A Look into Liver Mitochondrial Dysfunction as a Hallmark in Progression of Brain Energy Crisis and Development of Neurologic Symptoms in Hepatic Encephalopathy



Abstract
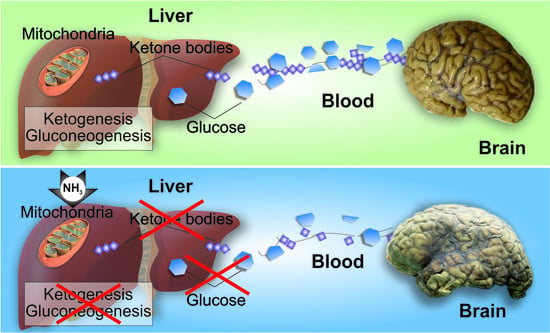
:
1. Introduction
2. Experimental Section
2.1. Experimental Design
2.2. Animals
2.3. Preparative and Analytical Methods
2.4. Freeze-Clamped Liver and Brain Neocortex
2.5. Isolation of Liver Mitochondria Using a Self-Generated Percoll Gradient
2.6. Mitochondrial Purity Assessment
2.7. Study of Mitochondrial Respiration
2.8. Determination of Pyruvate Carboxylase Activity (EC 6.4.1.1)
2.9. Determination of Phosphoenolpyruvate Carboxykinase Activity (EC 4.1.1.32)
2.10. Microsome Isolation and Glucose-6-Phosphatase Activity (EC 3.1.3.9)
2.11. Activity of Enzymes of Ketone Body Synthesis in the Liver
2.12. Acetoacetyl-CoA Thiolase (EC 2.3.1.9)
2.13. HMG-CoA Synthase (E.C.4. l.3.5)
2.14. HMG-CoA Lyase (E.C.4.1.3.4)
2.15. 3-Hydroxybutyrate Dehydrogenase (E.C.1.1.1.30)
2.16. Analysis of Ketogenesis in Mitochondria
2.17. Statistical Analysis
3. Results
3.1. Metabolic Changes in the Liver and Blood during Starvation
3.2. Effect of Acute Ammonia Intoxication on Levels of Liver and Blood Glucose, Ketone Bodies, FFA, and Ammonia
3.3. Intactness and Coupling Efficiency of Isolated Mitochondria
3.4. Oxidative Phosphorylation in Hyperammonemia
3.5. Adenine Nucleotides and NAD/NADH Ratio in Liver Mitochondria
3.6. Effect of Ammonia on Pyruvate Carboxylase, Phosphoenolpyruvate Carboxykinase, and Glucose-6-Phosphatase Activity
3.7. Effect of Ammonia on Enzyme Activities of the HMG-CoA Pathway and Acetoacetate Production in the Liver Mitochondria of Starved Rats
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Von Frerichs, F.T. A Clinical Treatise on Diseases of the Liver by Dr Friedrich Theodor Frerichs; The New Sydenham Society: London, UK, 1860; Volume 1, pp. 193–246. [Google Scholar]
- Hahn, M.; Massen, O.; Nencki, M.; Pawlow, J. Die Eck’sche fistel zwischen der unteren hohlvene und der pfortader und ihre folgen fur den organismus. Arch. Exp. Pathol. Pharmakol. 1893, 32, 161–210. [Google Scholar] [CrossRef] [Green Version]
- Butterworth, R.F.; Giguère, J.F.; Michaud, J.; Lavoie, J.; Layrargues, G.P. Ammonia: Key factor in the pathogenesis of hepatic encephalopathy. Neurochem. Pathol. 1987, 6, 1–12. [Google Scholar] [CrossRef]
- Rolando, N.; Wade, J.; Davalos, M.; Wendon, J.; Philpott-Howard, J.; Williams, R. The systemic inflammatory response syndrome in acute liver failure. Hepatology 2000, 32, 734–739. [Google Scholar] [CrossRef]
- Souba, W.W. Interorgan ammonia metabolism in health and disease: A surgeon’s view. J. Parenter. Enter. Nutr. 1987, 11, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.O.; Schenker, S. Pathogenesis of hepatic encephalopathy--with special reference to the role of ammonia. Am. J. Clin. Nutr. 1970, 23, 619–632. [Google Scholar] [CrossRef]
- Lockwood, A.H. Hepatic encephalopathy: Experimental approaches to human metabolic encephalopathy. Crit. Rev. Neurobiol. 1987, 3, 105–133. [Google Scholar]
- Cooper, A.J.; Plum, F. Biochemistry and physiology of brain ammonia. Physiol. Rev. 1987, 67, 440–519. [Google Scholar] [CrossRef] [PubMed]
- Kosenko, E.; Felipo, V.; Montoliu, C.; Grisolía, S.; Kaminsky, Y. Effects of acute hyperammonemia in vivo on oxidative metabolism in nonsynaptic rat brain mitochondria. Metab. Brain Dis. 1997, 12, 69–82. [Google Scholar] [CrossRef]
- Kosenko, E.; Kaminsky, Y.; Stavroskaya, I.G.; Felipo, V. Alteration of mitochondrial calcium homeostasis by ammonia-induced activation of NMDA receptors in rat brain in vivo. Brain Res. 2000, 880, 139–146. [Google Scholar] [CrossRef]
- Norenberg, M.D.; Huo, Z.; Neary, J.T.; Roig-Cantesano, A. The glial glutamate transporter in hyperammonemia and hepatic encephalopathy: Relation to energy metabolism and glutamatergic neurotransmission. Glia 1997, 21, 124–133. [Google Scholar] [CrossRef]
- Kosenko, E.; Kaminsky, Y.; Grau, E.; Miñana, M.D.; Marcaida, G.; Grisolía, S.; Felipo, V. Brain ATP depletion induced by acute ammonia intoxication in rats is mediated by activation of the NMDA receptor and Na+,K(+)-ATPase. J. Neurochem. 1994, 63, 2172–2178. [Google Scholar] [CrossRef] [PubMed]
- Kosenko, E.; Kaminsky, Y.; Kaminsky, A.; Valencia, M.; Lee, L.; Hermenegildo, C.; Felipo, V. Superoxide production and antioxidant enzymes in ammonia intoxication in rats. Free Radic. Res. 1997, 27, 637–644. [Google Scholar] [CrossRef]
- Norenberg, M.D. Oxidative and nitrosative stress in ammonia neurotoxicity. Hepatology 2003, 37, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Aldridge, D.R.; Tranah, E.J.; Shawcross, D.L. Pathogenesis of hepatic encephalopathy: Role of ammonia and systemic inflammation. J. Clin. Exp. Hepatol. 2015, 5, S7–S20. [Google Scholar] [CrossRef] [Green Version]
- Bjerring, P.N.; Gluud, L.L.; Larsen, F.S. Cerebral blood flow and metabolism in hepatic encephalopathy—A meta-analysis. J. Clin. Exp. Hepatol. 2018, 8, 286–293. [Google Scholar] [CrossRef] [Green Version]
- Alexander, B.; Aslam, M.; Benjamin, I.S. An investigation of the relationship between the liver and brain using an isolated perfused rat brain preparation. J. Pharmacol. Toxicol. Methods 1999, 42, 31–37. [Google Scholar] [CrossRef]
- Amaral, A.I. Effects of hypoglycaemia on neuronal metabolism in the adult brain: Role of alternative substrates to glucose. J. Inherit. Metab. Dis. 2013, 36, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, T.; Shimahara, Y.; Takada, Y.; Ino, K.; Mori, K.; Kobayashi, N.; Yamaoka, Y.; Ozawa, K. Evaluation of ketogenesis in seriously reduced hepatic mitochondrial redox state. An analysis of survivors and non-survivors in critically ill hepatectomized patients. Scand. J. Gastroenterol. 1992, 27, 472–478. [Google Scholar] [CrossRef]
- Owen, O.E.; Felig, P.; Morgan, A.P.; Wahren, J.; Cahill, G.F. Liver and kidney metabolism during prolonged starvation. J. Clin. Invest. 1969, 48, 574–583. [Google Scholar] [CrossRef]
- Mizock, B.A. Nutritional support in hepatic encephalopathy. Nutrition 1999, 15, 220–228. [Google Scholar] [CrossRef]
- Woll, P.J.; Record, C.O. Lactate elimination in man: Effects of lactate concentration and hepatic dysfunction. Eur. J. Clin. Invest. 1979, 9, 397–404. [Google Scholar] [CrossRef]
- Owen, O.E.; Reichle, F.A.; Mozzoli, M.A.; Kreulen, T.; Patel, M.S.; Elfenbein, I.B.; Golsorkhi, M.; Chang, K.H.; Rao, N.S.; Sue, H.S.; et al. Hepatic, gut, and renal substrate flux rates in patients with hepatic cirrhosis. J. Clin. Investig. 1981, 68, 240–252. [Google Scholar] [CrossRef] [Green Version]
- Sarna, G.S.; Bradbury, M.W.; Cremer, J.E.; Lai, J.C.; Teal, H.M. Brain metabolism and specific transport at the blood-brain barrier after portocaval anastomosis in the rat. Brain Res. 1979, 160, 69–83. [Google Scholar] [CrossRef]
- Mans, A.M.; Biebuyck, J.F.; Davis, D.W.; Hawkins, R.A. Portacaval anastomosis: Brain and plasma metabolite abnormalities and the effect of nutritional therapy. J. Neurochem. 1984, 43, 697–705. [Google Scholar] [CrossRef]
- Miñana, M.D.; Felipo, V.; Wallace, R.; Grisolía, S. Hyperammonemia decreases body fat content in rat. FEBS Lett. 1989, 249, 261–263. [Google Scholar] [CrossRef] [Green Version]
- Kosenko, E.; Felipo, V.; Miñana, M.D.; Grau, E.; Grisolía, S. Ammonium ingestion prevents depletion of hepatic energy metabolites induced by acute ammonium intoxication. Arch. Biochem. Biophys. 1991, 290, 484–488. [Google Scholar] [CrossRef]
- Lichtenstein, G.R.; Yang, Y.X.; Nunes, F.A.; Lewis, J.D.; Tuchman, M.; Tino, G.; Kaiser, L.R.; Palevsky, H.I.; Kotloff, R.M.; Furth, E.E.; et al. Fatal hyperammonemia after orthotopic lung transplantation. Ann. Intern. Med. 2000, 132, 283–287. [Google Scholar] [CrossRef]
- Snyder, M.J.; Bradford, W.D.; Kishnani, P.S.; Hale, L.P. Idiopathic hyperammonemia following an unrelated cord blood transplant for mucopolysaccharidosis I. Pediatr. Dev. Pathol. Off. J. Soc. Pediatr. Pathol. Paediatr. Pathol. Soc. 2003, 6, 78–83. [Google Scholar] [CrossRef]
- Williamson, J.R.; Jákob, A.; Scholz, R. Energy cost of gluconeogenesis in rat liver. Metabolism 1971, 20, 13–26. [Google Scholar] [CrossRef]
- Kosenko, E.; Montoliu, C.; Giordano, G.; Kaminsky, Y.; Venediktova, N.; Buryanov, Y.; Felipo, V. Acute ammonia intoxication induces an NMDA receptor-mediated increase in poly(ADP-ribose) polymerase level and NAD metabolism in nuclei of rat brain cells. J. Neurochem. 2004, 89, 1101–1110. [Google Scholar] [CrossRef]
- Kosenko, E.A.; Venediktova, N.I.; Kudryavtsev, A.A.; Ataullakhanov, F.I.; Kaminsky, Y.G.; Felipo, V.; Montoliu, C. Encapsulation of glutamine synthetase in mouse erythrocytes: A new procedure for ammonia detoxification. Biochem. Cell Biol. Biochim. Biol. Cell. 2008, 86, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Graham, J.M. Purification of a crude mitochondrial fraction by density-gradient centrifugation. Curr. Protoc. Cell Biol. 2001. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar]
- Kosenko, E.; Venediktova, N.; Kaminsky, Y.; Montoliu, C.; Felipo, V. Preparation and handling of brain mitochondria useful to study uptake and release of calcium. Brain Res. Brain Res. Protoc. 2001, 7, 248–254. [Google Scholar] [CrossRef]
- Graham, J.M. The identification of subcellular fractions from mammalian cells. Methods Mol. Biol. 1993, 19, 1–18. [Google Scholar] [CrossRef]
- Graham, J.M. Isolation of mitochondria from tissues and cells by differential centrifugation. Curr. Protoc. Cell Biol. 2001. [Google Scholar] [CrossRef] [PubMed]
- Scrutton, M.C.; Olmstead, M.R.; Utter, M.F. Pyruvate Carboxylase from Chicken Liver. In Methods in Enzymology; Lowenstein, J.M., Ed.; Academic Press: New York, NY, USA, 1969; Volume 13, pp. 235–250. [Google Scholar]
- Petrescu, I.; Bojan, O.; Saied, M.; Bârzu, O.; Schmidt, F.; Kühnle, H.F. Determination of phosphoenolpyruvate carboxykinase activity with deoxyguanosine 5′-diphosphate as nucleotide substrate. Anal. Biochem. 1979, 96, 279–281. [Google Scholar] [CrossRef]
- Arion, W.J. Measurement of intactness of rat liver endoplasmic reticulum. Methods Enzymol. 1989, 174, 58–67. [Google Scholar] [CrossRef]
- Baginski, E.S.; Foa, P.P.; Zak, B. Glucose 6-phosphatase. In Methods of Enzymatic Analysis; Bergmeyer, H.U., Ed.; Academic Press: New York, NY, USA, 1974; Volume 2, pp. 876–880. [Google Scholar]
- Williamson, D.H.; Bates, M.W.; Krebs, H.A. Activity and intracellular distribution of enzymes of ketone-body metabolism in rat liver. Biochem. J. 1968, 108, 353–361. [Google Scholar] [CrossRef] [Green Version]
- Quant, P.A.; Tubbs, P.K.; Brand, M.D. Treatment of rats with glucagon or mannoheptulose increases mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase activity and decreases succinyl-CoA content in liver. Biochem. J. 1989, 262, 159–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miziorko, H.M.; Clinkenbeard, K.D.; Reed, W.D.; Lane, M.D. 3-Hydroxy-3-methylglutaryl coenzyme A synthase. Evidence for an acetyl-S-enzyme intermediate and identification of a cysteinyl sulfhydryl as the site of acetylation. J. Biol. Chem. 1975, 250, 5768–5773. [Google Scholar] [PubMed]
- Clinkenbeard, K.D.; Reed, W.D.; Mooney, R.A.; Lane, M.D. Intracellular localization of the 3-hydroxy-3-methylglutaryl coenzme A cycle enzymes in liver. Separate cytoplasmic and mitochondrial 3-hydroxy-3-methylglutaryl coenzyme A generating systems for cholesterogenesis and ketogenesis. J. Biol. Chem. 1975, 250, 3108–3116. [Google Scholar]
- Wanders, R.J.; Zoeters, P.H.; Schutgens, R.B.; de Klerk, J.B.; Duran, M.; Wadman, S.K.; van Sprang, F.J.; Hemmes, A.M.; Voorbrood, B.S. Rapid diagnosis of 3-hydroxy-3-methylglutaryl-coenzyme A lyase deficiency via enzyme activity measurements in leukocytes or platelets using a simple spectrophotometric method. Clin. Chim. Acta Int. J. Clin. Chem. 1990, 189, 327–334. [Google Scholar] [CrossRef]
- Williamson, D.H.; Mellanby, J. Determination of Ketone Bodies. In Methods of Enzymatic Analysis; Bergmeyer, H.U., Ed.; Verlag Chemie: Weinheim, Germany, 1974; Volume 4, pp. 1836–1843. [Google Scholar]
- Kosenko, E.; Kaminsky, Y.; Lopata, O.; Muravyov, N.; Kaminsky, A.; Hermenegildo, C.; Felipo, V. Nitroarginine, an inhibitor of nitric oxide synthase, prevents changes in superoxide radical and antioxidant enzymes induced by ammonia intoxication. Metab. Brain Dis. 1998, 13, 29–41. [Google Scholar] [CrossRef]
- Kaminsky, Y.G.; Kosenko, E.A. Diurnal changes in succinate and D-3-hydroxybutyrate dehydrogenase activities of rat liver mitochondria after chronic alcohol consumption and withdrawal. Comp. Biochem. Physiol. C 1988, 90, 79–82. [Google Scholar] [CrossRef]
- Takeyama, N.; Itoh, Y.; Kitazawa, Y.; Tanaka, T. Altered hepatic mitochondrial fatty acid oxidation and ketogenesis in endotoxic rats. Am. J. Physiol. 1990, 259, E498–E505. [Google Scholar] [CrossRef]
- Kosenko, E.A.; Venediktova, N.I.; Kaminskiĭ, I.G. Calcium and ammonia stimulate monoamine oxidase A activity in brain mitochondria. Izv. Akad. Nauk. Ser. Biol. 2003, 542–546. [Google Scholar] [CrossRef]
- Kaminsky, Y.G.; Kosenko, E.A. Calculation of the concentration of metabolites freely and non-freely penetrating the cell membranes in the liver cylosol and mitochondria. Izv SSRR Ser. Biol. 1987, 2, 196–202. [Google Scholar]
- Walter, P.; Stucki, J.W. Regulation of pyruvate carboxylase in rat liver mitochondria by adenine nucleotides and short chain fatty acids. Eur. J. Biochem. 1970, 12, 508–519. [Google Scholar] [CrossRef]
- Williamson, D.H.; Lund, P.; Krebs, H.A. The redox state of free nicotinamide-adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Biochem. J. 1967, 103, 514–527. [Google Scholar] [CrossRef]
- Clifford, A.J.; Prior, R.L.; Visek, W.J. Depletion of reduced pyridine nucleotides in liver and blood with ammonia. Am. J. Physiol. 1969, 217, 1269–1272. [Google Scholar] [CrossRef] [Green Version]
- Blackshear, P.J.; Holloway, P.A.; Aberti, K.G. The effects of inhibition of gluconeogenesis on ketogenesis in starved and diabetic rats. Biochem. J. 1975, 148, 353–362. [Google Scholar] [CrossRef] [Green Version]
- Kosenko, E.; Venediktova, N.; Kaminsky, Y.; Montoliu, C.; Felipo, V. Sources of oxygen radicals in brain in acute ammonia intoxication in vivo. Brain Res. 2003, 981, 193–200. [Google Scholar] [CrossRef]
- Cryer, P.E.; Gerich, J.E. Glucose counterregulation, hypoglycemia, and intensive insulin therapy in diabetes mellitus. N. Engl. J. Med. 1985, 313, 232–241. [Google Scholar] [CrossRef]
- Kosenko, E.A. Energy Metabolism in Normal and in Pathology. In The Role of Stimulating Neurotransmitter; Lenand: Moscow, Russia, 2014. [Google Scholar]
- Worcel, A.; Erecinska, M. Mechanism of inhibitory action of ammonia on the respiration of rat-liver mitochondria. Biochim. Biophys. Acta 1962, 65, 27–33. [Google Scholar] [CrossRef]
- Wojtczak, L. Effect of long-chain fatty acids and acyl-CoA on mitochondrial permeability, transport, and energy-coupling processes. J. Bioenerg. Biomembr. 1976, 8, 293–311. [Google Scholar] [CrossRef]
- Rao, K.V.; Mawal, Y.R.; Qureshi, I.A. Progressive decrease of cerebral cytochrome C oxidase activity in sparse-fur mice: Role of acetyl-L-carnitine in restoring the ammonia-induced cerebral energy depletion. Neurosci. Lett. 1997, 224, 83–86. [Google Scholar] [CrossRef]
- Kosenko, E.; Kaminsky, Y.; Solomadin, I.; Marov, N.; Venediktova, N.; Felipo, V.; Montoliu, C. Acute ammonia neurotoxicity in vivo involves increase in cytoplasmic protein P53 without alterations in other markers of apoptosis. J. Neurosci. Res. 2007, 85, 2491–2499. [Google Scholar] [CrossRef] [PubMed]
- Kumashiro, N.; Beddow, S.A.; Vatner, D.F.; Majumdar, S.K.; Cantley, J.L.; Guebre-Egziabher, F.; Fat, I.; Guigni, B.; Jurczak, M.J.; Birkenfeld, A.L.; et al. Targeting pyruvate carboxylase reduces gluconeogenesis and adiposity and improves insulin resistance. Diabetes 2013, 62, 2183–2194. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | V3 | V4 | Vu | RCI | ADP/O |
|---|---|---|---|---|---|
| Control Animals Starved for 24 h | |||||
| Pyruvate + malate | 30.24 ± 1.099 | 10.11 ± 0.11 | 36.2 ± 2.23 | 2.99 ± 0.16 | 3.021 ± 0.13 |
| Glutamate + malate | 88.52 ± 4.37 | 12.24 ± 0.98 | 95.3 ± 4.54 | 7.12 ± 0.54 | 2.87 ± 0.21 |
| Succinate + rotenone | 117 ± 5.45 | 22.8 ± 0.87 | 141 ± 5.31 | 5.13 ± 0.36 | 1.64 ± 0.043 |
| PalmitoyI-L-carnitine | 45.16 ± 1.73 | 10.33 ± 1.81 | 81.04 ± 5.33 | 4.37 ± 0.29 | 1.83 ± 0.44 |
| Octanoate Na | 24.2 ± 0.9 | 4.70 ± 0.11 | 53.7 ± 3.41 | 5.15 ± 0.28 | 2.02 ± 0.06 |
| Oleic acid +L- carnitine | 18.5 ± 0.61 | 6.17 ± 0.23 | 54.4 ± 3.36 | 3.0 ± 0.17 | 2.07 ± 0.19 |
| Ammonia-Treated Animals Starved for 24 h | |||||
| Pyruvate + malate | 17.84 ± 2.43 *** | 9.32 ± 1.13 | 33.96 ± 2.63 | 1.91 ± 0.21 ** | 2.54 ± 0.09 * |
| Glutamate + malate | 56.98 ± 3.61 *** | 10.32 ± 2.12 | 96.2 ± 3.52 | 5.52 ± 0.27 * | 2.46 ± 0.31 |
| Succinate + rotenone | 72.3 ± 1.33 *** | 19.11 ± 3.33 | 134 ± 6.20 | 3.78 ± 0.53 | 1.77 ± 0.075 |
| PalmitoyI-L-carnitine | 23.9 ± 1.7 *** | 12.1 ± 1 | 102 ± 11 | 1.98 ± 0.12 *** | 2.45 ± 0.15 * |
| Na Octanoate | 14 ± 0.52 *** | 5.5 ± 0.29 * | 49.8 ± 1.5 | 2.55 ± 0.14 *** | 2.7 ± 0.2 ** |
| Oleic acid + L- carnitine | 10.6 ± 0.55 *** | 7.1 ± 1.45 | 54.5 ± 7.5 | 1.8 ± 0.25 ** | 2.7 ± 0.11 ** |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kosenko, E.; Tikhonova, L.; Alilova, G.; Montoliu, C. A Look into Liver Mitochondrial Dysfunction as a Hallmark in Progression of Brain Energy Crisis and Development of Neurologic Symptoms in Hepatic Encephalopathy. J. Clin. Med. 2020, 9, 2259. https://doi.org/10.3390/jcm9072259
Kosenko E, Tikhonova L, Alilova G, Montoliu C. A Look into Liver Mitochondrial Dysfunction as a Hallmark in Progression of Brain Energy Crisis and Development of Neurologic Symptoms in Hepatic Encephalopathy. Journal of Clinical Medicine. 2020; 9(7):2259. https://doi.org/10.3390/jcm9072259
Chicago/Turabian StyleKosenko, Elena, Lyudmila Tikhonova, Gubidat Alilova, and Carmina Montoliu. 2020. "A Look into Liver Mitochondrial Dysfunction as a Hallmark in Progression of Brain Energy Crisis and Development of Neurologic Symptoms in Hepatic Encephalopathy" Journal of Clinical Medicine 9, no. 7: 2259. https://doi.org/10.3390/jcm9072259