Risk Stratification in Hypertrophic Cardiomyopathy. Insights from Genetic Analysis and Cardiopulmonary Exercise Testing

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Methods
2.1. Study Sample
2.2. Patients Clinical and Functional Assessment
2.3. Genetic Testing
2.4. Clinical Outcomes
2.5. Statistical Analysis
3. Results
3.1. Genetic Results
3.2. Clinical and Functional Characteristics
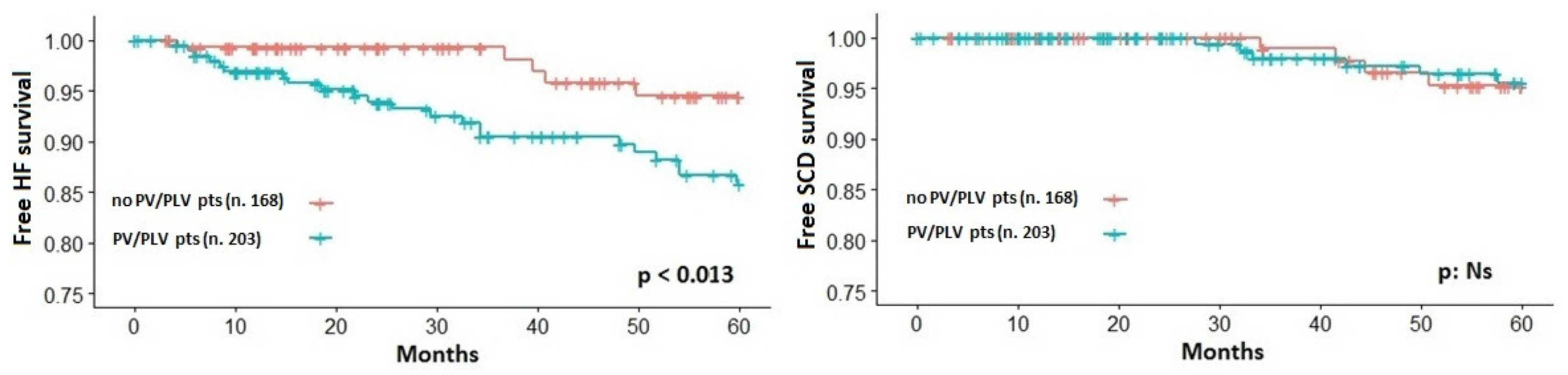
3.3. End-Point Analysis
4. Discussion
5. Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Maron, B.J.; Maron, M.S. Hypertrophic cardiomyopathy. Lancet 2013, 381, 242–255. [Google Scholar] [CrossRef]
- Gersh, B.J.; Maron, B.J.; Bonow, R.O.; Dearani, J.A.; Fifer, M.A.; Link, M.S.; Naidu, S.S.; Nishimura, R.A.; Ommen, S.R.; Rakowski, H.; et al. 2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy: Executive Summary: A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2011, 124, 2761–2796. [Google Scholar] [CrossRef]
- Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; Mahrholdt, H.; et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar] [CrossRef]
- Maron, B.J.; Ommen, S.R.; Semsarian, C.; Spirito, P.; Olivotto, I.; Maron, M.S. Hypertrophic cardiomyopathy: Present and future, with translation into contemporary cardiovascular medicine. J. Am. Coll. Cardiol. 2014, 64, 83–99. [Google Scholar] [CrossRef] [Green Version]
- Musumeci, M.B.; Russo, D.; Limite, L.R.; Canepa, M.; Tini, G.; Casenghi, M.; Francia, P.; Adduci, C.; Pagannone, E.; Magrì, D.; et al. Long-Term Left Ventricular Remodeling of Patients with Hypertrophic Cardiomyopathy. Am. J. Cardiol. 2018, 122, 1924–1931. [Google Scholar] [CrossRef]
- Marian, A.J.; Braunwald, E. Hypertrophic Cardiomyopathy: Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circ. Res. 2017, 121, 749–770. [Google Scholar] [CrossRef]
- Rubattu, S.; Bozzao, C.; Pennacchini, E.; Pagannone, E.; Musumeci, M.B.; Piane, M.; Germani, A.; Savio, C.; Francia, P.; Volpe, M.; et al. A Next-Generation Sequencing Approach to Identify Gene Mutations in Early- and Late-Onset Hypertrophic Cardiomyopathy Patients of an Italian Cohort. Int. J. Mol. Sci. 2016, 17, 1239. [Google Scholar] [CrossRef] [Green Version]
- Olivotto, I.; Girolami, F.; Ackerman, M.J.; Nistri, S.; Bos, J.M.; Zachara, E.; Ommen, S.R.; Theis, J.L.; Vaubel, R.A.; Re, F.; et al. Myofilament protein gene mutation screening and outcome of patients with hypertrophic cardiomyopathy. Mayo Clin. Proc. 2008, 83, 630–638. [Google Scholar] [CrossRef] [Green Version]
- Selvi Rani, D.; Nallari, P.; Dhandapany, P.S.; Rani, J.; Meraj, K.; Ganesan, M.; Narasimhan, C.; Thangaraj, K. Coexistence of Digenic Mutations in Both Thin (TPM1) and Thick (MYH7) Filaments of Sarcomeric Genes Leads to Severe Hypertrophic Cardiomyopathy in a South Indian FHCM. DNA Cell Biol. 2015, 34, 350–359. [Google Scholar] [CrossRef]
- Li, Q.; Gruner, C.; Chan, R.H.; Care, M.; Siminovitch, K.; Williams, L.; Woo, A.; Rakowski, H. Genotype-positive status in patients with hypertrophic cardiomyopathy is associated with higher rates of heart failure events. Circ. Cardiovasc. Genet. 2014, 7, 416–422. [Google Scholar] [CrossRef] [Green Version]
- Lopes, L.R.; Syrris, P.; Guttmann, O.P.; O’Mahony, C.; Tang, H.C.; Dalageorgou, C.; Jenkins, S.; Hubank, M.; Monserrat, L.; McKenna, W.J.; et al. Novel genotype-phenotype associations demonstrated by high-throughput sequencing in patients with hypertrophic cardiomyopathy. Heart 2015, 101, 294–301. [Google Scholar] [CrossRef]
- Van Velzen, H.G.; Vriesendorp, P.A.; Oldenburg, R.A.; van Slegtenhorst, M.A.; van der Velden, J.; Schinkel, A.F.L.; Michels, M. Value of Genetic Testing for the Prediction of Long-Term Outcome in Patients with Hypertrophic Cardiomyopathy. Am. J. Cardiol. 2016, 118, 881–887. [Google Scholar] [CrossRef]
- Ho, C.Y.; Day, S.M.; Ashley, E.A.; Michel, M.; Pereira, A.C.; Jacoby, D.; Cirino, A.L.; Fox, J.C.; Lakdawala, N.K.; Ware, J.S.; et al. Genotype and Lifetime Burden of Disease in Hypertrophic Cardiomyopathy: Insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe). Circulation 2018, 138, 1387–1398. [Google Scholar] [CrossRef]
- Masri, A.; Pierson, L.M.; Smedira, N.G.; Agarwal, S.; Lytle, B.W.; Naji, P.; Thamilarasan, M.; Lever, H.M.; Cho, L.S.; Desai, M.Y. Predictors of longterm outcomes in patients with hypertrophic cardiomyopathy undergoing cardiopulmonary stress testing and echocardiography. Am. Heart J. 2015, 169, 684–692. [Google Scholar] [CrossRef]
- Coats, C.J.; Rantell, K.; Bartnik, A.; Patel, A.; Mist, B.; McKenna, W.J.; Elliott, P.M. Cardiopulmonary exercise testing and prognosis in hypertrophic cardiomyopathy. Circ. Heart Fail. 2015, 8, 1022–1031. [Google Scholar] [CrossRef]
- Finocchiaro, G.; Haddad, F.; Knowles, J.W.; Caleshu, C.; Pavlovic, A.; Homburger, J.; Shmargad, Y.; Sinagra, G.; Magavern, E.; Wong, M.; et al. Cardiopulmonary responses and prognosis in hypertrophic cardiomyopathy: A potential role for comprehensive noninvasive hemodynamic assessment. JACC Heart Fail. 2015, 3, 408–418. [Google Scholar] [CrossRef]
- Magrì, D.; Re, F.; Limongelli, G.; Agostoni, P.; Zachara, E.; Correale, M.; Mastromarino, V.; Santolamazza, C.; Casenghi, M.; Pacileo, G.; et al. Heart Failure Progression in Hypertrophic Cardiomyopathy—Possible Insights From Cardiopulmonary Exercise Testing. Circ. J. 2016, 80, 2204–2211. [Google Scholar] [CrossRef] [Green Version]
- Magrì, D.; Limongelli, G.; Re, F.; Agostoni, P.; Zachara, E.; Correale, M.; Mastromarino, V.; Santolamazza, C.; Casenghi, M.; Pacileo, G.; et al. Cardiopulmonary exercise test and sudden cardiac death risk in hypertrophic cardiomyopathy. Heart 2016, 102, 602–609. [Google Scholar] [CrossRef]
- Lang, R.M.; Badano, L.P.; Mor-Avi, V.; Afilalo, J.; Armstrong, A.; Ernande, L.; Flachskampf, F.A.; Foster, E.; Goldstein, S.A.; Kuznetsova, T.; et al. Recommendations for cardiac chamber quantification by echocardiography in adults: An update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J. Am. Soc. Echocardiogr. 2015, 28, 1–39. [Google Scholar] [CrossRef] [Green Version]
- Autore, C.; Bernabò, P.; Barillà, C.S.; Bruzzi, P.; Spirito, P. The prognostic importance of left ventricular outflow obstruction in hypertrophic cardiomyopathy varies in relation to the severity of symptoms. J. Am. Coll. Cardiol. 2005, 45, 1076–1080. [Google Scholar] [CrossRef] [Green Version]
- Agostoni, P.; Bianchi, M.; Moraschi, A.; Palermo, P.; Cattadori, G.; La Gioia, R.; Bussotti, M.; Wasserman, K. Work-rate affects cardiopulmonary exercise test results in heart failure. Eur. J. Heart Fail. 2005, 7, 498–504. [Google Scholar] [CrossRef] [Green Version]
- Magrì, D.; Agostoni, P.; Sinagra, G.; Re, F.; Correale, M.; Limongelli, G.; Zachara, E.; Mastromarino, V.; Santolamazza, C.; Casenghi, M.; et al. Clinical and prognostic impact of chronotropic incompetence in patients with hypertrophic cardiomyopathy. Int. J. Cardiol. 2018, 271, 125–131. [Google Scholar] [CrossRef]
- Corrà, U.; Mezzani, A.; Giordano, A.; Bosimini, E.; Giannuzzi, P. Exercise haemodynamic variables rather than ventilatory efficiency indexes contribute to risk assessment in chronic heart failure patients treated with carvedilol. Eur. Heart J. 2009, 30, 3000–3006. [Google Scholar] [CrossRef] [Green Version]
- Wasserman, K.; Hansen, J.E.; Sue, D.Y.; Stringer, W.; Whipp, B.J. Normal Values. In Principles of Exercise Testing and Interpretation, 4th ed.; Weinberg, R., Ed.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2005; pp. 160–182. [Google Scholar]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Givertz, M.M.; Ho, C.Y.; Judge, D.P.; Kantor, P.; McBride, K.L.; Morales, A.; Taylor, M.R.G.; Vatta, M.; Ware, S.M.; et al. Genetic evaluation of cardiomyopathy: A clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2018, 20, 899–909. [Google Scholar] [CrossRef] [Green Version]
- O’Mahony, C.; Jichi, F.; Pavlou, M.; Monserrat, L.; Anastasakis, A.; Rapezzi, C.; Biagini, E.; Gimeno, J.R.; Limongelli, G.; McKenna, W.J.; et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM Risk-SCD). Eur. Heart J. 2014, 35, 2010–2020. [Google Scholar] [CrossRef]
- O’Mahony, C.; Akhtar, M.M.; Anastasiou, Z.; Guttmann, O.P.; Vriesendorp, P.A.; Michels, M.; Magrì, D.; Autore, C.; Fernández, A.; Ochoa, J.P.; et al. Effectiveness of the 2014 European Society of Cardiology guideline on sudden cardiac death in hypertrophic cardiomyopathy: A systematic review and meta-analysis. Heart 2019, 105, 623–631. [Google Scholar] [CrossRef]
- Biagini, E.; Olivotto, I.; Iascone, M.; Parodi, M.I.; Girolami, F.; Frisso, G.; Autore, C.; Limongelli, G.; Cecconi, M.; Maron, B.J.; et al. Significance of sarcomere gene mutations analysis in the end-stage phase of hypertrophic cardiomyopathy. Am. J. Cardiol. 2014, 114, 769–776. [Google Scholar] [CrossRef]
- Magrì, D.; Santolamazza, C. Cardiopulmonary Exercise Test in Hypertrophic Cardiomyopathy. Ann. Am. Thorac. Soc. 2017, 14 (Suppl. 1), 102–109. [Google Scholar] [CrossRef]
- Magrì, D.; Agostoni, P.; Cauti, F.M.; Musumeci, B.; Egidy Assenza, G.; De Cecco, C.N.; Muscogiuri, G.; Maruotti, A.; Ricotta, A.; Pagannone, E.; et al. Determinants of peak oxygen uptake in patients with hypertrophic cardiomyopathy: A single-center study. Intern. Emerg. Med. 2014, 9, 293–302. [Google Scholar] [CrossRef]
- Magrì, D. Peak oxygen uptake in heart failure: Look behind the number! Eur. J. Prev. Cardiol. 2018, 25, 1934–1936. [Google Scholar] [CrossRef] [Green Version]
- Agostoni, P.; Corrà, U.; Cattadori, G.; Veglia, F.; La Gioia, R.; Scardovi, A.B.; Emdin, M.; Metra, M.; Sinagra, G.; Limongelli, G.; et al. Metabolic exercise test data combined with cardiac and kidney indexes, the MECKI score: A multiparametric approach to heart failure prognosis. Int. J. Cardiol. 2013, 167, 2710–2718. [Google Scholar] [CrossRef]
- Paolillo, S.; Veglia, F.; Salvioni, E.; Corrà, U.; Piepoli, M.; Lagioia, R.; Limongelli, G.; Sinagra, G.; Cattadori, G.; Scardovi, A.B.; et al. Heart failure prognosis over time: How the prognostic role of oxygen consumption and ventilatory efficiency during exercise has changed in the last 20 years. Eur. J. Heart Fail. 2019, 21, 208–217. [Google Scholar] [CrossRef] [Green Version]
- Ciampi, Q.; Betocchi, S.; Lombardi, R.; Manganelli, F.; Storto, G.; Losi, M.A.; Pezzella, E.; Finizio, F.; Cuocolo, A.; Chiariello, M. Hemodynamic determinants of exercise-induced abnormal blood pressure response in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2002, 40, 278–284. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, T.; Azuma, A.; Kuribayashi, T.; Akakabe, Y.; Yamano, M.; Miki, S.; Sawada, T.; Kamitani, T.; Matsubara, H.; Sugihara, H. Vagal enhancement due to subendocardial ischemia as a cause of abnormal blood pressure response in hypertrophic cardiomyopathy. Int. J. Cardiol. 2008, 129, 59–64. [Google Scholar] [CrossRef]
- Arena, R.; Owens, D.S.; Arevalo, J.; Smith, K.; Mohiddin, S.A.; McAreavey, D.; Ulisney, K.L.; Tripodi, D.; Fananapazir, L.; Plehn, J.F. Ventilatory efficiency and resting hemodynamics in hypertrophic cardiomyopathy. Med. Sci. Sports Exerc. 2008, 40, 799–805. [Google Scholar] [CrossRef]
- Salvioni, E.; Corrà, U.; Piepoli, M.; Rovai, S.; Correale, M.; Paolillo, S.; Pasquali, M.; Magrì, D.; Vitale, G.; Fusini, L.; et al. Gender and age normalization and ventilation efficiency during exercise in heart failure with reduced ejection fraction. ESC Heart Fail. 2020, 7, 371–380. [Google Scholar] [CrossRef] [Green Version]
- Musumeci, M.B.; Mastromarino, V.; Casenghi, M.; Tini, G.; Francia, P.; Maruotti, A.; Romaniello, A.; Magrì, D.; Lillo, R.; Adduci, C.; et al. Pulmonary hypertension and clinical correlates in hypertrophic cardiomyopathy. Int. J. Cardiol. 2017, 248, 326–332. [Google Scholar] [CrossRef]
- Maron, B.J.; Maron, M.S.; Semsarian, C. Genetics of hypertrophic cardiomyopathy after 20 years: Clinical perspectives. J. Am. Coll. Cardiol. 2012, 60, 705–715. [Google Scholar] [CrossRef] [Green Version]
- Homburger, J.R.; Green, E.M.; Caleshu, C.; Sunitha, M.S.; Taylor, R.E.; Ruppel, K.M.; Prasad Rao Metpally, R.; Colan, S.D.; Michels, M.; Day, S.M.; et al. Multidimensional Structure-Function Relationships in Human β-Cardiac Myosin from Population-Scale Genetic Variation. Proc. Natl. Acad. Sci. USA 2016, 113, 6701–6706. [Google Scholar] [CrossRef] [Green Version]
- Alamo, L.; Ware, J.S.; Pinto, A.; Gillilan, R.E.; Seidman, J.G.; Seidman, C.E.; Padrón, R. Effects of Myosin Variants on Interacting-Heads Motif Explain Distinct Hypertrophic and Dilated Cardiomyopathy Phenotypes. eLife 2017, 6, e24634. [Google Scholar] [CrossRef]
- García-Giustiniani, D.; Arad, M.; Ortíz-Genga, M.; Barriales-Villa, R.; Fernández, X.; Rodríguez-García, I.; Mazzanti, A.; Veira, E.; Maneiro, E.; Rebolo, P.; et al. Phenotype and Prognostic Correlations of the Converter Region Mutations Affecting the β Myosin Heavy Chain. Heart 2015, 101, 1047–1053. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| General Data | |
|---|---|
| Age, years | 49 ± 16 |
| Male, n (%) | 238 (64) |
| Age at diagnosis, years | 40 ± 19 |
| NYHA III–IV, n (%) | 24 (6) |
| ICD, n (%) | 43 (12) |
| Previous myectomy, n (%) | 20 (5) |
| SCD risk factors | |
| NSVT, n (%) | 121 (32) |
| FH-SCD, n (%) | 48 (13) |
| MWT > 30 mm, n (%) | 26 (7) |
| Unexplained syncope, n (%) | 56 (15) |
| ABPRE, n (%) | 45 (16) |
| Echocardiographic data | |
| LVEDd, mm | 45 ± 5 |
| LAd, mm | 43 ± 7 |
| MWT, mm | 20 ± 5 |
| LVOT obstruction, n (%) | 125 (33) |
| LVOTGmax, mmHg | 11 (6–39) |
| LVEF, % | 62 ± 7 |
| CPET data | |
| Peak HR, % of predicted | 79 ± 13 |
| Peak VO2, mL/kg/min | 23 ± 7 |
| Peak VO2, % of predicted | 77 ± 18 |
| Peak SBP, mmHg | 164 ± 27 |
| CP%, % of predicted * mmHg | 12,937 ± 4300 |
| VE/VCO2 slope | 28.4 ± 5.5 |
| Medical treatment | |
| β-blocker, n (%) | 228 (61) |
| Nondihydropyridine CCB, n (%) | 31 (8) |
| ACE-I/ARB, n (%) | 107 (29) |
| Diuretics, n (%) | 78 (21) |
| Amiodaron, n (%) | 33 (9) |
| General Data | No Variants and VUS (n = 168) | LP/P Variants (n = 203) | p-Values |
|---|---|---|---|
| Age, years | 53 ± 18 | 45 ± 16 | <0.001 |
| Male, n (%) | 112 (67) | 124(61) | NS |
| Age at diagnosis, years | 47 ± 20 | 35 ± 17 | <0.001 |
| NYHA III-IV, n (%) | 10 (6) | 14 (7) | NS |
| ICD, n (%) | 11 (6) | 32 (16) | 0.019 |
| Previous myectomy, n (%) | 9 (5) | 11 (5) | NS |
| SCD risk factors | |||
| NSVT, n (%) | 48 (28) | 73 (36) | NS |
| FH-SCD, n (%) | 16 (9) | 32 (16) | 0.049 |
| MWT > 30 mm, n (%) | 10 (6) | 16 (8) | NS |
| Unexplained syncope, n (%) | 29 (17) | 26 (13) | NS |
| ABPRE, n (%) | 11 (7) | 34 (17) | 0.003 |
| Echocardiographic data | |||
| LVEDd, mm | 46 ± 4 | 45 ± 6 | NS |
| LAd, mm | 43 ± 7 | 43 ± 7 | NS |
| MWT, mm | 20 ± 5 | 20 ± 5 | NS |
| LVOT obstruction, n (%) | 79 (47) | 45 (22) | <0.001 |
| LVOTGmax, mmHg | 16 (9–39) | 10 (5–33) | 0.023 |
| LVEF, % | 63 ± 4 | 61 ± 6 | <0.001 |
| CPET data | |||
| Peak HR, % of predicted | 78 ± 12 | 79 ± 13 | NS |
| Peak VO2, mL/kg/min | 23 ± 7 | 23 ± 7 | NS |
| Peak VO2, % of predicted | 79 ± 18 | 75 ± 18 | 0.032 |
| Peak SBP, mmHg | 175 ± 26 | 157 ± 26 | <0.001 |
| CP%, % of predicted * mmHg | 14,070 ± 4269 | 12,015 ± 4020 | <0.001 |
| VE/VCO2 slope | 27.5 ± 4.9 | 29.1 ± 6.0 | 0.019 |
| Medical treatment | |||
| β-blocker, n (%) | 105 (62) | 122 (60) | NS |
| Non dihydropyridine CCB, n (%) | 11 (7) | 20 (10) | NS |
| ACE-I/ARB, n (%) | 58 (34) | 48 (24) | 0.014 |
| Diuretics, n (%) | 37 (22) | 41 (21) | NS |
| Amiodaron, n (%) | 16 (9) | 17 (8) | NS |
| HF Endpoint (n = 52) | SCD Endpoint (n = 14) | |||||
|---|---|---|---|---|---|---|
| H.R. (95% C.I.) | p-Values | C-Index | H.R. (95% C.I.) | p-Values | C-Index | |
| Age at CPET | – | NS | – | 0.964 (0.934–0.996) | 0.038 | 0.613 |
| Male sex | – | NS | – | – | NS | – |
| Age at diagnosis | – | NS | – | 0.944 (0.906–0.983) | 0.006 | 0.729 |
| FH-SCD | 1.869 (1.010–3.460) | 0.046 | 0.522 | 2.830 (0.892–8.979) | 0.077 | 0.607 |
| Unexplained Syncope | – | NS | – | – | NS | – |
| NSVT | 1.917 (1.102–3.333) | 0.021 | 0.548 | – | NS | – |
| ABPRE | 3.418 (1.769–6.605) | <0.001 | 0.641 | – | NS | – |
| MWT > 30 mm | – | NS | – | 3.956 (1.210–12.940) | 0.023 | 0.569 |
| MWT | – | NS | – | 1.100 (1.012–1.195) | 0.025 | 0.593 |
| LVOTO | 2.110 (1.215–3.664) | <0.01 | 0.641 | – | NS | – |
| LAd | 1.077 (1.039–1.116) | <0.001 | 0.704 | 1.054 (0.984–1.129) | 0.112 | 0.660 |
| LVOTGmax | 1.016 (1.008–1.024) | <0.001 | 0.672 | 0.971 (0.937–1.007) | 0.123 | 0.577 |
| LVEF | 0.929 (0.899–0.959) | <0.001 | 0.587 | – | NS | – |
| pVO2, mL/kg/min | 0.851 (0.799–0.905) | <0.001 | 0.739 | – | NS | – |
| pVO2, % of predicted | 0.851 (0.799–0.905) | <0.001 | 0.749 | – | NS | – |
| VE/VCO2 slope | 1.017 (1.069–1.146) | <0.001 | 0.724 | – | NS | – |
| CP% | 0.998 (0.997–0.999) | <0.001 | 0.778 | 0.998 (0.997–1.000) | 0.052 | 0.705 |
| LP or P variants | 2.395 (1.171–4.856) | 0.013 | 0.609 | – | NS | – |
| Multivariate Cox Proportional Survival Analysis | ||||||
|---|---|---|---|---|---|---|
| HF Endpoint | SCD Endpoint | |||||
| H.R. (95% C.I.) | p-Values | C-Index | H.R. (95% C.I.) | p-Values | C-Index | |
| LAd | 1.083 (1.039–1.130) | <0.001 | 0.839 | 1.078 (1.005–1.163) | 0.0485 | 0.738 |
| CP% | 0.998 (0.997–0.999) | <0.001 | 0.998 (0.9996–1.000) | 0.0488 | ||
| VE/VCO2 slope | 1.044 (0.999–1.090) | 0.05 | ||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magrì, D.; Mastromarino, V.; Gallo, G.; Zachara, E.; Re, F.; Agostoni, P.; Giordano, D.; Rubattu, S.; Forte, M.; Cotugno, M.; et al. Risk Stratification in Hypertrophic Cardiomyopathy. Insights from Genetic Analysis and Cardiopulmonary Exercise Testing. J. Clin. Med. 2020, 9, 1636. https://doi.org/10.3390/jcm9061636
Magrì D, Mastromarino V, Gallo G, Zachara E, Re F, Agostoni P, Giordano D, Rubattu S, Forte M, Cotugno M, et al. Risk Stratification in Hypertrophic Cardiomyopathy. Insights from Genetic Analysis and Cardiopulmonary Exercise Testing. Journal of Clinical Medicine. 2020; 9(6):1636. https://doi.org/10.3390/jcm9061636
Chicago/Turabian StyleMagrì, Damiano, Vittoria Mastromarino, Giovanna Gallo, Elisabetta Zachara, Federica Re, Piergiuseppe Agostoni, Dario Giordano, Speranza Rubattu, Maurizio Forte, Maria Cotugno, and et al. 2020. "Risk Stratification in Hypertrophic Cardiomyopathy. Insights from Genetic Analysis and Cardiopulmonary Exercise Testing" Journal of Clinical Medicine 9, no. 6: 1636. https://doi.org/10.3390/jcm9061636