Oxidative Stress and New Pathogenetic Mechanisms in Endothelial Dysfunction: Potential Diagnostic Biomarkers and Therapeutic Targets

 , ,
, ,
Abstract
:1. Introduction
2. Oxidative Stress and Endothelial Dysfunction and CVD
2.1. NADPH Oxidase (NOX) Activity and ROS Production
2.2. Antioxidant Enzymes in Endothelial Dysfunction
2.2.1. Superoxide Dismutase
2.2.2. Glutathione Peroxidase
2.2.3. Catalase
2.2.4. Thioredoxin
2.2.5. Peroxiredoxin
2.3. Redox-Regulated Transcription Factors
3. Oxidative Stress and Cellular Biomarkers for Prevention and Diagnosis of Endothelial Dysfunction
3.1. Lipid Peroxidation
3.2. Peroxynitrite and NO
3.3. Glutathione, Glutamyltransferase, Guanine and Low-Density Lipoprotein
3.4. Endothelial Dysfunction Markers Associated with Inflammation
3.5. Endothelial Progenitor Cells
3.6. Endoglin
3.7. Uric Acid
4. Epigenetic Regulation in Endothelial Dysfunction and Vascular Damage
4.1. DNA Methylation
4.2. Histone Modifications
4.3. MicroRNAs (miRNAs)
4.4. Long Non-Coding RNAs (lncRNAs)
5. New Therapeutic Approaches Targeted Endothelial Dysfunction
5.1. Nanotechnologies for Drug Delivery Systems
5.2. Mitochondrial and Oxidative Stress-Targeted Therapies
5.2.1. Antioxidant Therapies
Mitochondrial Targets
Antioxidant Enzyme Mimics
VEGF-VEGFR2 Signaling Targets
5.3. NOX-Targeted Therapies
5.4. Gene Targeted Therapies
6. Future Perspectives and Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| GSH | glutathione reduced |
| GSSG | glutathione disulfide oxidized |
| EPCs | endothelial progenitor cells |
| EMPs | endothelial microparticles |
| CECs | circulating endothelial cells |
| EC | endothelial cell |
| VSMC | vascular smooth muscle cell |
| T2D | type 2 diabetes mellitus |
| CAD | coronary artery diseases |
| CVD | cardiovascular diseases |
References
- Moghaddam, A.S.; Afshari, J.T.; Esmaeili, S.A.; Saburi, E.; Joneidi, Z.; Momtazi-Borojeni, A.A. Cardioprotective microRNAs: Lessons from stem cell-derived exosomal microRNAs to treat cardiovascular disease. Atherosclerosis 2019, 285, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buttar, H.S.; Li, T.; Ravi, N. Prevention of cardiovascular diseases: Role of exercise, dietary interventions, obesity and smoking cessation. Exp. Clin. Cardiol. 2005, 10, 229. [Google Scholar] [PubMed]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Sena, C.M.; Pereira, A.M.; Seica, R. Endothelial dysfunction—A major mediator of diabetic vascular disease. Biochim. Biophys. Acta 2013, 1832, 2216–2231. [Google Scholar] [CrossRef] [Green Version]
- Flammer, A.J.; Anderson, T.; Celermajer, D.S.; Creager, M.A.; Deanfield, J.; Ganz, P.; Hamburg, N.M.; Luscher, T.F.; Shechter, M.; Taddei, S.; et al. The assessment of endothelial function: From research into clinical practice. Circulation 2012, 126, 753–767. [Google Scholar] [CrossRef] [PubMed]
- Hadi, H.A.; Carr, C.S.; Al Suwaidi, J. Endothelial dysfunction: Cardiovascular risk factors, therapy, and outcome. Vasc. Health Risk Manag. 2005, 1, 183. [Google Scholar]
- Jamwal, S.; Sharma, S. Vascular endothelium dysfunction: A conservative target in metabolic disorders. Inflamm. Res. 2018, 67, 391–405. [Google Scholar] [CrossRef]
- Sitia, S.; Tomasoni, L.; Atzeni, F.; Ambrosio, G.; Cordiano, C.; Catapano, A.; Tramontana, S.; Perticone, F.; Naccarato, P.; Camici, P.; et al. From endothelial dysfunction to atherosclerosis. Autoimmun. Rev. 2010, 9, 830–834. [Google Scholar] [CrossRef]
- Heitzer, T.; Schlinzig, T.; Krohn, K.; Meinertz, T.; Munzel, T. Endothelial dysfunction, oxidative stress, and risk of cardiovascular events in patients with coronary artery disease. Circulation 2001, 104, 2673–2678. [Google Scholar] [CrossRef] [Green Version]
- Virdis, A.; Masi, S.; Colucci, R.; Chiriacò, M.; Uliana, M.; Puxeddu, I.; Bernardini, N.; Blandizzi, C.; Taddei, S. Microvascular endothelial dysfunction in patients with obesity. Curr. Hypertens. Rep. 2019, 21, 32. [Google Scholar] [CrossRef]
- Yudkin, J.S. Adipose tissue, insulin action and vascular disease: Inflammatory signals. Int. J. Obes. 2003, 27, S25–S28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodd-o, J.M.; Welsh, L.E.; Salazar, J.D.; Walinsky, P.L.; Peck, E.A.; Shake, J.G.; Caparrelli, D.J.; Ziegelstein, R.C.; Zweier, J.L.; Baumgartner, W.A.; et al. Effect of NADPH oxidase inhibition on cardiopulmonary bypass-induced lung injury. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H927–H936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Assar, M.; Angulo, J.; Santos-Ruiz, M.; Ruiz de Adana, J.C.; Pindado, M.L.; Sánchez-Ferrer, A.; Hernández, A.; Rodríguez-Mañas, L. Asymmetric dimethylarginine (ADMA) elevation and arginase up-regulation contribute to endothelial dysfunction related to insulin resistance in rats and morbidly obese humans. J. Physiol. 2016, 594, 3045–3060. [Google Scholar] [CrossRef] [PubMed]
- Masi, S.; Colucci, R.; Duranti, E.; Nannipieri, M.; Anselmino, M.; Ippolito, C.; Tirotta, E.; Georgiopoulos, G.; Garelli, F.; Nericcio, A.; et al. Aging Modulates the influence of arginase on endothelial dysfunction in obesity. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2474–2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.; Yang, T.; Chen, H.; Fu, D.; Hu, Y.; Wang, J.; Yuan, Q.; Yu, H.; Xu, W.; Xie, X. New insights into oxidative stress and inflammation during diabetes mellitus-accelerated atherosclerosis. Redox Biol. 2019, 20, 247–260. [Google Scholar] [CrossRef]
- Yu, E.P.; Bennett, M.R. The role of mitochondrial DNA damage in the development of atherosclerosis. Free Radic. Biol. Med. 2016, 100, 223–230. [Google Scholar] [CrossRef]
- Evans, J.L.; Goldfine, I.D. A new road for treating the vascular complications of diabetes: So let’s step on the gas. Diabetes 2016, 65, 346–348. [Google Scholar] [CrossRef] [Green Version]
- Suryavanshi, S.V.; Kulkarni, Y.A. NF-κβ: A potential target in the management of vascular complications of diabetes. Front. Pharmacol. 2017, 8, 798. [Google Scholar] [CrossRef] [Green Version]
- de Lucia, C.; Komici, K.; Borghetti, G.; Femminella, G.D.; Bencivenga, L.; Cannavo, A.; Corbi, G.; Ferrara, N.; Houser, S.R.; Koch, W.J.; et al. microRNA in cardiovascular aging and age-related cardiovascular diseases. Front. Med. 2017, 4, 74. [Google Scholar] [CrossRef] [Green Version]
- Brennan, E.; Wang, B.; McClelland, A.; Mohan, M.; Marai, M.; Beuscart, O.; Derouiche, S.; Gray, S.; Pickering, R.; Tikellis, C.; et al. Protective Effect of let-7 miRNA Family in Regulating Inflammation in Diabetes-Associated Atherosclerosis. Diabetes 2017, 66, 2266–2277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. The role of miR-126 in embryonic angiogenesis, adult vascular homeostasis, and vascular repair and its alterations in atherosclerotic disease. J. Mol. Cell. Cardiol. 2016, 97, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Magenta, A.; Ciarapica, R.; Capogrossi, M.C. The emerging role of miR-200 family in cardiovascular diseases. Circ. Res. 2017, 120, 1399–1402. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Araki, E. Impact of mitochondrial ROS production in the pathogenesis of diabetes mellitus and its complications. Antioxid. Redox Signal. 2007, 9, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.L.; Goldfine, I.D.; Maddux, B.A.; Grodsky, G.M. Oxidative stress and stress-activated signaling pathways: A unifying hypothesis of type 2 diabetes. Endocr. Rev. 2002, 23, 599–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xian, H.M.; Che, H.; Qin, Y.; Yang, F.; Meng, S.Y.; Li, X.G.; Bai, Y.L.; Wang, L.H. Coriolus versicolor aqueous extract ameliorates insulin resistance with PI3K/Akt and p38 MAPK signaling pathways involved in diabetic skeletal muscle. Phytother. Res. 2018, 32, 551–560. [Google Scholar] [CrossRef]
- Sawamura, T.; Kume, N.; Aoyama, T.; Moriwaki, H.; Hoshikawa, H.; Aiba, Y.; Tanaka, T.; Miwa, S.; Katsura, Y.; Kita, T.; et al. An endothelial receptor for oxidized low-density lipoprotein. Nature 1997, 386, 73–77. [Google Scholar] [CrossRef]
- Kaplan, M.; Aviram, M.; Hayek, T. Oxidative stress and macrophage foam cell formation during diabetes mellitus-induced atherogenesis: Role of insulin therapy. Pharmacol. Ther. 2012, 136, 175–185. [Google Scholar] [CrossRef]
- Chen, J.; Mehta, J.L.; Haider, N.; Zhang, X.; Narula, J.; Li, D. Role of caspases in Ox-LDL-induced apoptotic cascade in human coronary artery endothelial cells. Circ. Res. 2004, 94, 370–376. [Google Scholar] [CrossRef] [Green Version]
- Ndrepepa, G. Uric acid and cardiovascular disease. Clin. Chim. Acta Int. J. Clin. Chem. 2018, 484, 150–163. [Google Scholar] [CrossRef]
- Messner, B.; Bernhard, D. Smoking and cardiovascular disease: Mechanisms of endothelial dysfunction and early atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 509–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzik, T.J.; Touyz, R.M. Oxidative Stress, Inflammation, and Vascular Aging in Hypertension. Hypertension 2017, 70, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Saleh, M.A.; Kirabo, A.; Itani, H.A.; Montaniel, K.R.; Xiao, L.; Chen, W.; Mernaugh, R.L.; Cai, H.; Bernstein, K.E.; et al. Immune activation caused by vascular oxidation promotes fibrosis and hypertension. J. Clin. Investig. 2016, 126, 50–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goncharov, N.V.; Nadeev, A.D.; Jenkins, R.O.; Avdonin, P.V. Markers and Biomarkers of Endothelium: When Something Is Rotten in the State. Oxidative Med. Cell. Longev. 2017, 2017, 9759735. [Google Scholar] [CrossRef]
- Deng, F.; Wang, S.; Zhang, L. Endothelial microparticles act as novel diagnostic and therapeutic biomarkers of circulatory hypoxia-related diseases: A literature review. J. Cell. Mol. Med. 2017, 21, 1698–1710. [Google Scholar] [CrossRef]
- Nguyen Dinh Cat, A.; Montezano, A.C.; Burger, D.; Touyz, R.M. Angiotensin II, NADPH oxidase, and redox signaling in the vasculature. Antioxid. Redox Signal. 2013, 19, 1110–1120. [Google Scholar] [CrossRef] [Green Version]
- Huie, R.E.; Padmaja, S. The reaction of no with superoxide. Free Radic. Res. Commun. 1993, 18, 195–199. [Google Scholar] [CrossRef]
- Gao, X.; Yang, T.; Liu, M.; Peleli, M.; Zollbrecht, C.; Weitzberg, E.; Lundberg, J.O.; Persson, A.E.; Carlström, M. NADPH oxidase in the renal microvasculature is a primary target for blood pressure-lowering effects by inorganic nitrate and nitrite. Hypertension 2015, 65, 161–170. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, J.; Lu, L.; Chen, S.S.; Quinn, M.T.; Weber, K.T. Aldosterone-induced inflammation in the rat heart: Role of oxidative stress. Am. J. Pathol. 2002, 161, 1773–1781. [Google Scholar] [CrossRef]
- Seals, D.R.; Jablonski, K.L.; Donato, A.J. Aging and vascular endothelial function in humans. Clin. Sci. 2011, 120, 357–375. [Google Scholar] [CrossRef] [Green Version]
- Briet, M.; Barhoumi, T.; Mian, M.O.R.; Coelho, S.C.; Ouerd, S.; Rautureau, Y.; Coffman, T.M.; Paradis, P.; Schiffrin, E.L. Aldosterone-induced vascular remodeling and endothelial dysfunction require functional angiotensin type 1a receptors. Hypertension 2016, 67, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Donato, A.J.; Morgan, R.G.; Walker, A.E.; Lesniewski, L.A. Cellular and molecular biology of aging endothelial cells. J. Mol. Cell. Cardiol. 2015, 89, 122–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dedkova, E.N.; Ji, X.; Lipsius, S.L.; Blatter, L.A. Mitochondrial calcium uptake stimulates nitric oxide production in mitochondria of bovine vascular endothelial cells. Am. J. Physiol. Cell Physiol. 2004, 286, C406–C415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajith, T.A.; Jayakumar, T.G. Mitochondria-targeted agents: Future perspectives of mitochondrial pharmaceutics in cardiovascular diseases. World J. Cardiol. 2014, 6, 1091. [Google Scholar] [CrossRef]
- Ott, M.; Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria, oxidative stress and cell death. Apoptosis Int. J. Program. Cell Death 2007, 12, 913–922. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 14205. [Google Scholar] [CrossRef]
- Camici, G.G.; Schiavoni, M.; Francia, P.; Bachschmid, M.; Martin-Padura, I.; Hersberger, M.; Tanner, F.C.; Pelicci, P.; Volpe, M.; Anversa, P.; et al. Genetic deletion of p66(Shc) adaptor protein prevents hyperglycemia-induced endothelial dysfunction and oxidative stress. Proc. Natl. Acad. Sci. USA 2007, 104, 5217–5222. [Google Scholar] [CrossRef] [Green Version]
- Paneni, F.; Mocharla, P.; Akhmedov, A.; Costantino, S.; Osto, E.; Volpe, M.; Lüscher, T.F.; Cosentino, F. Gene silencing of the mitochondrial adaptor p66(Shc) suppresses vascular hyperglycemic memory in diabetes. Circ Res. 2012, 111, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S. P66Shc and vascular endothelial function. Biosci. Rep. 2019, 39, BSR20182134. [Google Scholar] [CrossRef] [Green Version]
- Ho, E.; Karimi Galougahi, K.; Liu, C.C.; Bhindi, R.; Figtree, G.A. Biological markers of oxidative stress: Applications to cardiovascular research and practice. Redox Biol. 2013, 1, 483–491. [Google Scholar] [CrossRef] [Green Version]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, J.B. Vascular endothelial dysfunction and pharmacological treatment. World J. Cardiol. 2015, 7, 719. [Google Scholar] [CrossRef] [PubMed]
- Okumura, N.; Kinoshita, S.; Koizumi, N. The role of rho kinase inhibitors in corneal endothelial dysfunction. Curr. Pharm. Des. 2017, 23, 660–666. [Google Scholar] [PubMed]
- Dong, M.; Yan, B.P.; Liao, J.K.; Lam, Y.Y.; Yip, G.W.; Yu, C.M. Rho-kinase inhibition: A novel therapeutic target for the treatment of cardiovascular diseases. Drug Discov. Today 2010, 15, 622–629. [Google Scholar] [CrossRef] [Green Version]
- Walsh, S.K.; English, F.A.; Crocker, I.P.; Johns, E.J.; Kenny, L.C. Contribution of PARP to endothelial dysfunction and hypertension in a rat model of pre-eclampsia. Br. J. Pharmacol. 2012, 166, 2109–2216. [Google Scholar] [CrossRef] [Green Version]
- Vercauteren, M.; Remy, E.; Devaux, C.; Dautreaux, B.; Henry, J.P.; Bauer, F.; Mulder, P.; Hooft van Huijsduijnen, R.; Bombrun, A.; Thuillez, C.; et al. Improvement of peripheral endothelial dysfunction by protein tyrosine phosphatase inhibitors in heart failure. Circulation 2006, 114, 2498–2507. [Google Scholar] [CrossRef] [Green Version]
- Hyndman, K.A.; Ho, D.H.; Sega, M.F.; Pollock, J.S. Histone deacetylase 1 reduces NO production in endothelial cells via lysine deacetylation of NO synthase 3. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H803–H809. [Google Scholar] [CrossRef] [Green Version]
- Hung, J.; Miscianinov, V.; Sluimer, J.C.; Newby, D.E.; Baker, A.H. Targeting Non-coding RNA in Vascular Biology and Disease. Front. Physiol. 2018, 9, 1655. [Google Scholar] [CrossRef] [Green Version]
- Taneja, G.; Sud, A.; Pendse, N.; Panigrahi, B.; Kumar, A.; Sharma, A.K. Nano-medicine and vascular endothelial dysfunction: Options and delivery strategies. Cardiovasc. Toxicol. 2019, 19, 1–12. [Google Scholar] [CrossRef]
- Yla-Herttuala, S.; Baker, A.H. Cardiovascular gene therapy: Past, present, and future. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 1095–1106. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.A.; Hale, S.L.; Baines, C.P.; del Rio, C.L.; Hamlin, R.L.; Yueyama, Y.; Kijtawornrat, A.; Yeh, S.T.; Frasier, C.R.; Stewart, L.M.; et al. Reduction of early reperfusion injury with the mitochondria-targeting peptide bendavia. J. Cardiovasc. Pharmacol. Ther. 2014, 19, 121–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Gao, G. State-of-the-art human gene therapy: Part II. Gene therapy strategies and clinical applications. Discov. Med. 2014, 18, 151. [Google Scholar] [PubMed]
- Gao, C.Z.; Ma, Q.Q.; Wu, J.; Liu, R.; Wang, F.; Bai, J.; Yang, X.J.; Fu, Q.; Wei, P. Comparison of the effects of ticagrelor and clopidogrel on inflammatory factors, vascular endothelium functions and short-term prognosis in patients with acute st-segment elevation myocardial infarction undergoing emergency percutaneous coronary intervention: A pilot study. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 48, 385–396. [Google Scholar]
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. NADPH Oxidases in Vascular Pathology. Antioxid. Redox Signal. 2014, 20, 2794–2814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morawietz, H. Endothelial NADPH Oxidases: Friends or Foes? Basic Res Cardiol. 2011, 106, 521–525. [Google Scholar] [CrossRef] [Green Version]
- Brandes, R.P.; Weissmann, N.; Schröder, K. NADPH oxidases in cardiovascular disease. Free Radic. Biol. Med. 2010, 49, 687–706. [Google Scholar] [CrossRef]
- Langbein, H.; Brunssen, C.; Hofmann, A.; Cimalla, P.; Brux, M.; Bornstein, S.R.; Deussen, A.; Koch, E.; Morawietz, H. NADPH Oxidase 4 Protects Against Development of Endothelial Dysfunction and Atherosclerosis in LDL Receptor Deficient Mice. Eur. Heart J. 2016, 37, 1753–1761. [Google Scholar] [CrossRef] [Green Version]
- Violi, F.; Loffredo, L.; Carnevale, R.; Pignatelli, P.; Pastori, D. Atherothrombosis and oxidative stress: Mechanisms and management in elderly. Antioxid. Redox Signal. 2017, 27, 1083–1124. [Google Scholar] [CrossRef]
- Murdoch, C.E.; Alom-Ruiz, S.P.; Wang, M.; Zhang, M.; Walker, S.; Yu, B.; Brewer, A.; Shah, A.M. Role of Endothelial Nox2 NADPH Oxidase in Angiotensin II-induced Hypertension and Vasomotor Dysfunction. Basic Res. Cardiol. 2011, 106, 527–538. [Google Scholar] [CrossRef] [Green Version]
- Guzik, T.J.; Sadowski, J.; Guzik, B.; Jopek, A.; Kapelak, B.; Przybylowski, P.; Wierzbicki, K.; Korbut, R.; Harrison, D.G.; Channon, K.M. Coronary Artery Superoxide Production and Nox Isoform Expression in Human Coronary Artery Disease. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Kleinschnitz, C.; Grund, H.; Wingler, K.; Armitage, M.E.; Jones, E.; Mittal, M.; Barit, D.; Schwarz, T.; Geis, C.; Kraft, P.; et al. Post-stroke inhibition of induced NADPH oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biol. 2010, 8, e1000479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielli, A.; Scioli, M.G.; Mazzaglia, D.; Doldo, E.; Orlandi, A. Antioxidants and vascular health. Life Sci. 2015, 143, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Poznyak, A.V.; Grechko, A.V.; Orekhova, V.A.; Chegodaev, Y.S.; Wu, W.K.; Orekhov, A.N. Oxidative stress and antioxidants in atherosclerosis development and treatment. Biology 2020, 9, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wassmann, S.; Wassmann, K.; Nickenig, G. Modulation of oxidant and antioxidant enzyme expression and function in vascular cells. Hypertension 2004, 44, 381–386. [Google Scholar] [CrossRef]
- Marrotte, E.J.; Chen, D.D.; Hakim, J.S.; Chen, A.F. Manganese superoxide dismutase expression in endothelial progenitor cells accelerates wound healing in diabetic mice. J. Clin. Investig. 2010, 120, 4207–4219. [Google Scholar] [CrossRef]
- Brown, K.A.; Chu, Y.; Lund, D.D.; Heistad, D.D.; Faraci, F.M. Gene transfer of extracellular superoxide dismutase protects against vascular dysfunction with aging. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H2600–H2605. [Google Scholar] [CrossRef] [Green Version]
- Awad, M.A.; Aldosari, S.R.; Abid, M.R. Genetic Alterations in Oxidant and Anti-Oxidant Enzymes in the Vascular System. Front. Cardiovasc. Med. 2018, 5, 107. [Google Scholar] [CrossRef] [Green Version]
- Torzewski, M.; Ochsenhirt, V.; Kleschyov, A.L.; Oelze, M.; Daiber, A.; Li, H.; Rossmann, H.; Tsimikas, S.; Reifenberg, K.; Cheng, F.; et al. Deficiency of glutathione peroxidase-1 accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 850–857. [Google Scholar] [CrossRef] [Green Version]
- Kozakowska, M.; Pietraszek-Gremplewicz, K.; Jozkowicz, A.; Dulak, J. The role of oxidative stress in skeletal muscle injury and regeneration: Focus on antioxidant enzymes. J. Muscle Res. Cell Motil. 2015, 36, 377–393. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.H.; Lee, D.J.; Lee, K.W.; Park, Y.S.; Lee, J.Y.; Lee, S.H.; Koh, Y.J.; Koh, G.Y.; Choi, C.; Yu, D.Y.; et al. Peroxiredoxin II is an essential antioxidant enzyme that prevents the oxidative inactivation of VEGF receptor-2 in vascular endothelial cells. Mol. Cell 2011, 44, 545–558. [Google Scholar] [CrossRef] [Green Version]
- Kümin, A.; Schäfer, M.; Epp, N.; Bugnon, P.; Born-Berclaz, C.; Oxenius, A.; Klippel, A.; Bloch, W.; Werner, S. Peroxiredoxin 6 is required for blood vessel integrity in wounded skin. J. Cell Biol. 2007, 179, 747–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mowbray, A.L.; Kang, D.H.; Rhee, S.G.; Kang, S.W.; Jo, H. Laminar shear stress up-regulates peroxiredoxins (PRX) in endothelial cells: PRX 1 as a mechanosensitive antioxidant. J. Biol. Chem. 2008, 283, 1622–1627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohlgrüber, S.; Upadhye, A.; Dyballa-Rukes, N.; McNamara, C.A.; Altschmied, J. Regulation of Transcription Factors by Reactive Oxygen Species and Nitric Oxide in Vascular Physiology and Pathology. Antioxid. Redox Signal. 2017, 26, 679–699. [Google Scholar] [CrossRef] [PubMed]
- Ganesh Yerra, V.; Negi, G.; Sharma, S.S.; Kumar, A. Potential therapeutic effects of the simultaneous targeting of the Nrf2 and NF-κB pathways in diabetic neuropathy. Redox Biol. 2013, 1, 394–397. [Google Scholar] [CrossRef] [Green Version]
- Bonaterra, G.A.; Schwarzbach, H.; Kelber, O.; Weiser, D.; Kinscherf, R. Anti-inflammatory effects of Phytodolor® (STW 1) and components (poplar, ash and goldenrod) on human monocytes/macrophages. Phytomedicine 2019, 58, 152868. [Google Scholar] [CrossRef]
- Yu, J.; Ming, H.; Li, H.Y.; Yu, B.; Chu, M.; Zhu, H.; Zhu, X. IMM-H007, a novel small molecule inhibitor for atherosclerosis, represses endothelium inflammation by regulating the activity of NF-κB and JNK/AP1 signaling. Toxicol. Appl. Pharmacol. 2019, 381, 114732. [Google Scholar] [CrossRef]
- Mumby, S.; Gambaryan, N.; Meng, C.; Perros, F.; Humbert, M.; Wort, S.J.; Adcock, I.M. Bromodomain and extra-terminal protein mimic JQ1 decreases inflammation in human vascular endothelial cells: Implications for pulmonary arterial hypertension. Respirology 2017, 22, 157–164. [Google Scholar] [CrossRef]
- Sena, C.M.; Leandro, A.; Azul, L.; Seiça, R.; Perry, G. Vascular oxidative stress: Impact and therapeutic approaches. Front. Physiol. 2018, 9, 1668. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Lu, Y.; Chen, Y.; Cheng, J. The Role of Nrf2 in oxidative stress-induced endothelial injuries. J. Endocrinol. 2015, 225, R83–R99. [Google Scholar] [CrossRef] [Green Version]
- Angulo, J.; El Assar, M.; Sevilleja-Ortiz, A.; Fernández, A.; Sánchez-Ferrer, A.; Romero-Otero, J.; Martínez-Salamanca, J.I.; La Fuente, J.M.; Rodríguez-Mañas, L. Short-term pharmacological activation of Nrf2 ameliorates vascular dysfunction in aged rats and in pathological human vasculature. A potential target for therapeutic intervention. Redox Biol. 2019, 26, 101271. [Google Scholar] [CrossRef]
- Mason, D.; Chen, Y.Z.; Krishnan, H.V.; Sant, S. Cardiac gene therapy: Recent advances and future directions. J. Control. Release Off. J. Control. Release Soc. 2015, 215, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Jain, T.; Nikolopoulou, E.A.; Xu, Q.; Qu, A. Hypoxia inducible factor as a therapeutic target for atherosclerosis. Pharmacol. Ther. 2018, 183, 22–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchida, L.; Tanaka, T.; Saito, H.; Sugahara, M.; Wakashima, T.; Fukui, K.; Nangaku, M. Effects of a prolyl hydroxylase inhibitor on kidney and cardiovascular complications in a rat model of chronic kidney disease. Am. J. Physiol. Renal Physiol. 2020, 318, F388–F401. [Google Scholar] [CrossRef] [PubMed]
- Ham, O.; Jin, W.; Lei, L.; Huang, H.H.; Tsuji, K.; Huang, M.; Roh, J.; Rosenzweig, A.; Lu, H.A.J. Pathological cardiac remodeling occurs early in CKD mice from unilateral urinary obstruction, and is attenuated by Enalapril. Sci. Rep. 2018, 8, 16087. [Google Scholar] [CrossRef]
- Ito, F.; Sono, Y.; Ito, T. Measurement and clinical significance of lipid peroxidation as a biomarker of oxidative stress: Oxidative stress in diabetes, atherosclerosis, and chronic inflammation. Antioxidants 2019, 8, 72. [Google Scholar] [CrossRef] [Green Version]
- Stanisavljevic, N.; Stojanovich, L.; Marisavljevic, D.; Djokovic, A.; Dopsaj, V.; Kotur-Stevuljevic, J.; Martinovic, J.; Memon, L.; Radovanovic, S.; Todic, B.; et al. Lipid peroxidation as risk factor for endothelial dysfunction in antiphospholipid syndrome patients. Clin. Rheumatol. 2016, 35, 2485–2493. [Google Scholar] [CrossRef]
- Ferroni, P.; Basili, S.; Paoletti, V.; Davi, G. Endothelial dysfunction and oxidative stress in arterial hypertension. Nutr. Metab. Cardiovasc. Dis. NMCD 2006, 16, 222–233. [Google Scholar] [CrossRef]
- Kim, J.A.; Montagnani, M.; Chandrasekran, S.; Quon, M.J. Role of lipotoxicity in endothelial dysfunction. Heart Fail. Clin. 2012, 8, 589–607. [Google Scholar] [CrossRef] [Green Version]
- Frijhoff, J.; Winyard, P.G.; Zarkovic, N.; Davies, S.S.; Stocker, R.; Cheng, D.; Knight, A.R.; Taylor, E.L.; Oettrich, J.; Ruskovska, T.; et al. Clinical relevance of biomarkers of oxidative stress. Antioxid. Redox Signal. 2015, 23, 1144–1170. [Google Scholar] [CrossRef] [Green Version]
- Cipak Gasparovic, A.; Zarkovic, N.; Zarkovic, K.; Semen, K.; Kaminskyy, D.; Yelisyeyeva, O.; Bottari, S.P. Biomarkers of oxidative and nitro-oxidative stress: Conventional and novel approaches. Br. J. Pharmacol. 2017, 174, 1771–1783. [Google Scholar] [CrossRef] [Green Version]
- Pereira, E.C.; Ferderbar, S.; Bertolami, M.C.; Faludi, A.A.; Monte, O.; Xavier, H.T.; Pereira, T.V.; Abdalla, D.S. Biomarkers of oxidative stress and endothelial dysfunction in glucose intolerance and diabetes mellitus. Clin. Biochem. 2008, 41, 1454–1460. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Harrison, D.G. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ. Res. 2000, 87, 840–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Wang, K.; Shang, J.; Cao, C.; Zhen, P.; Liu, X.; Wang, W.; Zhang, H.; Du, Y.; Liu, H. Anti-peroxynitrite treatment ameliorated vasorelaxation of resistance arteries in aging rats: Involvement with NO-sGC-cGKs pathway. PLoS ONE 2014, 9, e104788. [Google Scholar] [CrossRef] [PubMed]
- Ashfaq, S.; Abramson, J.L.; Jones, D.P.; Rhodes, S.D.; Weintraub, W.S.; Hooper, W.C.; Vaccarino, V.; Alexander, R.W.; Harrison, D.G.; Quyyumi, A.A. Endothelial function and aminothiol biomarkers of oxidative stress in healthy adults. Hypertension 2008, 52, 80–85. [Google Scholar] [CrossRef]
- Bradley, R.D.; Fitzpatrick, A.L.; Jacobs, D.R., Jr.; Lee, D.H.; Jenny, N.S.; Herrington, D. Associations between gamma-glutamyltransferase (GGT) and biomarkers of atherosclerosis: The Multi-ethnic Study of Atherosclerosis (MESA). Atherosclerosis 2014, 233, 387–393. [Google Scholar] [CrossRef] [Green Version]
- Ndrepepa, G.; Kastrati, A. Gamma-glutamyl transferase and cardiovascular disease. Ann. Transl. Med. 2016, 4, 481. [Google Scholar] [CrossRef]
- Maschirow, L.; Khalaf, K.; Al-Aubaidy, H.A.; Jelinek, H.F. Inflammation, coagulation, endothelial dysfunction and oxidative stress in prediabetes—Biomarkers as a possible tool for early disease detection for rural screening. Clin. Biochem. 2015, 48, 581–585. [Google Scholar] [CrossRef] [Green Version]
- Xuan, Y.; Gao, X.; Holleczek, B.; Brenner, H.; Schottker, B. Prediction of myocardial infarction, stroke and cardiovascular mortality with urinary biomarkers of oxidative stress: Results from a large cohort study. Int. J. Cardiol. 2018, 273, 223–229. [Google Scholar] [CrossRef]
- Arifin, R.; Kyi, W.M.; Che Yaakob, C.A.; Yaacob, N.M. Increased circulating oxidised low-density lipoprotein and antibodies to oxidised low-density lipoprotein in preeclampsia. J. Obstet. Gynaecol. J. Inst. Obstet. Gynaecol. 2017, 37, 580–584. [Google Scholar] [CrossRef]
- Jiang, Y.H.; Peng, C.H.; Liu, H.T.; Kuo, H.C. Increased pro-inflammatory cytokines, C-reactive protein and nerve growth factor expressions in serum of patients with interstitial cystitis/bladder pain syndrome. PLoS ONE 2013, 8, e76779. [Google Scholar] [CrossRef]
- Sproston, N.R.; El Mohtadi, M.; Slevin, M.; Gilmore, W.; Ashworth, J.J. The effect of C-reactive protein isoforms on nitric oxide production by U937 monocytes/macrophages. Front. Immunol. 2018, 9, 1500. [Google Scholar] [CrossRef] [PubMed]
- Aljwaid, H.; White, D.L.; Collard, K.J.; Moody, A.J.; Pinkney, J.H. Non-transferrin-bound iron is associated with biomarkers of oxidative stress, inflammation and endothelial dysfunction in type 2 diabetes. J. Diabetes Its Complicat. 2015, 29, 943–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onat, D.; Brillon, D.; Colombo, P.C.; Schmidt, A.M. Human vascular endothelial cells: A model system for studying vascular inflammation in diabetes and atherosclerosis. Curr. Diabetes Rep. 2011, 11, 193–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, C.Y.; Huang, P.H.; Chiang, C.H.; Leu, H.B.; Huang, C.C.; Chen, J.W.; Lin, S.J. Increased circulating endothelial apoptotic microparticle to endothelial progenitor cell ratio is associated with subsequent decline in glomerular filtration rate in hypertensive patients. PLoS ONE 2013, 8, e68644. [Google Scholar] [CrossRef] [Green Version]
- Gallardo-Vara, E.; Tual-Chalot, S.; Botella, L.M.; Arthur, H.M.; Bernabeu, C. Soluble endoglin regulates expression of angiogenesis-related proteins and induction of arteriovenous malformations in a mouse model of hereditary hemorrhagic telangiectasia. Dis. Models Mech. 2018, 11, dmm034397. [Google Scholar] [CrossRef] [Green Version]
- Nemeckova, I.; Serwadczak, A.; Oujo, B.; Jezkova, K.; Rathouska, J.; Fikrova, P.; Varejckova, M.; Bernabeu, C.; Lopez-Novoa, J.M.; Chlopicki, S.; et al. High soluble endoglin levels do not induce endothelial dysfunction in mouse aorta. PLoS ONE 2015, 10, e0119665. [Google Scholar] [CrossRef] [Green Version]
- Vitverova, B.; Blazickova, K.; Najmanova, I.; Vicen, M.; Hyspler, R.; Dolezelova, E.; Nemeckova, I.; Tebbens, J.D.; Bernabeu, C.; Pericacho, M.; et al. Soluble endoglin and hypercholesterolemia aggravate endothelial and vessel wall dysfunction in mouse aorta. Atherosclerosis 2018, 271, 15–25. [Google Scholar] [CrossRef]
- Blazquez-Medela, A.M.; Garcia-Ortiz, L.; Gomez-Marcos, M.A.; Recio-Rodriguez, J.I.; Sanchez-Rodriguez, A.; Lopez-Novoa, J.M.; Martinez-Salgado, C. Increased plasma soluble endoglin levels as an indicator of cardiovascular alterations in hypertensive and diabetic patients. BMC Med. 2010, 8, 86. [Google Scholar] [CrossRef] [Green Version]
- Berry, C.; Hamilton, C.A.; Brosnan, M.J.; Magill, F.G.; Berg, G.A.; McMurray, J.J.; Dominiczak, A.F. Investigation into the sources of superoxide in human blood vessels: Angiotensin II increases superoxide production in human internal mammary arteries. Circulation 2000, 101, 2206–2212. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Hong, Q.; Zhang, X.; Xiao, W.; Wang, L.; Cui, S.; Feng, Z.; Lv, Y.; Cai, G.; Chen, X.; et al. Aldose reductase mediates endothelial cell dysfunction induced by high uric acid concentrations. Cell Commun. Signal. CCS 2017, 15, 3. [Google Scholar] [CrossRef] [Green Version]
- Meitzler, J.L.; Antony, S.; Wu, Y.; Juhasz, A.; Liu, H.; Jiang, G.; Lu, J.; Roy, K.; Doroshow, J.H. NADPH oxidases: A perspective on reactive oxygen species production in tumor biology. Antioxid. Redox Signal. 2014, 20, 2873–2889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feig, D.I.; Kang, D.H.; Johnson, R.J. Uric acid and cardiovascular risk. N. Engl. J. Med. 2008, 359, 1811–1821. [Google Scholar] [CrossRef] [PubMed]
- Dawson, J.; Walters, M. Uric acid and xanthine oxidase: Future therapeutic targets in the prevention of cardiovascular disease? Br. J. Clin. Pharmacol. 2006, 62, 633–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higgins, P.; Dawson, J.; Walters, M. The potential for xanthine oxidase inhibition in the prevention and treatment of cardiovascular and cerebrovascular disease. Cardiovasc. Psychiatry Neurol. 2009, 2009, 282059. [Google Scholar] [CrossRef] [Green Version]
- Crotty, G.F.; Ascherio, A.; Schwarzschild, M.A. Targeting urate to reduce oxidative stress in Parkinson disease. Exp. Neurol. 2017, 298, 210–224. [Google Scholar] [CrossRef]
- Glantzounis, G.K.; Tsimoyiannis, E.C.; Kappas, A.M.; Galaris, D.A. Uric acid and oxidative stress. Curr. Pharm. Des. 2005, 11, 4145–4151. [Google Scholar] [CrossRef]
- Abi Khalil, C. The emerging role of epigenetics in cardiovascular disease. Ther. Adv. Chronic Dis. 2014, 5, 178–187. [Google Scholar] [CrossRef] [Green Version]
- Levy, E.; Spahis, S.; Bigras, J.L.; Delvin, E.; Borys, J.M. The epigenetic machinery in vascular dysfunction and hypertension. Curr. Hypertens. Rep. 2017, 19, 52. [Google Scholar] [CrossRef]
- Lorenzen, J.M.; Martino, F.; Thum, T. Epigenetic modifications in cardiovascular disease. Basic Res. Cardiol. 2012, 107, 245. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Gou, L.; Chen, L.; Zhong, X.; Zhang, D.; Zhu, H.; Lu, X.; Zeng, T.; Deng, X.; Li, Y. NADPH oxidase 4 and endothelial nitric oxide synthase contribute to endothelial dysfunction mediated by histone methylations in metabolic memory. Free Radic. Biol. Med. 2018, 115, 383–394. [Google Scholar] [CrossRef]
- Ounzain, S.; Crippa, S.; Pedrazzini, T. Small and long non-coding RNAs in cardiac homeostasis and regeneration. Biochim. Biophys. Acta 2013, 1833, 923–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carthew, R.W.; Sontheimer, E.J. Origins and Mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamaluddin, M.S.; Weakley, S.M.; Zhang, L.; Kougias, P.; Lin, P.H.; Yao, Q.; Chen, C. miRNAs: Roles and clinical applications in vascular disease. Expert Rev. Mol. Diagn. 2011, 11, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Improta Caria, A.C.; Nonaka, C.K.V.; Pereira, C.S.; Soares, M.B.P.; Macambira, S.G.; Souza, B.S.F. Exercise Training-Induced Changes in MicroRNAs: Beneficial Regulatory Effects in Hypertension, Type 2 Diabetes, and Obesity. Int. J. Mol. Sci. 2018, 19, 3608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladomery, M.R.; Maddocks, D.G.; Wilson, I.D. MicroRNAs: Their discovery, biogenesis, function and potential use as biomarkers in non-invasive prenatal diagnostics. Int. J. Mol. Epidemiol. Genet. 2011, 2, 253. [Google Scholar]
- Espinosa-Diez, C.; Wilson, R.; Chatterjee, N.; Hudson, C.; Ruhl, R.; Hipfinger, C.; Helms, E.; Khan, O.F.; Anderson, D.G.; Anand, S. MicroRNA regulation of the MRN complex impacts DNA damage, cellular senescence, and angiogenic signaling. Cell Death Dis. 2018, 9, 632. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Z.; Zhang, D.Y.; Zhu, J.; Zhang, T.; Wang, C. microRNA 126 inhibits the transition of endothelial progenitor cells to mesenchymal cells via the PIK3R2-PI3K/Akt signalling pathway. PLoS ONE 2013, 8, e83294. [Google Scholar] [CrossRef]
- Wang, S.; Aurora, A.B.; Johnson, B.A.; Qi, X.; McAnally, J.; Hill, J.A.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev. Cell 2008, 15, 261–271. [Google Scholar] [CrossRef] [Green Version]
- Harris, T.A.; Yamakuchi, M.; Ferlito, M.; Mendell, J.T.; Lowenstein, C.J. MicroRNA-126 regulates endothelial expression of vascular cell adhesion molecule 1. Proc. Natl. Acad. Sci. USA 2008, 105, 1516–1521. [Google Scholar] [CrossRef] [Green Version]
- Hua, Z.; Lv, Q.; Ye, W.; Wong, C.K.; Cai, G.; Gu, D.; Ji, Y.; Zhao, C.; Wang, J.; Yang, B.B.; et al. MiRNA-directed regulation of VEGF and other angiogenic factors under hypoxia. PLoS ONE 2006, 1, e116. [Google Scholar] [CrossRef] [Green Version]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Jia, C.; Wang, P.; Xiong, M.; Cui, J.; Li, L.; Wang, W.; Wu, Q.; Chen, Y.; Zhang, T. MicroRNA-505 identified from patients with essential hypertension impairs endothelial cell migration and tube formation. Int. J. Cardiol. 2014, 177, 925–934. [Google Scholar] [CrossRef]
- Palao, T.; Sward, K.; Jongejan, A.; Moerland, P.D.; de Vos, J.; van Weert, A.; Arribas, S.M.; Groma, G.; vanBavel, E.; Bakker, E.N. Gene expression and microrna expression analysis in small arteries of spontaneously hypertensive rats. Evidence for ER stress. PLoS ONE 2015, 10, e0137027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, C.A.; Carneiro, F.S.; Matsumoto, T.; Tostes, R.C. Bonus effects of antidiabetic drugs: Possible beneficial effects on endothelial dysfunction, vascular inflammation and atherosclerosis. Basic Clin. Pharmacol. Toxicol. 2018, 123, 523–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.X.; Zeng, D.Y.; Li, R.T.; Pang, R.P.; Yang, H.; Hu, Y.L.; Zhang, Q.; Jiang, Y.; Huang, L.Y.; Tang, Y.B.; et al. Essential role of microRNA-155 in regulating endothelium-dependent vasorelaxation by targeting endothelial nitric oxide synthase. Hypertension 2012, 60, 1407–1414. [Google Scholar] [CrossRef] [Green Version]
- Pankratz, F.; Bemtgen, X.; Zeiser, R.; Leonhardt, F.; Kreuzaler, S.; Hilgendorf, I.; Smolka, C.; Helbing, T.; Hoefer, I.; Esser, J.S.; et al. MicroRNA-155 exerts cell-specific antiangiogenic but proarteriogenic effects during adaptive neovascularization. Circulation 2015, 131, 1575–1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, X.; Wang, X.; Wang, Y.; Tang, Z.; Cui, Q.; Xi, J.; Li, Y.S.; Chien, S.; Wang, N. MicroRNA-19a mediates the suppressive effect of laminar flow on cyclin D1 expression in human umbilical vein endothelial cells. Proc. Natl. Acad. Sci. USA 2010, 107, 3240–3244. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Zhang, Y.C.; Chen, Y.; Xiang, Y.; Shen, C.X.; Li, Y.G. The role of miR-19b in the inhibition of endothelial cell apoptosis and its relationship with coronary artery disease. Sci. Rep. 2015, 5, 15132. [Google Scholar] [CrossRef]
- White, K.; Dempsie, Y.; Caruso, P.; Wallace, E.; McDonald, R.A.; Stevens, H.; Hatley, M.E.; Van Rooij, E.; Morrell, N.W.; MacLean, M.R.; et al. Endothelial apoptosis in pulmonary hypertension is controlled by a microRNA/programmed cell death 4/caspase-3 axis. Hypertension 2014, 64, 185–194. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Liang, H.; Liu, H.; Li, D.; Chen, X.; Li, L.; Zhang, C.Y.; Zen, K. Platelet-secreted microRNA-223 promotes endothelial cell apoptosis induced by advanced glycation end products via targeting the insulin-like growth factor 1 receptor. J. Immunol. (Baltimore Md. 1950) 2014, 192, 437–446. [Google Scholar] [CrossRef] [Green Version]
- Dickinson, B.A.; Semus, H.M.; Montgomery, R.L.; Stack, C.; Latimer, P.A.; Lewton, S.M.; Lynch, J.M.; Hullinger, T.G.; Seto, A.G.; van Rooij, E. Plasma microRNAs serve as biomarkers of therapeutic efficacy and disease progression in hypertension-induced heart failure. Eur. J. Heart Fail. 2013, 15, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Pacurari, M.; Tchounwou, P.B. Role of MicroRNAs in Renin-Angiotensin-Aldosterone System-Mediated Cardiovascular Inflammation and Remodeling. Int. J. Inflamm. 2015, 2015, 101527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalil, N.; O’Connor, R.N.; Unruh, H.W.; Warren, P.W.; Flanders, K.C.; Kemp, A.; Bereznay, O.H.; Greenberg, A.H. Increased production and immunohistochemical localization of transforming growth factor-b in idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 1991, 5, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Boettger, T.; Beetz, N.; Kostin, S.; Schneider, J.; Kruger, M.; Hein, L.; Braun, T. Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster. J. Clin. Investig. 2009, 119, 2634–2647. [Google Scholar] [CrossRef] [Green Version]
- Kemp, J.R.; Unal, H.; Desnoyer, R.; Yue, H.; Bhatnagar, A.; Karnik, S.S. Angiotensin II-regulated microRNA 483-3p directly targets multiple components of the renin-angiotensin system. J. Mol. Cell. Cardiol. 2014, 75, 25–39. [Google Scholar] [CrossRef] [Green Version]
- Ceolotto, G.; Papparella, I.; Bortoluzzi, A.; Strapazzon, G.; Ragazzo, F.; Bratti, P.; Fabricio, A.S.; Squarcina, E.; Gion, M.; Palatini, P.; et al. Interplay between miR-155, AT1R A1166C polymorphism, and AT1R expression in young untreated hypertensives. Am. J. Hypertens. 2011, 24, 241–246. [Google Scholar] [CrossRef]
- Wu, W.H.; Hu, C.P.; Chen, X.P.; Zhang, W.F.; Li, X.W.; Xiong, X.M.; Li, Y.J. MicroRNA-130a mediates proliferation of vascular smooth muscle cells in hypertension. Am. J. Hypertens. 2011, 24, 1087–1093. [Google Scholar] [CrossRef]
- Kawai-Kowase, K.; Owens, G.K. Multiple repressor pathways contribute to phenotypic switching of vascular smooth muscle cells. American journal of physiology. Cell Physiol. 2007, 292, C59–C69. [Google Scholar]
- Davis, B.N.; Hilyard, A.C.; Nguyen, P.H.; Lagna, G.; Hata, A. Induction of microRNA-221 by platelet-derived growth factor signaling is critical for modulation of vascular smooth muscle phenotype. J. Biol. Chem. 2009, 284, 3728–3738. [Google Scholar] [CrossRef] [Green Version]
- Mandraffino, G.; Imbalzano, E.; Sardo, M.A.; D’Ascola, A.; Mamone, F.; Lo Gullo, A.; Alibrandi, A.; Loddo, S.; Mormina, E.; David, A.; et al. Circulating progenitor cells in hypertensive patients with different degrees of cardiovascular involvement. J. Hum. Hypertens. 2014, 28, 543–550. [Google Scholar] [CrossRef]
- Kontaraki, J.E.; Marketou, M.E.; Zacharis, E.A.; Parthenakis, F.I.; Vardas, P.E. Differential expression of vascular smooth muscle-modulating microRNAs in human peripheral blood mononuclear cells: Novel targets in essential hypertension. J. Hum. Hypertens. 2014, 28, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Elia, L.; Quintavalle, M.; Zhang, J.; Contu, R.; Cossu, L.; Latronico, M.V.; Peterson, K.L.; Indolfi, C.; Catalucci, D.; Chen, J.; et al. The knockout of miR-143 and -145 alters smooth muscle cell maintenance and vascular homeostasis in mice: Correlates with human disease. Cell Death Differ. 2009, 16, 1590–1598. [Google Scholar] [CrossRef] [PubMed]
- Torella, D.; Iaconetti, C.; Catalucci, D.; Ellison, G.M.; Leone, A.; Waring, C.D.; Bochicchio, A.; Vicinanza, C.; Aquila, I.; Curcio, A.; et al. MicroRNA-133 controls vascular smooth muscle cell phenotypic switch in vitro and vascular remodeling in vivo. Circ. Res. 2011, 109, 880–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Zheng, C.; Ye, H.; Teng, Y.; Zheng, B.; Yang, X.; Zhang, J. MicroRNA-365 inhibits vascular smooth muscle cell proliferation through targeting cyclin D1. Int. J. Med Sci. 2014, 11, 765. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Zhang, C. MicroRNA-21 in cardiovascular disease. J. Cardiovasc. Transl. Res. 2010, 3, 251–255. [Google Scholar] [CrossRef] [Green Version]
- Vegter, E.L.; Ovchinnikova, E.S.; van Veldhuisen, D.J.; Jaarsma, T.; Berezikov, E.; van der Meer, P.; Voors, A.A. Low circulating microRNA levels in heart failure patients are associated with atherosclerotic disease and cardiovascular-related rehospitalizations. Clin. Res. Cardiol. Off. J. Ger. Card. Soc. 2017, 106, 598–609. [Google Scholar] [CrossRef] [Green Version]
- Schober, A.; Nazari-Jahantigh, M.; Wei, Y.; Bidzhekov, K.; Gremse, F.; Grommes, J.; Megens, R.T.; Heyll, K.; Noels, H.; Hristov, M.; et al. MicroRNA-126-5p promotes endothelial proliferation and limits atherosclerosis by suppressing Dlk1. Nat. Med. 2014, 20, 368–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Kim, C.W.; Simmons, R.D.; Jo, H. Role of flow-sensitive microRNAs in endothelial dysfunction and atherosclerosis: Mechanosensitive athero-miRs. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2206–2216. [Google Scholar] [CrossRef] [Green Version]
- Chamorro-Jorganes, A.; Araldi, E.; Suarez, Y. MicroRNAs as pharmacological targets in endothelial cell function and dysfunction. Pharmacol. Res. 2013, 75, 15–27. [Google Scholar] [CrossRef] [Green Version]
- Orlandi, A.; Bochaton-Piallat, M.L.; Gabbiani, G.; Spagnoli, L.G. Aging, smooth muscle cells and vascular pathobiology: Implications for atherosclerosis. Atherosclerosis 2006, 188, 221–230. [Google Scholar] [CrossRef]
- Kiss, T.; Giles, C.B.; Tarantini, S.; Yabluchanskiy, A.; Balasubramanian, P.; Gautam, T.; Csipo, T.; Nyúl-Tóth, Á.; Lipecz, A.; Szabo, C.; et al. Nicotinamide mononucleotide (NMN) supplementation promotes anti-aging miRNA expression profile in the aorta of aged mice, predicting epigenetic rejuvenation and anti-atherogenic effects. Geroscience 2019, 41, 419–439. [Google Scholar] [CrossRef]
- Menghini, R.; Casagrande, V.; Cardellini, M.; Martelli, E.; Terrinoni, A.; Amati, F.; Vasa-Nicotera, M.; Ippoliti, A.; Novelli, G.; Melino, G.; et al. MicroRNA 217 modulates endothelial cell senescence via silent information regulator 1. Circulation 2009, 120, 1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasa-Nicotera, M.; Chen, H.; Tucci, P.; Yang, A.L.; Saintigny, G.; Menghini, R.; Mahe, C.; Agostini, M.; Knight, R.A.; Melino, G.; et al. miR-146a is modulated in human endothelial cell with aging. Atherosclerosis 2011, 217, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Guduric-Fuchs, J.; O’Connor, A.; Cullen, A.; Harwood, L.; Medina, R.J.; O’Neill, C.L.; Stitt, A.W.; Curtis, T.M.; Simpson, D.A. Deep sequencing reveals predominant expression of miR-21 amongst the small non-coding RNAs in retinal microvascular endothelial cells. J. Cell. Biochem. 2012, 113, 2098–2111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Yang, P.; Xiong, Q.; Song, X.; Yang, X.; Liu, L.; Yuan, W.; Rui, Y.C. MicroRNA-125a/b-5p inhibits endothelin-1 expression in vascular endothelial cells. J. Hypertens. 2010, 28, 1646–1654. [Google Scholar] [CrossRef] [PubMed]
- Tahamtan, A.; Teymoori-Rad, M.; Nakstad, B.; Salimi, V. Anti-inflammatory MicroRNAs and their potential for inflammatory diseases treatment. Front. Immunol. 2018, 9, 1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valastyan, S.; Weinberg, R.A. Roles for microRNAs in the regulation of cell adhesion molecules. J. Cell Sci. 2011, 124, 999–1006. [Google Scholar] [CrossRef] [Green Version]
- Bueno, M.J.; Malumbres, M. MicroRNAs and the cell cycle. Biochim. Biophys. Acta 2011, 1812, 592–601. [Google Scholar] [CrossRef] [Green Version]
- Hwang, H.W.; Mendell, J.T. MicroRNAs in cell proliferation, cell death, and tumorigenesis. Br. J. Cancer 2006, 94, 776–780. [Google Scholar] [CrossRef]
- Su, Z.; Yang, Z.; Xu, Y.; Chen, Y.; Yu, Q. MicroRNAs in apoptosis, autophagy and necroptosis. Oncotarget 2015, 6, 8474. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Williams, D.; Sur, S.; Wang, J.Y.; Jo, H. Role of flow-sensitive microRNAs and long noncoding RNAs in vascular dysfunction and atherosclerosis. Vasc. Pharmacol. 2019, 114, 76–92. [Google Scholar] [CrossRef] [PubMed]
- Nigi, L.; Grieco, G.E.; Ventriglia, G.; Brusco, N.; Mancarella, F.; Formichi, C.; Dotta, F.; Sebastiani, G. MicroRNAs as Regulators of Insulin Signaling: Research Updates and Potential Therapeutic Perspectives in Type 2 Diabetes. Int. J. Mol. Sci. 2018, 19, 3705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barutta, F.; Bruno, G.; Matullo, G.; Chaturvedi, N.; Grimaldi, S.; Schalkwijk, C.; Stehouwer, C.D.; Fuller, J.H.; Gruden, G. MicroRNA-126 and micro-/macrovascular complications of type 1 diabetes in the EURODIAB Prospective Complications Study. Acta Diabetol. 2017, 54, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Zampetaki, A.; Kiechl, S.; Drozdov, I.; Willeit, P.; Mayr, U.; Prokopi, M.; Mayr, A.; Weger, S.; Oberhollenzer, F.; Bonora, E.; et al. Plasma microRNA profiling reveals loss of endothelial miR-126 and other microRNAs in type 2 diabetes. Circ. Res. 2010, 107, 810–817. [Google Scholar] [CrossRef]
- Fichtlscherer, S.; De Rosa, S.; Fox, H.; Schwietz, T.; Fischer, A.; Liebetrau, C.; Weber, M.; Hamm, C.W.; Roxe, T.; Muller-Ardogan, M.; et al. Circulating microRNAs in patients with coronary artery disease. Circ. Res. 2010, 107, 677–684. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.F.; Yang, L.X.; Guo, R.W.; Liu, H.; Shi, Y.K.; Ye, J.S.; Yang, Z.H. microRNA-155 is inversely associated with severity of coronary stenotic lesions calculated by the Gensini score. Coron. Artery Dis. 2014, 25, 304–310. [Google Scholar] [CrossRef]
- Luo, L.; Chen, B.; Li, S.; Wei, X.; Liu, T.; Huang, Y.; Lin, X. Plasma miR-10a: A Potential Biomarker for Coronary Artery Disease. Dis. Markers 2016, 2016, 3841927. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Lin, J.; Zhang, Y.; Kang, S.; Belkin, N.; Wara, A.K.; Icli, B.; Hamburg, N.M.; Li, D.; Feinberg, M.W. MicroRNA-181b Improves Glucose Homeostasis and Insulin Sensitivity by Regulating Endothelial Function in White Adipose Tissue. Circ. Res. 2016, 118, 810–821. [Google Scholar] [CrossRef] [Green Version]
- Njock, M.S.; Fish, J.E. Endothelial miRNAs as Cellular Messengers in Cardiometabolic Diseases. Trends Endocrinol. Metab. 2017, 28, 237–246. [Google Scholar] [CrossRef]
- Romaine, S.P.; Tomaszewski, M.; Condorelli, G.; Samani, N.J. MicroRNAs in cardiovascular disease: An introduction for clinicians. Heart (Br. Card. Soc.) 2015, 101, 921–928. [Google Scholar] [CrossRef]
- Gomes, C.P.C.; Spencer, H.; Ford, K.L.; Michel, L.Y.M.; Baker, A.H.; Emanueli, C.; Balligand, J.L.; Devaux, Y. The Function and Therapeutic Potential of Long Non-coding RNAs in Cardiovascular Development and Disease. Molecular therapy. Nucleic Acids 2017, 8, 494–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Z.Q.; Xu, J.; Li, L.; Han, Y.S. Down-regulation of SENCR promotes smooth muscle cells proliferation and migration in db/db mice through up-regulation of FoxO1 and TRPC6. Biomed. Pharmacother. 2015, 74, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Boulberdaa, M.; Scott, E.; Ballantyne, M.; Garcia, R.; Descamps, B.; Angelini, G.D.; Brittan, M.; Hunter, A.; McBride, M.; McClure, J.; et al. A role for the long noncoding RNA SENCR in commitment and function of endothelial cells. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 978–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, J.X. LncRNA H19 promotes atherosclerosis by regulating MAPK and NF-kB signaling pathway. Eur. Rev. Med Pharmacol. Sci. 2017, 21, 322–328. [Google Scholar] [PubMed]
- Tang, Z.; Zhang, J.; Lu, X.; Wang, W.; Chen, H.; Robinson, M.K.; Cheng, J.; Tang, G.; Medeiros, L.J. Coexistent genetic alterations involving ALK, RET, ROS1 or MET in 15 cases of lung adenocarcinoma. Mod. Pathol. 2018, 31, 307–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; Meng, K.; Jiang, L.; Zhong, Y.; Yang, Y.; Lan, Y.; Zeng, Q.; Cheng, L. Thymic stromal lymphopoietin-induced HOTAIR activation promotes endothelial cell proliferation and migration in atherosclerosis. Biosci. Rep. 2017, 37, BSR20170351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haemmig, S.; Simion, V.; Yang, D.; Deng, Y.; Feinberg, M.W. Long noncoding RNAs in cardiovascular disease, diagnosis, and therapy. Curr. Opin. Cardiol. 2017, 32, 776. [Google Scholar] [CrossRef]
- Mrakic-Sposta, S.; Di Santo, S.G.; Franchini, F.; Arlati, S.; Zangiacomi, A.; Greci, L.; Moretti, S.; Jesuthasan, N.; Marzorati, M.; Rizzo, G.; et al. Effects of Combined Physical and Cognitive Virtual Reality-Based Training on Cognitive Impairment and Oxidative Stress in MCI Patients: A Pilot Study. Front. Aging Neurosci. 2018, 10, 282. [Google Scholar] [CrossRef] [Green Version]
- Rotolo, O.; Zinzi, I.; Veronese, N.; Cisternino, A.M.; Reddavide, R.; Inguaggiato, R.; Leandro, G.; Notarnicola, M.; Tutino, V.; De Nunzio, V.; et al. Women in LOVe: Lacto-Ovo-Vegetarian Diet Rich in Omega-3 Improves Vasomotor Symptoms in Postmenopausal Women. An Exploratory Randomized Controlled Trial. Endocr. Metab. Immune. Disord. Drug Targets 2019, 19, 1232–1239. [Google Scholar] [CrossRef]
- Hunter, D.J.; James, L.; Hussey, B.; Wadley, A.J.; Lindley, M.R.; Mastana, S.S. Impact of aerobic exercise and fatty acid supplementation on global and gene-specific DNA methylation. Epigenetics 2019, 14, 294–309. [Google Scholar] [CrossRef] [Green Version]
- Venturini, D.; Simão, A.N.; Urbano, M.R.; Dichi, I. Effects of extra virgin olive oil and fish oil on lipid profile and oxidative stress in patients with metabolic syndrome. Nutrition 2015, 31, 834–840. [Google Scholar] [CrossRef] [PubMed]
- González-Hedström, D.; Amor, S.; de la Fuente-Fernández, M.; Tejera-Muñoz, A.; Priego, T.; Martín, A.I.; López-Calderón, A.; Inarejos-García, A.M.; García-Villalón, Á.L.; Granado, M. A Mixture of Algae and Extra Virgin Olive Oils Attenuates the Cardiometabolic Alterations Associated with Aging in Male Wistar Rats. Antioxidants 2020, 9, 483. [Google Scholar]
- Testai, L.; Piragine, E.; Piano, I.; Flori, L.; Da Pozzo, E.; Miragliotta, V.; Pirone, A.; Citi, V.; Di Cesare Mannelli, L.; Brogi, S.; et al. The Citrus Flavonoid Naringenin Protects the Myocardium from Ageing-Dependent Dysfunction: Potential Role of SIRT1. Oxidative Med. Cell. Longev. 2020, 2020, 4650207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Randriamboavonjy, J.I.; Heurtebise, S.; Pacaud, P.; Loirand, G.; Tesse, A. Moringa oleifera Seeds Improve Aging-Related Endothelial Dysfunction in Wistar Rats. Oxidative Med. Cell. Longev. 2019, 2019, 2567198. [Google Scholar] [CrossRef]
- de Picciotto, N.E.; Gano, L.B.; Johnson, L.C.; Martens, C.R.; Sindler, A.L.; Mills, K.F.; Imai, S.; Seals, D.R. Nicotinamide mononucleotide supplementation reverses vascular dysfunction and oxidative stress with aging in mice. Aging Cell. 2016, 15, 522–530. [Google Scholar] [CrossRef]
- Chauvierre, C.; Letourneur, D. The European project NanoAthero to fight cardiovascular diseases using nanotechnologies. Nanomedicine 2015, 10, 3391–3400. [Google Scholar] [CrossRef]
- Rhee, J.W.; Wu, J.C. Advances in nanotechnology for the management of coronary artery disease. Trends Cardiovasc. Med. 2013, 23, 39–45. [Google Scholar] [CrossRef] [Green Version]
- Broz, P.; Ben-Haim, N.; Grzelakowski, M.; Marsch, S.; Meier, W.; Hunziker, P. Inhibition of macrophage phagocytotic activity by a receptor-targeted polymer vesicle-based drug delivery formulation of pravastatin. J. Cardiovasc. Pharmacol. 2008, 51, 246–252. [Google Scholar] [CrossRef] [Green Version]
- Leuschner, F.; Dutta, P.; Gorbatov, R.; Novobrantseva, T.I.; Donahoe, J.S.; Courties, G.; Lee, K.M.; Kim, J.I.; Markmann, J.F.; Marinelli, B.; et al. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat. Biotechnol. 2011, 29, 1005–1010. [Google Scholar] [CrossRef]
- Blow, N. Small RNAs: Delivering the future. Nature 2007, 450, 1119–1120. [Google Scholar] [CrossRef]
- Lobatto, M.E.; Fayad, Z.A.; Silvera, S.; Vucic, E.; Calcagno, C.; Mani, V.; Dickson, S.D.; Nicolay, K.; Banciu, M.; Schiffelers, R.M.; et al. Multimodal clinical imaging to longitudinally assess a nanomedical anti-inflammatory treatment in experimental atherosclerosis. Mol. Pharm. 2010, 7, 2020–2029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, J.R.; Korngold, E.; Weissleder, R.; Jaffer, F.A. A light-activated theranostic nanoagent for targeted macrophage ablation in inflammatory atherosclerosis. Small (Weinh. Der Bergstr. Ger.) 2010, 6, 2041–2049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tassa, C.; Shaw, S.Y.; Weissleder, R. Dextran-coated iron oxide nanoparticles: A versatile platform for targeted molecular imaging, molecular diagnostics, and therapy. Acc. Chem. Res. 2011, 44, 842–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bharadwaj, A.S.; Appukuttan, B.; Wilmarth, P.A.; Pan, Y.; Stempel, A.J.; Chipps, T.J.; Benedetti, E.E.; Zamora, D.O.; Choi, D.; David, L.L.; et al. Role of the retinal vascular endothelial cell in ocular disease. Prog. Retin. Eye Res. 2013, 32, 102–180. [Google Scholar] [CrossRef] [Green Version]
- Mudau, M.; Genis, A.; Lochner, A.; Strijdom, H. Endothelial dysfunction: The early predictor of atherosclerosis. Cardiovasc. J. Afr. 2012, 23, 222. [Google Scholar] [CrossRef]
- Pacher, P.; Mabley, J.G.; Soriano, F.G.; Liaudet, L.; Komjati, K.; Szabo, C. Endothelial dysfunction in aging animals: The role of poly(ADP-ribose) polymerase activation. Br. J. Pharmacol. 2002, 135, 1347–1350. [Google Scholar] [CrossRef] [Green Version]
- Radovits, T.; Seres, L.; Gero, D.; Berger, I.; Szabo, C.; Karck, M.; Szabo, G. Single dose treatment with PARP-inhibitor INO-1001 improves aging-associated cardiac and vascular dysfunction. Exp. Gerontol. 2007, 42, 676–685. [Google Scholar] [CrossRef] [Green Version]
- Jagtap, P.; Szabo, C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat. Rev. Drug Discov. 2005, 4, 421–440. [Google Scholar] [CrossRef]
- Ferraris, D.V. Evolution of poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors. From concept to clinic. J. Med. Chem. 2010, 53, 4561–4584. [Google Scholar] [CrossRef]
- Ballak, D.B.; Brunt, V.E.; Sapinsley, Z.J.; Ziemba, B.P.; Richey, J.J.; Zigler, M.C.; Johnson, L.C.; Gioscia-Ryan, R.A.; Culp-Hill, R.; Eisenmesser, E.Z.; et al. Short-term interleukin-37 treatment improves vascular endothelial function, endurance exercise capacity, and whole-body glucose metabolism in old mice. Aging Cell 2020, 19, e13074. [Google Scholar] [CrossRef] [Green Version]
- Demirtaş Şahin, T.; Yazir, Y.; Utkan, T.; Gacar, G.; Furat Rençber, S.; Gocmez, S.S. TNF-α antagonism with etanercept enhances penile NOS expression, cavernosal reactivity, and testosterone levels in aged rats. Can. J. Physiol. Pharmacol. 2018, 96, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Idigo, W.O.; Reilly, S.; Zhang, M.H.; Zhang, Y.H.; Jayaram, R.; Carnicer, R.; Crabtree, M.J.; Balligand, J.L.; Casadei, B. Regulation of endothelial nitric-oxide synthase (NOS) S-glutathionylation by neuronal NOS: Evidence of a functional interaction between myocardial constitutive NOS isoforms. J. Biol. Chem. 2012, 287, 43665–43673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, D.I.; Singh, M. Effect of demethylasterriquinone b1 in hypertension associated vascular endothelial dysfunction. Int. J. Cardiol. 2007, 120, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.I.; Singh, M. Possible role of Akt to improve vascular endothelial dysfunction in diabetic and hyperhomocysteinemic rats. Mol. Cell. Biochem. 2007, 295, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.I.; Singh, M. Inhibition of protein tyrosin phosphatase improves vascular endothelial dysfunction. Vasc. Pharmacol. 2006, 44, 177–182. [Google Scholar] [CrossRef]
- Willsky, G.R.; Goldfine, A.B.; Kostyniak, P.J.; McNeill, J.H.; Yang, L.Q.; Khan, H.R.; Crans, D.C. Effect of vanadium(IV) compounds in the treatment of diabetes: In vivo and in vitro studies with vanadyl sulfate and bis(maltolato)oxovandium(IV). J. Inorg. Biochem. 2001, 85, 33–42. [Google Scholar] [CrossRef]
- Okumura, N.; Kinoshita, S.; Koizumi, N. Application of Rho Kinase Inhibitors for the Treatment of Corneal Endothelial Diseases. J. Ophthalmol. 2017, 2017, 2646904. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, S.; Koizumi, N.; Ueno, M.; Okumura, N.; Imai, K.; Tanaka, H.; Yamamoto, Y.; Nakamura, T.; Inatomi, T.; Bush, J.; et al. Injection of Cultured Cells with a ROCK Inhibitor for Bullous Keratopathy. N. Engl. J. Med. 2018, 378, 995–1003. [Google Scholar] [CrossRef]
- Chu, U.B.; Duellman, T.; Weaver, S.J.; Tao, Y.; Yang, J. Endothelial protective genes induced by statin are mimicked by ERK5 activation as triggered by a drug combination of FTI-277 and GGTI-298. Biochim. Biophys. Acta 2015, 1850, 1415–1425. [Google Scholar] [CrossRef] [Green Version]
- Stirban, A.; Gawlowski, T.; Roden, M. Vascular effects of advanced glycation endproducts: Clinical effects and molecular mechanisms. Mol. Metab. 2014, 3, 94–108. [Google Scholar] [CrossRef]
- Stirban, A.; Negrean, M.; Stratmann, B.; Gawlowski, T.; Horstmann, T.; Gotting, C.; Kleesiek, K.; Mueller-Roesel, M.; Koschinsky, T.; Uribarri, J.; et al. Benfotiamine prevents macro- and microvascular endothelial dysfunction and oxidative stress following a meal rich in advanced glycation end products in individuals with type 2 diabetes. Diabetes Care 2006, 29, 2064–2071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Bernatchez, P.N.; de Haan, J.B. Targeting Endothelial Dysfunction in Vascular Complications Associated With Diabetes. Int. J. Vasc. Med. 2012, 2012, 750126. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; Smith, R.A. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 629–656. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.; Adlam, V.J.; Blaikie, F.H.; Manas, A.R.; Porteous, C.M.; James, A.M.; Ross, M.F.; Logan, A.; Cocheme, H.M.; Trnka, J.; et al. Mitochondria-targeted antioxidants in the treatment of disease. Ann. N. Y. Acad. Sci. 2008, 1147, 105–111. [Google Scholar] [CrossRef]
- Gioscia-Ryan, R.A.; Battson, M.L.; Cuevas, L.M.; Eng, J.S.; Murphy, M.P.; Seals, D.R. Mitochondria-targeted Antioxidant Therapy With MitoQ Ameliorates Aortic Stiffening in Old Mice. J. Appl. Physiol. (1985) 2018, 124, 1194–1202. [Google Scholar] [CrossRef]
- Chatfield, K.C.; Sparagna, G.C.; Sucharov, C.C.; Miyamoto, S.D.; Grudis, J.E.; Sobus, R.D.; Hijmans, J.; Stauffer, B.L. Dysregulation of cardiolipin biosynthesis in pediatric heart failure. J. Mol. Cell. Cardiol. 2014, 74, 251–259. [Google Scholar] [CrossRef] [Green Version]
- Sparagna, G.C.; Chicco, A.J.; Murphy, R.C.; Bristow, M.R.; Johnson, C.A.; Rees, M.L.; Maxey, M.L.; McCune, S.A.; Moore, R.L. Loss of cardiac tetralinoleoyl cardiolipin in human and experimental heart failure. J. Lipid Res. 2007, 48, 1559–1570. [Google Scholar] [CrossRef] [Green Version]
- Sabbah, H.N.; Gupta, R.C.; Singh-Gupta, V.; Zhang, K.; Lanfear, D.E. Abnormalities of Mitochondrial Dynamics in the Failing Heart: Normalization Following Long-Term Therapy with Elamipretide. Cardiovasc. Drugs Ther. 2018, 32, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Bione, S.; D’Adamo, P.; Maestrini, E.; Gedeon, A.K.; Bolhuis, P.A.; Toniolo, D. A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat. Genet. 1996, 12, 385–389. [Google Scholar] [CrossRef]
- Petrosillo, G.; Matera, M.; Moro, N.; Ruggiero, F.M.; Paradies, G. Mitochondrial complex I dysfunction in rat heart with aging: Critical role of reactive oxygen species and cardiolipin. Free Radic. Biol. Med. 2009, 46, 88–94. [Google Scholar] [CrossRef]
- Petrosillo, G.; Matera, M.; Casanova, G.; Ruggiero, F.M.; Paradies, G. Mitochondrial dysfunction in rat brain with aging Involvement of complex I, reactive oxygen species and cardiolipin. Neurochem. Int. 2008, 53, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhao, H.; Cai, J.; Li, H.; Wu, Q.; Qiao, T.; Li, K. Chronic administration of mitochondrion-targeted peptide SS-31 prevents atherosclerotic development in ApoE knockout mice fed Western diet. PLoS ONE 2017, 12, e0185688. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.; Itani, H.; Richmond, B.; Vergeade, A.; Rahman, S.M.J.; Boutaud, O.; Blackwell, T.; Massion, P.P.; Harrison, D.G.; Dikalova, A. Tobacco smoking induces cardiovascular mitochondrial oxidative stress, promotes endothelial dysfunction, and enhances hypertension. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H639–H646. [Google Scholar] [CrossRef] [PubMed]
- Joshi, M.; Kotha, S.R.; Malireddy, S.; Selvaraju, V.; Satoskar, A.R.; Palesty, A.; McFadden, D.W.; Parinandi, N.L.; Maulik, N. Conundrum of pathogenesis of diabetic cardiomyopathy: Role of vascular endothelial dysfunction, reactive oxygen species, and mitochondria. Mol. Cell. Biochem. 2014, 386, 233–249. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Soong, Y.; Seshan, S.V.; Szeto, H.H. Novel cardiolipin therapeutic protects endothelial mitochondria during renal ischemia and mitigates microvascular rarefaction, inflammation, and fibrosis. Am. J. Physiol. Renal Physiol. 2014, 306, F970–F980. [Google Scholar] [CrossRef] [Green Version]
- Eirin, A.; Ebrahimi, B.; Kwon, S.H.; Fiala, J.A.; Williams, B.J.; Woollard, J.R.; He, Q.; Gupta, R.C.; Sabbah, H.N.; Prakash, Y.S.; et al. Restoration of Mitochondrial Cardiolipin Attenuates Cardiac Damage in Swine Renovascular Hypertension. J. Am. Heart Assoc. 2016, 5, e003118. [Google Scholar] [CrossRef] [Green Version]
- Zhao, K.; Luo, G.; Zhao, G.M.; Schiller, P.W.; Szeto, H.H. Transcellular transport of a highly polar 3+ net charge opioid tetrapeptide. J. Pharmacol. Exp. Ther. 2003, 304, 425–432. [Google Scholar] [CrossRef] [Green Version]
- Kloner, R.A.; Hale, S.L.; Dai, W.; Gorman, R.C.; Shuto, T.; Koomalsingh, K.J.; Gorman, J.H.; 3rd Sloan, R.C.; Frasier, C.R.; Watson, C.A.; et al. Reduction of ischemia/reperfusion injury with bendavia, a mitochondria-targeting cytoprotective Peptide. J. Am. Heart Assoc. 2012, 1, e001644. [Google Scholar] [CrossRef] [Green Version]
- Yuan, F.; Hedayat, A.F.; Ferguson, C.M.; Lerman, A.; Lerman, L.O.; Eirin, A. Mitoprotection attenuates myocardial vascular impairment in porcine metabolic syndrome. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H669–H680. [Google Scholar] [CrossRef]
- Sabbah, H.N.; Gupta, R.C.; Kohli, S.; Wang, M.; Hachem, S.; Zhang, K. Chronic Therapy With Elamipretide (MTP-131), a Novel Mitochondria-Targeting Peptide, Improves Left Ventricular and Mitochondrial Function in Dogs With Advanced Heart Failure. Circulation. Heart Fail. 2016, 9, e002206. [Google Scholar] [CrossRef] [Green Version]
- Eirin, A.; Ebrahimi, B.; Zhang, X.; Zhu, X.Y.; Woollard, J.R.; He, Q.; Textor, S.C.; Lerman, A.; Lerman, L.O. Mitochondrial protection restores renal function in swine atherosclerotic renovascular disease. Cardiovasc. Res. 2014, 103, 461–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eirin, A.; Hedayat, A.F.; Ferguson, C.M.; Textor, S.C.; Lerman, A.; Lerman, L.O. Mitoprotection preserves the renal vasculature in porcine metabolic syndrome. Exp. Physiol. 2018, 103, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Gibson, C.M.; Giugliano, R.P.; Kloner, R.A.; Bode, C.; Tendera, M.; Janosi, A.; Merkely, B.; Godlewski, J.; Halaby, R.; Korjian, S.; et al. EMBRACE STEMI study: A Phase 2a trial to evaluate the safety, tolerability, and efficacy of intravenous MTP-131 on reperfusion injury in patients undergoing primary percutaneous coronary intervention. Eur. Heart J. 2016, 37, 1296–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daubert, M.A.; Yow, E.; Dunn, G.; Marchev, S.; Barnhart, H.; Douglas, P.S.; O’Connor, C.; Goldstein, S.; Udelson, J.E.; Sabbah, H.N. Novel Mitochondria-Targeting Peptide in Heart Failure Treatment: A Randomized, Placebo-Controlled Trial of Elamipretide. Circ. Heart Fail. 2017, 10, e004389. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.; Khan, M.S.; Anker, S.D.; Fonarow, G.C.; Kim, R.J.; Nodari, S.; O’Connor, C.M.; Pieske, B.; Pieske-Kraigher, E.; Sabbah, H.N.; et al. Effects of Elamipretide on Left Ventricular Function in Patients with Heart Failure With Reduced Ejection Fraction: The PROGRESS-HF Phase 2 Trial. J. Card. Fail. 2020, 26, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Chew, P.; Yuen, D.Y.; Koh, P.; Stefanovic, N.; Febbraio, M.A.; Kola, I.; Cooper, M.E.; de Haan, J.B. Site-specific Antiatherogenic Effect of the Antioxidant Ebselen in the Diabetic Apolipoprotein E-deficient Mouse. Arter. Thromb. Vasc. Biol. 2009, 29, 823–830. [Google Scholar] [CrossRef] [Green Version]
- Hort, M.A.; Straliotto, M.R.; Netto, P.M.; da Rocha, J.B.; de Bem, A.F.; Ribeiro-do-Valle, R.M. Diphenyl Diselenide Effectively Reduces Atherosclerotic Lesions in LDLr -/- Mice by Attenuation of Oxidative Stress and Inflammation. J. Cardiovasc. Pharmacol. 2011, 58, 91–101. [Google Scholar] [CrossRef]
- Hilgers, R.H.; Kundumani-Sridharan, V.; Subramani, J.; Chen, L.C.; Cuello, L.G.; Rusch, N.J.; Das, K.C. Thioredoxin reverses age-related hypertension by chronically improving vascular redox and restoring eNOS function. Sci. Transl. Med. 2017, 9, eaaf6094. [Google Scholar] [CrossRef] [Green Version]
- Hemling, P.; Zibrova, D.; Strutz, J.; Sohrabi, Y.; Desoye, G.; Schulten, H.; Findeisen, H.; Heller, R.; Godfrey, R.; Waltenberger, J. Hyperglycemia-induced endothelial dysfunction is alleviated by thioredoxin mimetic peptides through the restoration of VEGFR-2-induced responses and improved cell survival. Int. J. Cardiol. 2020, 308, 73–81. [Google Scholar] [CrossRef]
- Chen, H.Z.; Wan, Y.Z.; Liu, D.P. Cross-talk between SIRT1 and p66Shc in vascular diseases. Trends Cardiovasc. Med. 2013, 23, 237–241. [Google Scholar] [CrossRef]
- Wang, L.; Ge, H.; Peng, L.; Wang, B. A meta-analysis of the relationship between VEGFR2 polymorphisms and atherosclerotic cardiovascular diseases. Clin. Cardiol. 2019, 42, 860–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kivelä, R.; Hemanthakumar, K.A.; Vaparanta, K.; Robciuc, M.; Izumiya, Y.; Kidoya, H.; Takakura, N.; Peng, X.; Sawyer, D.B.; Elenius, K.; et al. Endothelial cells regulate physiological cardiomyocyte growth via VEGFR2-mediated paracrine signaling. Circulation 2019, 139, 2570–2584. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Jiang, L. Oxidized Low-Density Lipoprotein Decreases VEGFR2 Expression in HUVECs and Impairs Angiogenesis. Exp. Ther. Med. 2016, 12, 3742–3748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edirisinghe, I.; Rahman, I. Cigarette smoke-mediated oxidative stress, shear stress, and endothelial dysfunction: Role of VEGFR2. Ann. N. Y. Acad. Sci. 2010, 1203, 66–72. [Google Scholar] [CrossRef]
- Edirisinghe, I.; Yang, S.R.; Yao, H.; Rajendrasozhan, S.; Caito, S.; Adenuga, D.; Wong, C.; Rahman, A.; Phipps, R.P.; Jin, Z.G.; et al. VEGFR-2 inhibition augments cigarette smoke-induced oxidative stress and inflammatory responses leading to endothelial dysfunction. FASEB J. 2008, 22, 2297–2310. [Google Scholar] [CrossRef] [Green Version]
- Warren, C.M.; Ziyad, S.; Briot, A.; Der, A.; Iruela-Arispe, M.L. A ligand-independent VEGFR2 signaling pathway limits angiogenic responses in diabetes. Sci. Signal. 2014, 7, ra1. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.J.; Tian, D.C.; Wang, F.W.; Zhang, M.H.; Fan, C.D.; Chen, W.; Wang, M.H.; Fu, X.Y.; Ma, J.K. Astaxanthin inhibits homocysteine-induced endothelial cell dysfunction via the regulation of the reactive oxygen species-dependent VEGF-VEGFR2-FAK signaling pathway. Mol. Med. Rep. 2019, 19, 4753–4760. [Google Scholar] [CrossRef] [Green Version]
- Santilli, F.; D’Ardes, D.; Davi, G. Oxidative stress in chronic vascular disease: From prediction to prevention. Vasc. Pharmacol. 2015, 74, 23–37. [Google Scholar] [CrossRef]
- Lu, W.J.; Li, J.Y.; Chen, R.J.; Huang, L.T.; Lee, T.Y.; Lin, K.H. VAS2870 and VAS3947 attenuate platelet activation and thrombus formation via a NOX-independent pathway downstream of PKC. Sci. Rep. 2019, 9, 18852. [Google Scholar] [CrossRef]
- El Assar, M.; Angulo, J.; Santos-Ruiz, M.; Moreno, P.; Novials, A.; Villanueva-Peñacarrillo, M.L.; Rodríguez-Mañas, L. Differential effect of amylin on endothelial-dependent vasodilation in mesenteric arteries from control and insulin resistant rats. PLoS ONE 2015, 10, e0120479. [Google Scholar] [CrossRef]
- Cha, J.J.; Min, H.S.; Kim, K.T.; Kim, J.E.; Ghee, J.Y.; Kim, H.W.; Lee, J.E.; Han, J.Y.; Lee, G.; Ha, H.J.; et al. APX-115, a first-in-class pan-NADPH oxidase (Nox) inhibitor, protects db/db mice from renal injury. Lab. Investig. 2017, 97, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.P.; Jha, J.C.; Kennedy, K.; van Bommel, E.; Chew, P.; Szyndralewiez, C.; Touyz, R.M.; Schmidt, H.; Cooper, M.E.; Jandeleit-Dahm, K.A.M. Combined NOX1/4 inhibition with GKT137831 in mice provides dose-dependent reno- and atheroprotection even in established micro- and macrovascular disease. Diabetologia 2017, 60, 927–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Y.; Wang, Y.; Li, G.; Ma, W.; Zhou, X.S.; Wang, J.; Liu, B. The Nox1/Nox4 inhibitor attenuates acute lung injury induced by ischemia-reperfusion in mice. PLoS ONE 2018, 13, e0209444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Cifuentes-Pagano, E.; DeVallance, E.R.; de Jesus, D.S.; Sahoo, S.; Meijles, D.N.; Koes, D.; Camacho, C.J.; Ross, M.; St Croix, C.; et al. NADPH oxidase 2 inhibitors CPP11G and CPP11H attenuate endothelial cell inflammation & vessel dysfunction and restore mouse hind-limb flow. Redox Biol. 2019, 22, 101143. [Google Scholar]
- Berger, M.; Wraith, K.; Woodward, C.; Aburima, A.; Raslan, Z.; Hindle, M.S.; Moellmann, J.; Febbraio, M.; Naseem, K.M. Dyslipidemia-associated atherogenic oxidized lipids induce platelet hyperactivity through phospholipase Cγ2-dependent reactive oxygen species generation. Platelets 2019, 30, 467–472. [Google Scholar] [CrossRef]
- Anvari, E.; Wikstrom, P.; Walum, E.; Welsh, N. The novel NADPH oxidase 4 inhibitor GLX351322 counteracts glucose intolerance in high-fat diet-treated C57BL/6 mice. Free Radic. Res. 2015, 49, 1308–1318. [Google Scholar] [CrossRef] [Green Version]
- Scioli, M.G.; Bielli, A.; Agostinelli, S.; Tarquini, C.; Arcuri, G.; Ferlosio, A.; Costanza, G.; Doldo, E.; Orlandi, A. Antioxidant Treatment Prevents Serum Deprivation- and TNF-alpha-Induced Endothelial Dysfunction through the Inhibition of NADPH Oxidase 4 and the Restoration of beta-Oxidation. J. Vasc. Res. 2014, 51, 327–337. [Google Scholar] [CrossRef]
- Scioli, M.G.; Cervelli, V.; Arcuri, G.; Gentile, P.; Doldo, E.; Bielli, A.; Bonanno, E.; Orlandi, A. High insulin-induced down-regulation of Erk-1/IGF-1R/FGFR-1 signaling is required for oxidative stress-mediated apoptosis of adipose-derived stem cells. J. Cell. Physiol. 2014, 229, 2077–2087. [Google Scholar] [CrossRef]
- Wang, W.; Wu, Q.H.; Sui, Y.; Wang, Y.; Qiu, X. Rutin protects endothelial dysfunction by disturbing Nox4 and ROS-sensitive NLRP3 inflammasome. Biomed. Pharmacother. 2017, 86, 32–40. [Google Scholar] [CrossRef]
- Stachowicz, A.; Olszanecki, R.; Suski, M.; Wisniewska, A.; Toton-Zuranska, J.; Madej, J.; Jawien, J.; Bialas, M.; Okon, K.; Gajda, M.; et al. Mitochondrial aldehyde dehydrogenase activation by Alda-1 inhibits atherosclerosis and attenuates hepatic steatosis in apolipoprotein E-knockout mice. J. Am. Heart Assoc. 2014, 3, e001329. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.Y.; Wang, Y.B.; Han, B.; Yang, B.; Qiang, Y.W.; Zhang, Y.; Wang, Z.; Huang, X.; Liu, J.; Chen, Y.D.; et al. Activation of aldehyde dehydrogenase 2 slows down the progression of atherosclerosis via attenuation of ER stress and apoptosis in smooth muscle cells. Acta Pharmacol. Sin. 2018, 39, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Zhang, C. MicroRNAs in vascular disease. J. Cardiovasc. Pharmacol. 2011, 57, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemecz, M.; Alexandru, N.; Tanko, G.; Georgescu, A. Role of MicroRNA in Endothelial Dysfunction and Hypertension. Curr. Hypertens. Rep. 2016, 18, 87. [Google Scholar] [CrossRef]
- Chen, Y.; Gao, D.Y.; Huang, L. In vivo delivery of miRNAs for cancer therapy: Challenges and strategies. Adv. Drug Deliv. Rev. 2015, 81, 128–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fluiter, K.; Mook, O.R.; Baas, F. The therapeutic potential of LNA-modified siRNAs: Reduction of off-target effects by chemical modification of the siRNA sequence. Methods Mol. Biol. (Clifton N.J.) 2009, 487, 189. [Google Scholar]
- Shah, M.Y.; Ferrajoli, A.; Sood, A.K.; Lopez-Berestein, G.; Calin, G.A. microRNA Therapeutics in Cancer—An Emerging Concept. EBioMedicine 2016, 12, 34–42. [Google Scholar] [CrossRef] [Green Version]
- Elmen, J.; Lindow, M.; Schutz, S.; Lawrence, M.; Petri, A.; Obad, S.; Lindholm, M.; Hedtjarn, M.; Hansen, H.F.; Berger, U.; et al. LNA-mediated microRNA silencing in non-human primates. Nature 2008, 452, 896–899. [Google Scholar] [CrossRef]
- Esau, C.; Davis, S.; Murray, S.F.; Yu, X.X.; Pandey, S.K.; Pear, M.; Watts, L.; Booten, S.L.; Graham, M.; McKay, R.; et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006, 3, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Krutzfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature 2005, 438, 685–689. [Google Scholar] [CrossRef]
- van Rooij, E.; Marshall, W.S.; Olson, E.N. Toward microRNA-based therapeutics for heart disease: The sense in antisense. Circ. Res. 2008, 103, 919–928. [Google Scholar] [CrossRef]
- Stenvang, J.; Petri, A.; Lindow, M.; Obad, S.; Kauppinen, S. Inhibition of microRNA function by antimiR oligonucleotides. Silence 2012, 3, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karakikes, I.; Chaanine, A.H.; Kang, S.; Mukete, B.N.; Jeong, D.; Zhang, S.; Hajjar, R.J.; Lebeche, D. Therapeutic cardiac-targeted delivery of miR-1 reverses pressure overload-induced cardiac hypertrophy and attenuates pathological remodeling. J. Am. Heart Assoc. 2013, 2, e000078. [Google Scholar] [CrossRef] [Green Version]
- Pan, Z.; Sun, X.; Shan, H.; Wang, N.; Wang, J.; Ren, J.; Feng, S.; Xie, L.; Lu, C.; Yuan, Y.; et al. MicroRNA-101 inhibited postinfarct cardiac fibrosis and improved left ventricular compliance via the FBJ osteosarcoma oncogene/transforming growth factor-beta1 pathway. Circulation 2012, 126, 840–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardo, B.C.; Nguyen, S.S.; Winbanks, C.E.; Gao, X.M.; Boey, E.J.; Tham, Y.K.; Kiriazis, H.; Ooi, J.Y.; Porrello, E.R.; Igoor, S.; et al. Therapeutic silencing of miR-652 restores heart function and attenuates adverse remodeling in a setting of established pathological hypertrophy. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2014, 28, 5097–5110. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Hu, Z.; Lin, Y.; Zhang, C.; Perez-Polo, J.R. Downregulation of microRNA-29 by antisense inhibitors and a PPAR-gamma agonist protects against myocardial ischaemia-reperfusion injury. Cardiovasc. Res. 2010, 87, 535–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, M.; Sepodes, B.; Martins, A.P. Regulatory and Scientific Advancements in Gene Therapy: State-of-the-Art of Clinical Applications and of the Supporting European Regulatory Framework. Front. Med. 2017, 4, 182. [Google Scholar] [CrossRef] [Green Version]
- Kastrup, J.; Jorgensen, E.; Ruck, A.; Tagil, K.; Glogar, D.; Ruzyllo, W.; Botker, H.E.; Dudek, D.; Drvota, V.; Hesse, B.; et al. Direct intramyocardial plasmid vascular endothelial growth factor-A165 gene therapy in patients with stable severe angina pectoris A randomized double-blind placebo-controlled study: The Euroinject One trial. J. Am. Coll. Cardiol. 2005, 45, 982–988. [Google Scholar] [CrossRef] [Green Version]
- Hedman, M.; Hartikainen, J.; Syvanne, M.; Stjernvall, J.; Hedman, A.; Kivela, A.; Vanninen, E.; Mussalo, H.; Kauppila, E.; Simula, S.; et al. Safety and feasibility of catheter-based local intracoronary vascular endothelial growth factor gene transfer in the prevention of postangioplasty and in-stent restenosis and in the treatment of chronic myocardial ischemia: Phase II results of the Kuopio Angiogenesis Trial (KAT). Circulation 2003, 107, 2677–2683. [Google Scholar]
- Stewart, D.J.; Hilton, J.D.; Arnold, J.M.; Gregoire, J.; Rivard, A.; Archer, S.L.; Charbonneau, F.; Cohen, E.; Curtis, M.; Buller, C.E.; et al. Angiogenic gene therapy in patients with nonrevascularizable ischemic heart disease: A phase 2 randomized, controlled trial of AdVEGF(121) (AdVEGF121) versus maximum medical treatment. Gene Ther. 2006, 13, 1503–1511. [Google Scholar] [CrossRef] [Green Version]
- Stewart, D.J.; Kutryk, M.J.; Fitchett, D.; Freeman, M.; Camack, N.; Su, Y.; Della Siega, A.; Bilodeau, L.; Burton, J.R.; Proulx, G.; et al. VEGF gene therapy fails to improve perfusion of ischemic myocardium in patients with advanced coronary disease: Results of the NORTHERN trial. Mol. Ther. J. Am. Soc. Gene Ther. 2009, 17, 1109–1115. [Google Scholar] [CrossRef] [Green Version]
- Kastrup, J.; Jorgensen, E.; Fuchs, S.; Nikol, S.; Botker, H.E.; Gyongyosi, M.; Glogar, D.; Kornowski, R. A randomised, double-blind, placebo-controlled, multicentre study of the safety and efficacy of BIOBYPASS (AdGVVEGF121.10NH) gene therapy in patients with refractory advanced coronary artery disease: The NOVA trial. Eurointervention J. Eur. Collab. Work. Group Interv. Cardiol. Eur. Soc. Cardiol. 2011, 6, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Ripa, R.S.; Wang, Y.; Jorgensen, E.; Johnsen, H.E.; Hesse, B.; Kastrup, J. Intramyocardial injection of vascular endothelial growth factor-A165 plasmid followed by granulocyte-colony stimulating factor to induce angiogenesis in patients with severe chronic ischaemic heart disease. Eur. Heart J. 2006, 27, 1785–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartikainen, J.; Hassinen, I.; Hedman, A.; Kivela, A.; Saraste, A.; Knuuti, J.; Husso, M.; Mussalo, H.; Hedman, M.; Rissanen, T.T.; et al. Adenoviral intramyocardial VEGF-DDeltaNDeltaC gene transfer increases myocardial perfusion reserve in refractory angina patients: A phase I/IIa study with 1-year follow-up. Eur. Heart J. 2017, 38, 2547–2555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaski, J.C.; Consuegra-Sanchez, L. Evaluation of ASPIRE trial: A Phase III pivotal registration trial, using intracoronary administration of Generx (Ad5FGF4) to treat patients with recurrent angina pectoris. Expert Opin. Biol. Ther. 2013, 13, 1749–1753. [Google Scholar] [CrossRef] [PubMed]
- Deev, R.V.; Bozo, I.Y.; Mzhavanadze, N.D.; Voronov, D.A.; Gavrilenko, A.V.; Chervyakov, Y.V.; Staroverov, I.N.; Kalinin, R.E.; Shvalb, P.G.; Isaev, A.A. pCMV-vegf165 Intramuscular Gene Transfer is an Effective Method of Treatment for Patients With Chronic Lower Limb Ischemia. J. Cardiovasc. Pharmacol. Ther. 2015, 20, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Zhang, J.; Guo, L.; Cui, S.; Li, X.; Ding, D.; Kim, J.M.; Ho, S.H.; Hahn, W.; Kim, S. A phase I clinical study of naked DNA expressing two isoforms of hepatocyte growth factor to treat patients with critical limb ischemia. J. Gene Med. 2011, 13, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Shishehbor, M.H.; Rundback, J.; Bunte, M.; Hammad, T.A.; Miller, L.; Patel, P.D.; Sadanandan, S.; Fitzgerald, M.; Pastore, J.; Kashyap, V.; et al. SDF-1 plasmid treatment for patients with peripheral artery disease (STOP-PAD): Randomized, double-blind, placebo-controlled clinical trial. Vasc. Med. 2019, 24, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Jaski, B.E.; Jessup, M.L.; Mancini, D.M.; Cappola, T.P.; Pauly, D.F.; Greenberg, B.; Borow, K.; Dittrich, H.; Zsebo, K.M.; Hajjar, R.J.; et al. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID Trial), a first-in-human phase 1/2 clinical trial. J. Card. Fail. 2009, 15, 171–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenberg, B.; Butler, J.; Felker, G.M.; Ponikowski, P.; Voors, A.A.; Desai, A.S.; Barnard, D.; Bouchard, A.; Jaski, B.; Lyon, A.R.; et al. Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): A randomised, multinational, double-blind, placebo-controlled, phase 2b trial. Lancet 2016, 387, 1178–1186. [Google Scholar] [CrossRef]


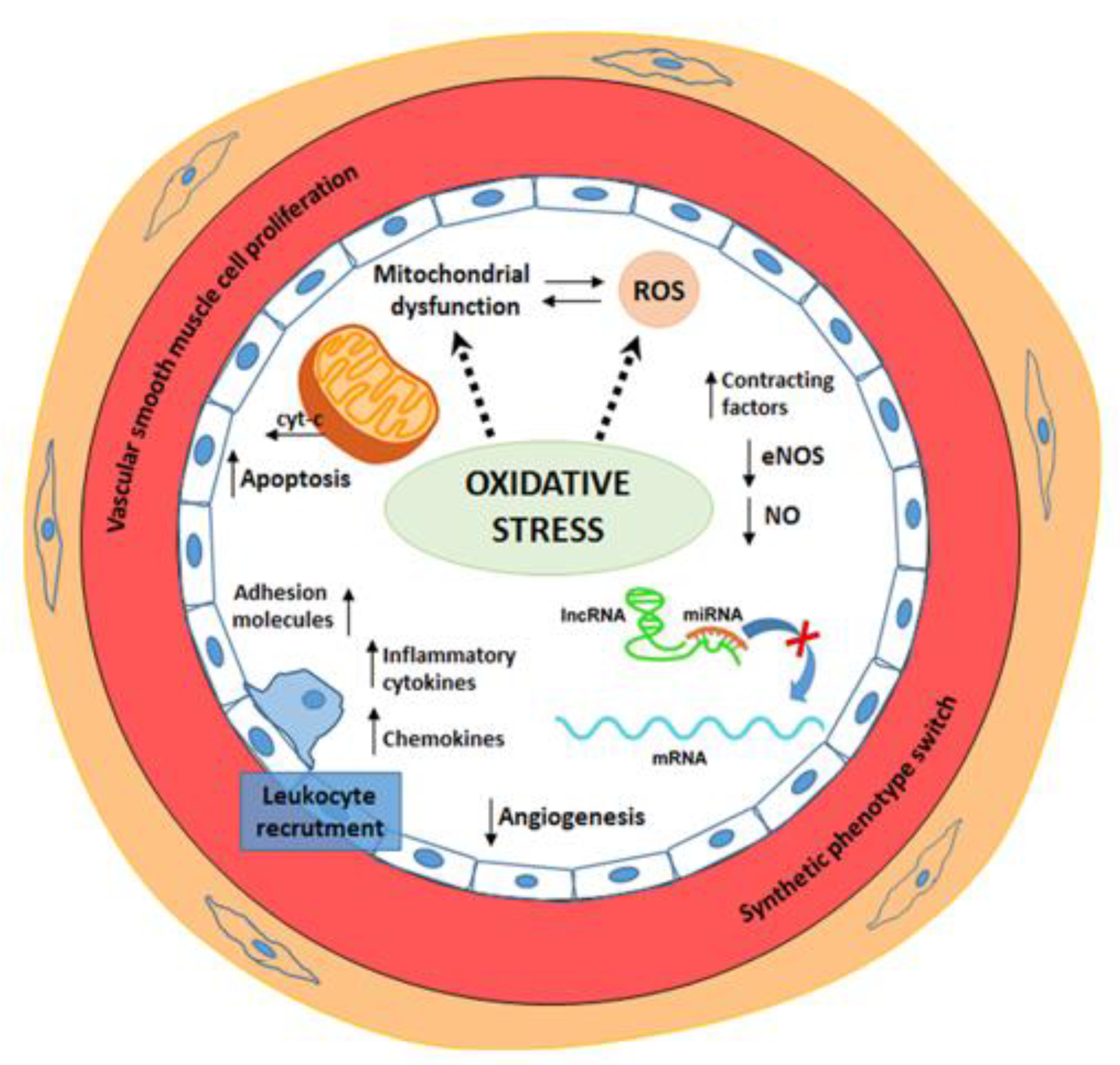{kind=link}
| Pathological Condition | Relevant Oxidative Stress Mechanisms | References |
|---|---|---|
| Obesity | Reactive oxygen species (ROS)-induced downregulation of Nitric Oxide (NO) activity. | [10] |
| Perivascular adipose tissue is responsible for tumor necrosis factor alpha (TNF-α) and interleukin (IL)-6 secretion, which sustain a low grade inflammatory state and an increased ROS production. | [11,12] | |
| In morbidly obese patients, arginase is up-regulated. Increased arginase activity competes with endothelial NO synthase (eNOS), whose main substrate for NO production is arginine; thus, reducing NO availability. | [13,14] | |
| Diabetes and hyperglycemic conditions | Hyperglycemia is involved in mitochondrial generation of superoxide anion (O−.) which contributes to diabetic endothelial dysfunction through four main pathways: the polyol pathway, increase in intracellular production of advanced glycation end products (AGEs), activation of the protein-kinase C (PKC) pathway, the hexosamine pathway | [15,16] |
| Hyperglycemia determines an increased ROS production that leads to mitochondrial DNA damage in endothelial cells. A so-determined increase in mitochondrial fission impairs electron-transport chain, which causes an altered nicotinamide adenine dinucleotide phosphate (NADH)\ flavin adenine dinucleotide (FADH2) ratio | [17] | |
| Hyperglycemia triggers a chronic vascular inflammatory state sustained by TNF-α, interleukin-1 beta (IL-1β), interleukin-6 (IL-6), cluster of differentiation 36 (CD36), monocyte chemoattractant protein-1 (MCP-1), and mediated through the up-regulation of the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway, which leads to endothelial cell apoptosis. | [18,19] | |
| In the pathogenesis of diabetes, an altered expression of several microRNAs has been demonstrated. A reduced lethal-7 (let-7) microRNA (miRNA) and miR-126 expression and an increased miR-200 expression have been linked to β-cells impairment, insulin resistance, chronic inflammation, and vascular oxidative stress. | [20,21,22,23] | |
| Insulin resistance is generated by ROS mediated activation of several pathways, such as p38 mitogen-activated protein kinase (p38 MAPK), extracellular signal-regulated kinase (ERK), IkB kinase (IKK). These pathways converge on phosphorylation of insulin receptor substrate proteins, which determine alterations in insulin signal transmission. | [24,25] | |
| High insulin levels upregulate the MAPK pathway, which is involved in plaque formation and vascular smooth muscle cells (VSMC) hypertrophy and proliferation. | [26] | |
| Dysmetabolic conditions | ROS have been associated to low-density lipoproteins (LDL) oxidation, generating oxidized low-density lipoproteins (ox-LDL), which are removed from the systemic circulation by binding to scavenger receptor on macrophage surface. This elicits inflammatory response through NF-κB pathway and determines formation of foam cells. | [27,28] |
| Ox-LDLs drive caspase activation and endothelial cell apoptosis. | [29] | |
| Hyperuricemia contributes to endothelial dysfunction mainly through NO depletion. The reaction between uric acid (UA) and NO generates 6-aminouracil, thus, reducing NO availability. UA is also responsible for increased arginine degradation which is the main eNOS substrate for NO synthesis. | [30] | |
| Cigarette Smoke | Cigarette smoke (CS) is a suppressor of endothelial NO synthase eNOS and determines reduction in NO bioavailability. It is also responsible for ROS production, tissue remodeling, and increased expression of adhesion molecules and prothrombotic factors. | [31] |
| Hypertension | Chronic inflammation produces inflammatory chemokines, such as IL-6, IL-1β, TNF-α, and interleukin-17 (IL-17), which promote oxidative stress and recruit macrophages, T and B lymphocytes that are responsible for ROS production and vascular fibrotic remodeling. | [32,33] |
| Damage to the endothelial cells that release increased levels of endothelial microparticles (EMP), which causes an impaired glomerular filtration rate and is implicated in vascular inflammation, thrombosis, angiogenesis, and atherosclerosis progression. | [34,35] | |
| Hypertensive inflammation induces NADPH oxidases (Nox), one of the most important sources of superoxide anion (O−) in endothelial dysfunction. Superoxide anion react with NO forming peroxynitrite, which oxidizes the 4-tetrahydrobiopterin (BH4), a cofactor of eNOS; thus, determining the uncoupling of eNOS and a decreased NO bioavailability. | [36,37] | |
| NO is able to modulate response to angiotensin II and to invert angiotensin II-induced arteriolar contraction. | [38] | |
| Aldosterone has proinflammatory effects that are mediated through the mineralocorticoid receptor (MR). Aberrant activation of MR mediates endothelial and organ damage, directly, or through angiotensin 1. | [39,40] | |
| Aging | Endothelium-dependent dilation (EDD) is decreased with aging because of a reduced NO bioavailability. NO production in older adults are reduced under baseline resting conditions. | [41] |
| Aging increases the production of reactive oxygen species in the face of unchanged or reduced antioxidant defenses. | [41] | |
| Age-related endothelial redox changes affect NF-kB, whose activation induces transcription of pro-inflammatory cytokines that can further suppress endothelial function, thus, creating a vicious feed-forward cycle. | [42] | |
| Genomic instability, telomere dysfunction or DNA damage has been shown to trigger cell senescence via the p53/p21 pathway and result in increased inflammatory signaling in arteries from older adults. | [42] |
| Type | Function | References |
|---|---|---|
| Lipid peroxidation | indicates reactive oxygen species (ROS) overproduction | [95] |
| NO | depleted in endothelial dysfunction | [101] |
| Glutathione | the reduced GSH/GSSG indicates oxidative stress | [104] |
| oxGua | indicates ROS overproduction | [108] |
| 8-isoprostane | indicates ROS overproduction | |
| oxLDL | indicates ROS overproduction | [109] |
| EPCs | increased in endothelial damage | [34] |
| EMPs | depleted in vascular inflammation, thrombosis, angiogenesis and atherosclerosis progression | [34,114] |
| CECs | [113] | |
| Endoglin | secreted in endothelial injury and inflammation process | [117] |
| Uric acid | indicates oxidative stress | [119] |
| GGT | increased in oxidative stress | [105] |
| JMJD2A | mediator of cardiac hypertrophy | [129] |
| jmjd3 | mediator of angiogenesis | |
| miR-126 | increased in angiogenesis and regulates vascular integrity | [137] |
| miR-15b | negative regulators of angiogenesis and vascular function | [140] |
| miR-16 | ||
| miR-505 | increased in hypertension | [142] |
| miR-17-3p | increased in vascular inflammation | [76,77] |
| miR-31 | [77] | |
| miR-155 | negative regulator of endothelium-dependent vasodilation | [143,146] |
| miR-19a | antiproliferative activity in ECs | [147] |
| miR-19b | counteracts EC apoptosis | [148] |
| Let-7g | possible role in EC apoptosis | [149,150] |
| miR-21 | possible role in EC apoptosis and positive regulator of VSMC proliferation and survival | [149,150,165] |
| miR-223 | possible role in EC apoptosis | [149,150,151] |
| miR-155 | involved in RAAS-mediated cardiovascular inflammation | [152,153,154,155,156] |
| miR-146a/b | ||
| miR-132/122 | ||
| miR-483-3p | ||
| miR-145 | inhibit ACE expression | [153,155] |
| miR-27a/b | ||
| miR-143/145 | negative regulator of ACE expression and of VSMC contractile phenotype | [154,161,162] |
| miR-221 | reduces VSMC contractile phenotype | [159] |
| miR-222 | increased in hypertension | [160] |
| miR-133 | negative regulator of VSMC proliferation | [163] |
| miR-365 | downregulates cyclin D1 | [164] |
| miR-18a-5p | decreased in atherosclerotic disease | [166] |
| miR-27a-3p | ||
| miR-199a-3p | ||
| miR-223-3p | ||
| miR-652-3p | ||
| miR-126-5p | promotes EC regeneration and turnover | [167,168] |
| miR-217 | negative regulator of SIRT1 expression | [172] |
| miR-10a | target genes involved in EC inflammation, adhesion molecules, cell cycle, proliferation, migration, apoptosis, and nitric oxide signaling | [176,177,178,179,180,181] |
| miR-23b | [178,180] | |
| miR-17~92 | [177,180,181] | |
| miR-663 | [180,181] | |
| miR-92a | [177,179,181] | |
| miR-101 | [180,181] | |
| miR-712 | [108,113] | |
| miR-205 | [177,181] | |
| miR-155 | [180,181] | |
| miR-15a | downregulated in T2D | [184] |
| miR-29b | ||
| miR-181b | decreased in patients with CAD and positive role in glucose homeostasis and insulin signaling | [187,188] |
| SENCR | regulates VSMC and EC differentiation, downregulated in T2D and CAD | [192,193] |
| H19 | increased in CVD | [125,126] |
| HOTAIR | reduced in ECs from atherosclerotic plaque | [196] |
| MALAT1 | induces EC proliferation and angiogenesis | [197] |
| Class | Type | Mechanism of Action/Results | Target | References |
|---|---|---|---|---|
| Nano-medicine | Liposomes Niosomes Polymers Carbon nanotubes | drug carrier and delivery | drugs | [59,206,207] |
| PARP inhibitors | PJ-34 | counteracts reactive oxygen/nitrogen species | poly (ADP-ribose) polymerase | [218] |
| INO 1001 | against T2D, hyperhomocysteinemia, hypertension | poly (ADP-ribose) polymerase | [219] | |
| DAQ B1 | reduces oxidative stress and prevents hypertension and T2D | Akt | [223,224] | |
| PTPase inhibitors | BMOV | regulates opening ATP-sensitive channels decreasing oxidative stress | eNOS | [226] |
| ROCK inhibitors | Y-27632 | counteracts corneal endothelial dysfunction | Rho-associated protein kinase | [227,228] |
| Geranylgeranyl transferase inhibitors | GGTI-298 | counteracts eNOS inactivation | Rho A and Rac1 | [229] |
| Transketolase activator | Benfotiamine | prevents vascular AGE accumulation and induction of pro-apoptotic caspace-3 | AGE | [231] |
| Mitochondria-targeted antioxidants | MitoQ | upregulates mitochondrial dynamics proteins Mfn2 and Drp-1 exerting a protective effect | Ubiquinol α-tocopherol | [234] |
| SIRT mimetics | reduction of p66Shc fundamental in the homeostasis of mitochondria | p66Shc | [88] | |
| ELAM | stabilizes the cardiolipin | cardiolipin | [253,254,255] | |
| NADPH inhibitors | GKT137831 | reduces oxidative stress and diabetic vasculopathy | NOX1, NOX4 | [272] |
| GLX351322 | reduces oxidative stress and T2D | NOX4 | [276] | |
| Gene-target therapies | EuroinjectOne | fails to improve perfusion in CAD | VEGF-A165 | [297] |
| KAT | improves perfusion in CAD | VEGF-A165 | [298] | |
| REVASC | improves perfusion in CAD | VEGF-A121 | [299] | |
| NOTHERN | fails to improve perfusion in CAD | VEGF-A165 | [300] | |
| NOVA | fails to improve perfusion in CAD | VEGF-A121 | [301] | |
| VEGF-Neupogen | fails to improve perfusion in CAD | VEGF-A165 | [302] | |
| KAT301 | improves perfusion in CAD | VEGF-D | [303] | |
| ASPIRE | N/A (CAD) | FGF4 | [304] | |
| Neovasculgen | improves perfusion in PAD | VEGF-A165 | [305] | |
| HGF-X7 | N/A (CAD) | HGF | [306] | |
| JVS-100 | N/A (PAD) | SDF-1 | [307] | |
| SERCA2a | improves left ventricular remodeling in severe heart failure | calcium ATPases | [308,309] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scioli, M.G.; Storti, G.; D’Amico, F.; Rodríguez Guzmán, R.; Centofanti, F.; Doldo, E.; Céspedes Miranda, E.M.; Orlandi, A. Oxidative Stress and New Pathogenetic Mechanisms in Endothelial Dysfunction: Potential Diagnostic Biomarkers and Therapeutic Targets. J. Clin. Med. 2020, 9, 1995. https://doi.org/10.3390/jcm9061995
Scioli MG, Storti G, D’Amico F, Rodríguez Guzmán R, Centofanti F, Doldo E, Céspedes Miranda EM, Orlandi A. Oxidative Stress and New Pathogenetic Mechanisms in Endothelial Dysfunction: Potential Diagnostic Biomarkers and Therapeutic Targets. Journal of Clinical Medicine. 2020; 9(6):1995. https://doi.org/10.3390/jcm9061995
Chicago/Turabian StyleScioli, Maria Giovanna, Gabriele Storti, Federico D’Amico, Roger Rodríguez Guzmán, Federica Centofanti, Elena Doldo, Ela María Céspedes Miranda, and Augusto Orlandi. 2020. "Oxidative Stress and New Pathogenetic Mechanisms in Endothelial Dysfunction: Potential Diagnostic Biomarkers and Therapeutic Targets" Journal of Clinical Medicine 9, no. 6: 1995. https://doi.org/10.3390/jcm9061995