minder, E.; Rehman, A.; Ott, S.J.; Steurer-Stey, C.; Frei, A.; Frei, P.; Scharl, M.; et al. Smoking cessation induces profound changes in the composition of the intestinal microbiota in humans. PLoS ONE 2013, 8, e59260. [Google Scholar] [CrossRef]
- Mutlu, E.A.; Gillevet, P.M.; Rangwala, H.; Sikaroodi, M.; Naqvi, A.; Engen, P.A.; Kwasny, M.; Lau, C.K.; Keshavarzian, A. Colonic microbiome is altered in alcoholism. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G966–G978. [Google Scholar] [CrossRef]
- Clarke, S.F.; Murphy, E.F.; Nilaweera, K.; Ross, P.R.; Shanahan, F.; O’Toole, P.W.; Cotter, P.D. The gut microbiota and its relationship to diet and obesity: New insights. Gut Microbes 2012, 3, 186–202. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojarvi, J.; Kootte, R.S.; Bartelsman, J.F.; Dallinga-Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012, 143, 913–916.e917. [Google Scholar] [CrossRef]
- Moreira, G.V.; Azevedo, F.F.; Ribeiro, L.M.; Santos, A.; Guadagnini, D.; Gama, P.; Liberti, E.A.; Saad, M.; Carvalho, C. Liraglutide modulates gut microbiota and reduces NAFLD in obese mice. J. Nutr. Biochem. 2018, 62, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Brandt, A.; Hernandez-Arriaga, A.; Kehm, R.; Sanchez, V.; Jin, C.J.; Nier, A.; Baumann, A.; Camarinha-Silva, A.; Bergheim, I. Metformin attenuates the onset of non-alcoholic fatty liver disease and affects intestinal microbiota and barrier in small intestine. Sci. Rep. 2019, 9, 6668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, W.; Wang, H.; Zhang, P.; Gao, C.; Tao, J.; Ge, Z.; Zhu, D.; Bi, Y. Modulation of gut microbiota contributes to curcumin-mediated attenuation of hepatic steatosis in rats. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1801–1812. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Chi, L.; Mahbub, R.; Bian, X.; Tu, P.; Ru, H.; Lu, K. Multi-Omics Reveals that Lead Exposure Disturbs Gut Microbiome Development, Key Metabolites, and Metabolic Pathways. Chem. Res. Toxicol. 2017, 30, 996–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, B.; Bian, X.; Mahbub, R.; Lu, K. Sex-Specific effects of organophosphate diazinon on the gut microbiome and its metabolic functions. Environ. Health Perspect. 2017, 125, 198–206. [Google Scholar] [CrossRef] [Green Version]
- Joly, C.; Gay-Queheillard, J.; Leke, A.; Chardon, K.; Delanaud, S.; Bach, V.; Khorsi-Cauet, H. Impact of chronic exposure to low doses of chlorpyrifos on the intestinal microbiota in the Simulator of the Human Intestinal Microbial Ecosystem (SHIME) and in the rat. Environ. Sci. Pollut. Res. Int. 2013, 20, 2726–2734. [Google Scholar] [CrossRef]
- Joly Condette, C.; Bach, V.; Mayeur, C.; Gay-Queheillard, J.; Khorsi-Cauet, H. Chlorpyrifos exposure during perinatal period affects intestinal microbiota associated with delay of maturation of digestive tract in rats. J. Pediatr. Gastroenterol. Nutr. 2015, 61, 30–40. [Google Scholar] [CrossRef]
- Hooper, L.V.; Littman, D.R.; Macpherson, A.J. Interactions between the microbiota and the immune system. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaiss, C.A.; Zmora, N.; Levy, M.; Elinav, E. The microbiome and innate immunity. Nature 2016, 535, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Britanova, L.; Diefenbach, A. Interplay of innate lymphoid cells and the microbiota. Immunol. Rev. 2017, 279, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, S.C.; Barbara, G.; Buurman, W.; Ockhuizen, T.; Schulzke, J.D.; Serino, M.; Tilg, H.; Watson, A.; Wells, J.M. Intestinal permeability—A new target for disease prevention and therapy. BMC Gastroenterol. 2014, 14, 189. [Google Scholar] [CrossRef] [Green Version]
- Kirpich, I.A.; Marsano, L.S.; McClain, C.J. Gut-liver axis, nutrition, and non-alcoholic fatty liver disease. Clin. Biochem. 2015, 48, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, B.O.; Birchenough, G.M.H.; Stahlman, M.; Arike, L.; Johansson, M.E.V.; Hansson, G.C.; Backhed, F. Bifidobacteria or fiber protects against diet-induced microbiota-mediated colonic mucus deterioration. Cell Host Microbe 2018, 23, 27–40.e7. [Google Scholar] [CrossRef] [Green Version]
- Luck, H.; Tsai, S.; Chung, J.; Clemente-Casares, X.; Ghazarian, M.; Revelo, X.S.; Lei, H.; Luk, C.T.; Shi, S.Y.; Surendra, A.; et al. Regulation of obesity-related insulin resistance with gut anti-inflammatory agents. Cell Metab. 2015, 21, 527–542. [Google Scholar] [CrossRef] [Green Version]
- Spruss, A.; Kanuri, G.; Wagnerberger, S.; Haub, S.; Bischoff, S.C.; Bergheim, I. Toll-like receptor 4 is involved in the development of fructose-induced hepatic steatosis in mice. Hepatology 2009, 50, 1094–1104. [Google Scholar] [CrossRef]
- Lambertz, J.; Weiskirchen, S.; Landert, S.; Weiskirchen, R. Fructose: A dietary sugar in crosstalk with microbiota contributing to the development and progression of non-alcoholic liver disease. Front. Immunol. 2017, 8, 1159. [Google Scholar] [CrossRef] [Green Version]
- Ray, K. NAFLD. Leaky guts: Intestinal permeability and NASH. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 123. [Google Scholar] [CrossRef]
- Miele, L.; Marrone, G.; Lauritano, C.; Cefalo, C.; Gasbarrini, A.; Day, C.; Grieco, A. Gut-liver axis and microbiota in NAFLD: Insight pathophysiology for novel therapeutic target. Curr. Pharm. Des. 2013, 19, 5314–5324. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Possemiers, S.; Van de Wiele, T.; Guiot, Y.; Everard, A.; Rottier, O.; Geurts, L.; Naslain, D.; Neyrinck, A.; Lambert, D.M.; et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 2009, 58, 1091–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, S.; Chi, M.M.; Scull, B.P.; Rigby, R.; Schwerbrock, N.M.; Magness, S.; Jobin, C.; Lund, P.K. High-fat diet: Bacteria interactions promote intestinal inflammation which precedes and correlates with obesity and insulin resistance in mouse. PLoS ONE 2010, 5, e12191. [Google Scholar] [CrossRef] [Green Version]
- Kavanagh, K.; Wylie, A.T.; Tucker, K.L.; Hamp, T.J.; Gharaibeh, R.Z.; Fodor, A.A.; Cullen, J.M. Dietary fructose induces endotoxemia and hepatic injury in calorically controlled primates. Am. J. Clin. Nutr. 2013, 98, 349–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bluemel, S.; Wang, L.; Martino, C.; Lee, S.; Wang, Y.; Williams, B.; Horvath, A.; Stadlbauer, V.; Zengler, K.; Schnabl, B. The role of intestinal c-type regenerating islet derived-3 lectins for nonalcoholic steatohepatitis. Hepatol. Commun. 2018, 2, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Willment, A.; Patel, S.S.; Sun, X.; Song, M.; Mannery, Y.O.; Kosters, A.; McClain, C.J.; Vos, M.B. Fructose induced endotoxemia in pediatric nonalcoholic Fatty liver disease. Int. J. Hepatol. 2014, 2014, 560620. [Google Scholar] [CrossRef]
- Bifulco, M. Mediterranean diet: The missing link between gut microbiota and inflammatory diseases. Eur. J. Clin. Nutr. 2015, 69, 1078. [Google Scholar] [CrossRef] [Green Version]
- Biolato, M.; Manca, F.; Marrone, G.; Cefalo, C.; Racco, S.; Miggiano, G.A.; Valenza, V.; Gasbarrini, A.; Miele, L.; Grieco, A. Intestinal permeability after Mediterranean diet and low-fat diet in non-alcoholic fatty liver disease. World J. Gastroenterol. WJG 2019, 25, 509–520. [Google Scholar] [CrossRef]
- Enomoto, N.; Yamashina, S.; Kono, H.; Schemmer, P.; Rivera, C.A.; Enomoto, A.; Nishiura, T.; Nishimura, T.; Brenner, D.A.; Thurman, R.G. Development of a new, simple rat model of early alcohol-induced liver injury based on sensitization of Kupffer cells. Hepatology 1999, 29, 1680–1689. [Google Scholar] [CrossRef]
- Pappo, I.; Bercovier, H.; Berry, E.M.; Haviv, Y.; Gallily, R.; Freund, H.R. Polymyxin B reduces total parenteral nutrition-associated hepatic steatosis by its antibacterial activity and by blocking deleterious effects of lipopolysaccharide. Jpn. J. Parenter. Enter. Nutr. 1992, 16, 529–532. [Google Scholar] [CrossRef]
- Pappo, I.; Becovier, H.; Berry, E.M.; Freund, H.R. Polymyxin B reduces cecal flora, TNF production and hepatic steatosis during total parenteral nutrition in the rat. J. Surg. Res. 1991, 51, 106–112. [Google Scholar] [CrossRef]
- Drenick, E.J.; Fisler, J.; Johnson, D. Hepatic steatosis after intestinal bypass—Prevention and reversal by metronidazole, irrespective of protein-calorie malnutrition. Gastroenterology 1982, 82, 535–548. [Google Scholar] [CrossRef]
- Alisi, A.; Bedogni, G.; Baviera, G.; Giorgio, V.; Porro, E.; Paris, C.; Giammaria, P.; Reali, L.; Anania, F.; Nobili, V. Randomised clinical trial: The beneficial effects of VSL#3 in obese children with non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2014, 39, 1276–1285. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yang, S.; Lin, H.; Huang, J.; Watkins, P.A.; Moser, A.B.; Desimone, C.; Song, X.Y.; Diehl, A.M. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology 2003, 37, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Dallio, M.; Masarone, M.; Errico, S.; Gravina, A.G.; Nicolucci, C.; Di Sarno, R.; Gionti, L.; Tuccillo, C.; Persico, M.; Stiuso, P.; et al. Role of bisphenol A as environmental factor in the promotion of non-alcoholic fatty liver disease: In vitro and clinical study. Aliment. Pharmacol. Ther. 2018, 47, 826–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, D.; Zhang, H.; Jiang, X.; Zou, J.; Li, Q.; Mai, H.; Su, D.; Ling, W.; Feng, X. Bisphenol A exposure induces gut microbiota dysbiosis and consequent activation of gut-liver axis leading to hepatic steatosis in CD-1 mice. Environ. Pollut. 2020, 265, 114880. [Google Scholar] [CrossRef]
- Wang, D.; Yan, J.; Teng, M.; Yan, S.; Zhou, Z.; Zhu, W. In utero and lactational exposure to BDE-47 promotes obesity development in mouse offspring fed a high-fat diet: Impaired lipid metabolism and intestinal dysbiosis. Arch. Toxicol. 2018, 92, 1847–1860. [Google Scholar] [CrossRef]
- Deierlein, A.L.; Rock, S.; Park, S. Persistent Endocrine-Disrupting Chemicals and Fatty Liver Disease. Curr. Environ. Health Rep. 2017, 4, 439–449. [Google Scholar] [CrossRef]
- Cave, M.; Appana, S.; Patel, M.; Falkner, K.C.; McClain, C.J.; Brock, G. Polychlorinated biphenyls, lead, and mercury are associated with liver disease in American adults: NHANES 2003–2004. Environ. Health Perspect. 2010, 118, 1735–1742. [Google Scholar] [CrossRef] [Green Version]
- Chi, Y.; Lin, Y.; Lu, Y.; Huang, Q.; Ye, G.; Dong, S. Gut microbiota dysbiosis correlates with a low-dose PCB126-induced dyslipidemia and non-alcoholic fatty liver disease. Sci. Total Environ. 2019, 653, 274–282. [Google Scholar] [CrossRef]
- Petriello, M.C.; Hoffman, J.B.; Vsevolozhskaya, O.; Morris, A.J.; Hennig, B. Dioxin-like PCB 126 increases intestinal inflammation and disrupts gut microbiota and metabolic homeostasis. Environ. Pollut. 2018, 242, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Lukowicz, C.; Ellero-Simatos, S.; Regnier, M.; Polizzi, A.; Lasserre, F.; Montagner, A.; Lippi, Y.; Jamin, E.L.; Martin, J.F.; Naylies, C.; et al. Metabolic effects of a chronic dietary exposure to a low-dose pesticide cocktail in mice: Sexual dimorphism and role of the constitutive androstane receptor. Environ. Health Perspect. 2018, 126, 067007. [Google Scholar] [CrossRef] [PubMed]
- Hyder, O.; Chung, M.; Cosgrove, D.; Herman, J.M.; Li, Z.; Firoozmand, A.; Gurakar, A.; Koteish, A.; Pawlik, T.M. Cadmium exposure and liver disease among US adults. J. Gastrointest. Surg. Off. J. Soc. Surg. Aliment. Tract 2013, 17, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Jin, Y.; Zeng, Z.; Liu, Z.; Fu, Z. Subchronic exposure of mice to cadmium perturbs their hepatic energy metabolism and gut microbiome. Chem. Res. Toxicol. 2015, 28, 2000–2009. [Google Scholar] [CrossRef] [PubMed]
- Ba, Q.; Li, M.; Chen, P.; Huang, C.; Duan, X.; Lu, L.; Li, J.; Chu, R.; Xie, D.; Song, H.; et al. Sex-Dependent effects of cadmium exposure in early life on gut microbiota and fat accumulation in mice. Environ. Health Perspect. 2017, 125, 437–446. [Google Scholar] [CrossRef] [PubMed]







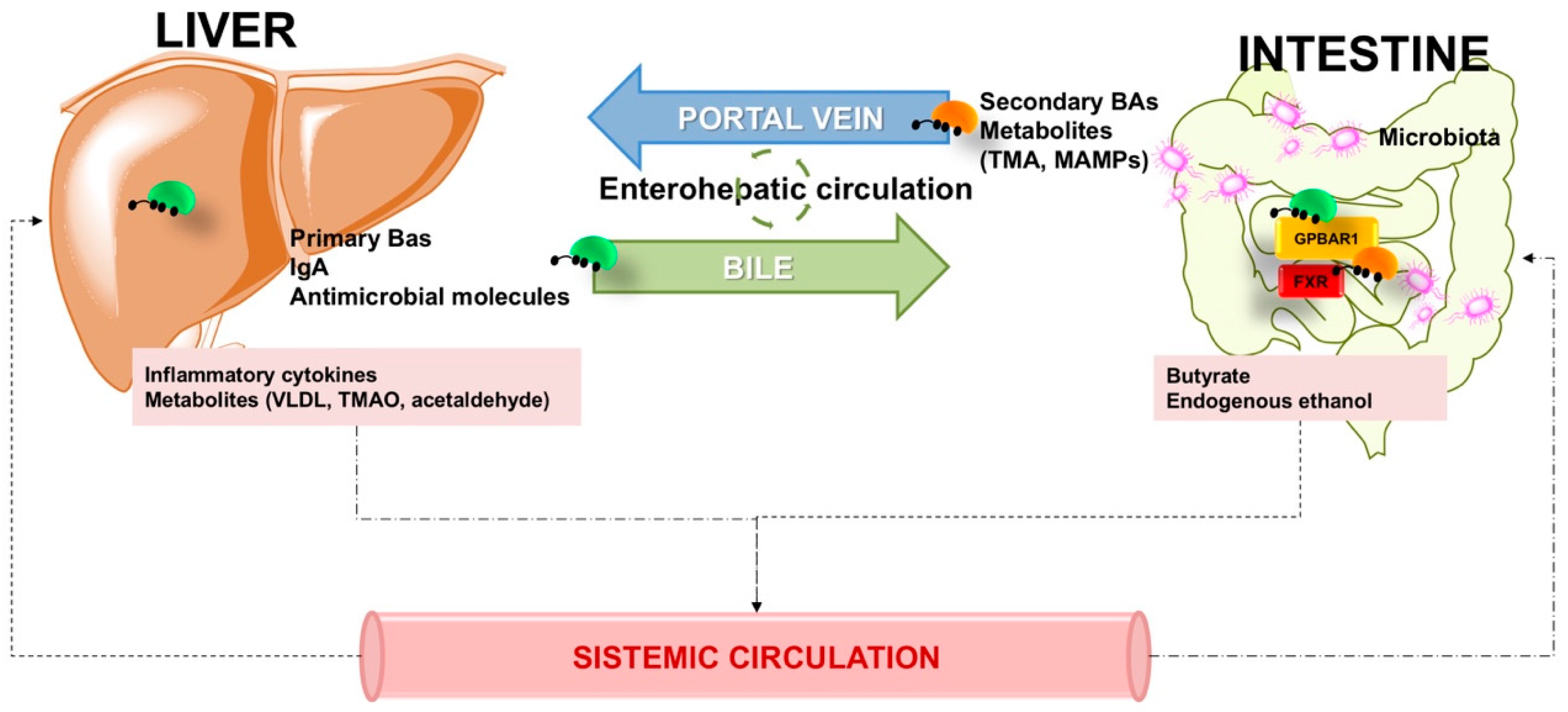{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolic: Nonalcoholic fatty liver (NAFLD) or metabolic-dysfunction-associated fatty liver disease (MAFLD) |
| Alcoholic liver disease (ALD) |
| Hepatitis C (in particular genotype 3) |
| Lipodystrophy |
| Parenteral nutrition |
| Starvation |
| Wilson disease |
| Abetalipoproteinemia |
| Drugs (e.g., methotrexate, tamoxifen, glucocorticoids, amiodarone, valproate, anti-retroviral agents for HIV) |
| Acute fatty liver of pregnancy |
| HELLP (hemolytic anemia, elevated liver enzymes, low platelet count) syndrome |
| Reye syndrome |
| Inborn errors of metabolism (LCAT deficiency, cholesterol ester storage disease, Wolman disease) |
| Drug-induced liver disease |
| Food contaminants of environmental origin (e.g., endocrine disrupting chemicals, persistent organic pollutants, metals) |
| Free Fatty Acids (FFA) |
|
| Triglycerides (TG) |
|
| Lysophosphatidylcholine (LPC) |
|
| Ceramides |
|
| Free Cholesterol |
|
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Ciaula, A.; Baj, J.; Garruti, G.; Celano, G.; De Angelis, M.; Wang, H.H.; Di Palo, D.M.; Bonfrate, L.; Wang, D.Q.-H.; Portincasa, P. Liver Steatosis, Gut-Liver Axis, Microbiome and Environmental Factors. A Never-Ending Bidirectional Cross-Talk. J. Clin. Med. 2020, 9, 2648. https://doi.org/10.3390/jcm9082648
Di Ciaula A, Baj J, Garruti G, Celano G, De Angelis M, Wang HH, Di Palo DM, Bonfrate L, Wang DQ-H, Portincasa P. Liver Steatosis, Gut-Liver Axis, Microbiome and Environmental Factors. A Never-Ending Bidirectional Cross-Talk. Journal of Clinical Medicine. 2020; 9(8):2648. https://doi.org/10.3390/jcm9082648
Chicago/Turabian StyleDi Ciaula, Agostino, Jacek Baj, Gabriella Garruti, Giuseppe Celano, Maria De Angelis, Helen H. Wang, Domenica Maria Di Palo, Leonilde Bonfrate, David Q-H Wang, and Piero Portincasa. 2020. "Liver Steatosis, Gut-Liver Axis, Microbiome and Environmental Factors. A Never-Ending Bidirectional Cross-Talk" Journal of Clinical Medicine 9, no. 8: 2648. https://doi.org/10.3390/jcm9082648