Major Lipids, Apolipoproteins, and Alterations of Gut Microbiota

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Methods
2.1. Study Subjects
2.2. Data Collection and Group Definitions
2.3. DNA Extraction from Fecal Samples and 16S rRNA Gene Sequencing
2.4. Statistical Analysis
3. Results
3.1. Subject Demographics
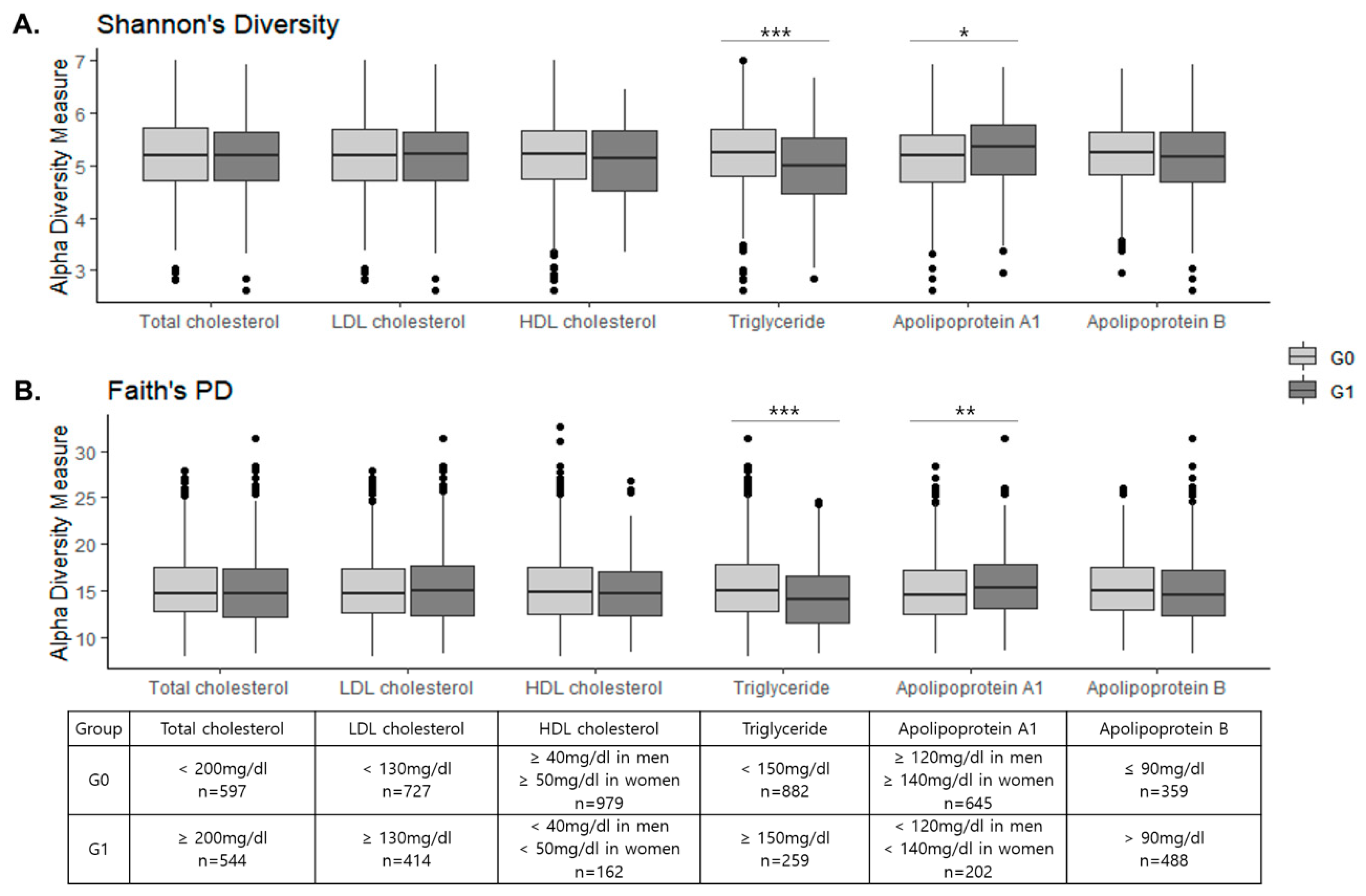
3.2. Fecal Microbial Diversity between the G0 and G1 Groups
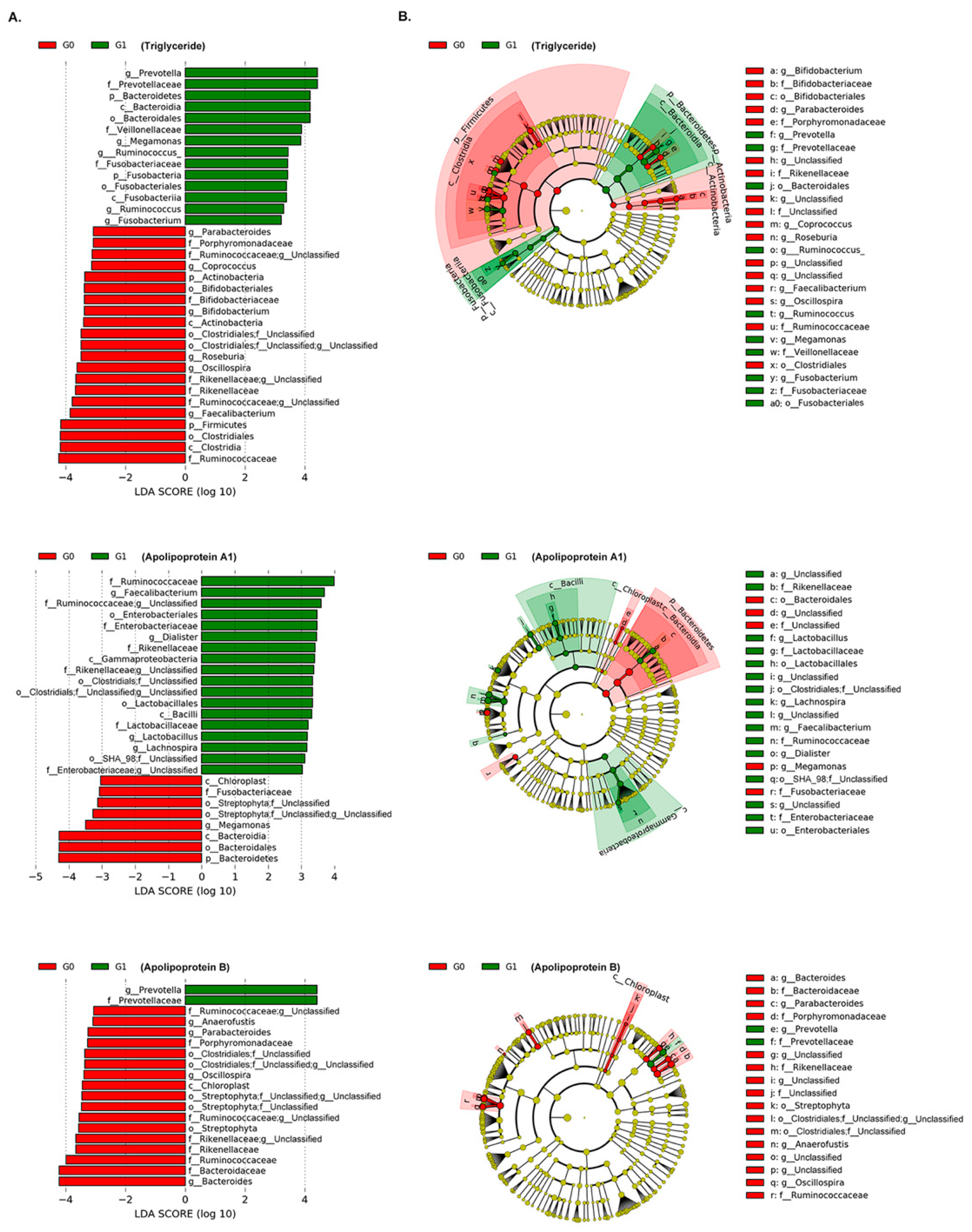
3.3. Association of Gut Microbiota with Lipid Measurements
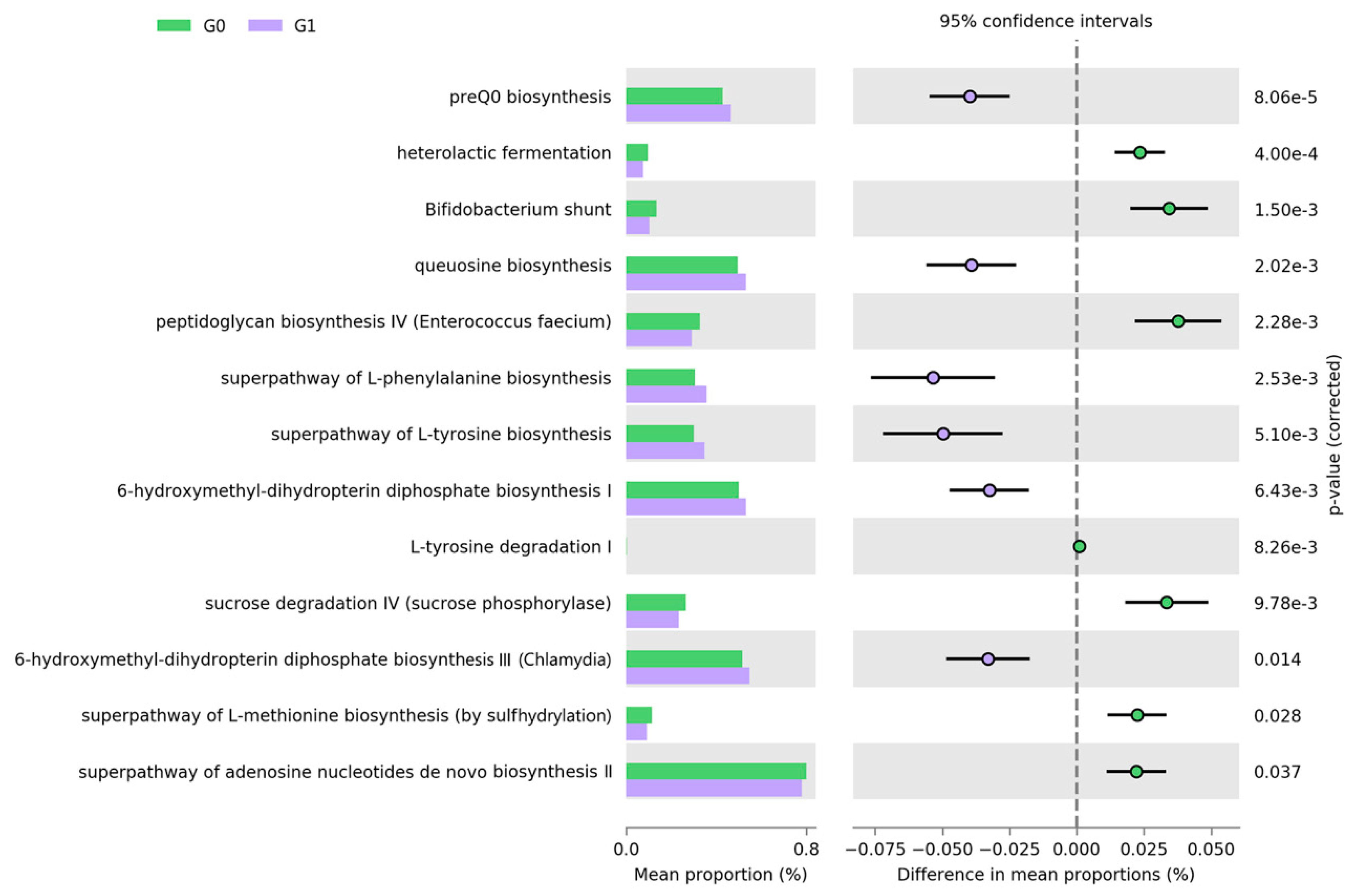
3.4. Functional Profiling Related to the Multiple Lipid Measurements
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Matey-Hernandez, M.L.; Williams, F.M.K.; Potter, T.; Valdes, A.M.; Spector, T.D.; Menni, C. Genetic and microbiome influence on lipid metabolism and dyslipidemia. Physiol. Genom. 2018, 50, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Klarin, D.; Damrauer, S.M.; Cho, K.; Sun, Y.V.; Teslovich, T.M.; Honerlaw, J.; Gagnon, D.R.; DuVall, S.L.; Li, J.; Peloso, G.M.; et al. Genetics of blood lipids among ~300,000 multi-ethnic participants of the Million Veteran Program. Nat. Genet. 2018, 50, 1514–1523. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, L.; Aldujaili, E.; Drummond, S. Effects of advice on dietary intake and/or physical activity on body composition, blood lipids and insulin resistance following a low-fat, sucrose-containing, high-carbohydrate, energy-restricted diet. Int. J. Food Sci. Nutr. 2007, 58, 383–397. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, A.A.; Ross, R.P.; Fitzgerald, G.F.; Caplice, N.; Stanton, C. Role of the gut in modulating lipoprotein metabolism. Curr. Cardiol. Rep. 2014, 16, 515. [Google Scholar] [CrossRef]
- Da Silva, P.M.; Duarte, J.S.; von Hafe, P.; Gil, V.; de Oliveira, J.N.; de Sousa, G. Standardization of laboratory and lipid profile evaluation: A call for action with a special focus in 2016 ESC/EAS dyslipidaemia guidelines—Full report. Atheroscler. Suppl. 2018, 31, e1–e12. [Google Scholar] [CrossRef]
- Clemente, J.C.; Ursell, L.K.; Parfrey, L.W.; Knight, R. The impact of the gut microbiota on human health: An integrative view. Cell 2012, 148, 1258–1270. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Raka, F.; Adeli, K. The Role of the Gut Microbiota in Lipid and Lipoprotein Metabolism. J. Clin. Med. 2019, 8, 2227. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Koonen, D.; Hofker, M.; Fu, J. Gut microbiome and lipid metabolism: From associations to mechanisms. Curr. Opin. Lipidol. 2016, 27, 216–224. [Google Scholar] [CrossRef]
- Leone, V.; Gibbons, S.M.; Martinez, K.; Hutchison, A.L.; Huang, E.Y.; Cham, C.M.; Pierre, J.F.; Heneghan, A.F.; Nadimpalli, A.; Hubert, N.; et al. Effects of diurnal variation of gut microbes and high-fat feeding on host circadian clock function and metabolism. Cell Host Microbe 2015, 17, 681–689. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Guryn, K.; Hubert, N.; Frazier, K.; Urlass, S.; Musch, M.W.; Ojeda, P.; Pierre, J.F.; Miyoshi, J.; Sontag, T.J.; Cham, C.M.; et al. Small Intestine Microbiota Regulate Host Digestive and Absorptive Adaptive Responses to Dietary Lipids. Cell Host Microbe 2018, 23, 458–469.e5. [Google Scholar] [CrossRef]
- Rebolledo, C.; Cuevas, A.; Zambrano, T.; Acuna, J.J.; Jorquera, M.A.; Saavedra, K.; Martinez, C.; Lanas, F.; Seron, P.; Salazar, L.A.; et al. Bacterial Community Profile of the Gut Microbiota Differs between Hypercholesterolemic Subjects and Controls. Biomed. Res. Int. 2017, 2017, 8127814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J.; Bonder, M.J.; Cenit, M.C.; Tigchelaar, E.F.; Maatman, A.; Dekens, J.A.; Brandsma, E.; Marczynska, J.; Imhann, F.; Weersma, R.K.; et al. The Gut Microbiome Contributes to a Substantial Proportion of the Variation in Blood Lipids. Circ. Res. 2015, 117, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.W.; Priya, S.; Blekhman, R.; Bordenstein, S.R. Gut microbiota diversity across ethnicities in the United States. PLoS Biol. 2018, 16, e2006842. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.; Cho, Y.K.; Kim, Y.; Sung, E.; Ahn, J.; Jung, H.S.; Yun, K.E.; Shin, H.; Ryu, S. Nonheavy Drinking and Worsening of Noninvasive Fibrosis Markers in Nonalcoholic Fatty Liver Disease: A Cohort Study. Hepatology 2019, 69, 64–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.; Kim, B.K.; Yun, K.E.; Cho, J.; Zhang, Y.; Rampal, S.; Zhao, D.; Jung, H.S.; Choi, Y.; Ahn, J.; et al. Metabolically-healthy obesity and coronary artery calcification. J. Am. Coll. Cardiol. 2014, 63, 2679–2686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feeman, W.E. Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA 2001, 285, 2486–2497. [Google Scholar]
- Brunzell, J.D.; Davidson, M.; Furberg, C.D.; Goldberg, R.B.; Howard, B.V.; Stein, J.H.; Witztum, J.L. Lipoprotein management in patients with cardiometabolic risk: Consensus conference report from the American Diabetes Association and the American College of Cardiology Foundation. J. Am. Coll. Cardiol. 2008, 51, 1512–1524. [Google Scholar] [CrossRef] [Green Version]
- Catapano, A.L.; Graham, I.; De Backer, G.; Wiklund, O.; Chapman, M.J.; Drexel, H.; Hoes, A.W.; Jennings, C.S.; Landmesser, U.; Pedersen, T.R.; et al. 2016 ESC/EAS Guidelines for the Management of Dyslipidaemias. Eur. Heart J. 2016, 37, 2999–3058. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.N.; Yun, Y.; Ryu, S.; Chang, Y.; Kwon, M.J.; Cho, J.; Shin, H.; Kim, H.L. Correlation between gut microbiota and personality in adults: A cross-sectional study. Brain Behav. Immun. 2018, 69, 374–385. [Google Scholar] [CrossRef]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolyen, E.; Rideout, J.; Dillon, M.; Bokulich, N.; Abnet, C.; Al-Ghalith, G.; Alexander, H.; Alm, E.; Arumugam, M.; Asnicar, F.; et al. QIIME 2: Reproducible, interactive, scalable, and extensible microbiome data science. PeerJ Prepr. 2018, 2, e27295. [Google Scholar] [CrossRef] [PubMed]
- Faith, D.P.; Baker, A.M. Phylogenetic diversity (PD) and biodiversity conservation: Some bioinformatics challenges. Evol. Bioinform. Online 2006, 2, 121–128. [Google Scholar] [CrossRef]
- Lozupone, C.; Lladser, M.E.; Knights, D.; Stombaugh, J.; Knight, R. UniFrac: An effective distance metric for microbial community comparison. ISME J. 2011, 5, 169–172. [Google Scholar] [CrossRef] [Green Version]
- Bray, J.R.; Curtis, J.T. An ordination of the upland forest communities of Southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Mandal, S.; Van Treuren, W.; White, R.A.; Eggesbo, M.; Knight, R.; Peddada, S.D. Analysis of composition of microbiomes: A novel method for studying microbial composition. Microb. Ecol. Health Dis. 2015, 26, 27663. [Google Scholar] [CrossRef] [Green Version]
- Morgan, X.C.; Tickle, T.L.; Sokol, H.; Gevers, D.; Devaney, K.L.; Ward, D.V.; Reyes, J.A.; Shah, S.A.; LeLeiko, N.; Snapper, S.B.; et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012, 13, R79. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G. PICRUSt2: An improved and extensible approach for metagenome inference. BioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Barbera, P.; Kozlov, A.M.; Czech, L.; Morel, B.; Darriba, D.; Flouri, T.; Stamatakis, A. EPA-ng: Massively Parallel Evolutionary Placement of Genetic Sequences. Syst. Biol. 2019, 68, 365–369. [Google Scholar] [CrossRef]
- Czech, L.; Stamatakis, A. Scalable methods for analyzing and visualizing phylogenetic placement of metagenomic samples. PLoS ONE 2019, 14, e0217050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caspi, R.; Billington, R.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Midford, P.E.; Ong, Q.; Ong, W.K.; et al. The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res. 2018, 46, D633–D639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allayee, H.; Hazen, S.L. Contribution of Gut Bacteria to Lipid Levels: Another Metabolic Role for Microbes? Circ. Res. 2015, 117, 750–754. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, J.K.; Waters, J.L.; Poole, A.C.; Sutter, J.L.; Koren, O.; Blekhman, R.; Beaumont, M.; Van Treuren, W.; Knight, R.; Bell, J.T.; et al. Human genetics shape the gut microbiome. Cell 2014, 159, 789–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escobar, J.S.; Klotz, B.; Valdes, B.E.; Agudelo, G.M. The gut microbiota of Colombians differs from that of Americans, Europeans and Asians. BMC Microbiol. 2014, 14, 311. [Google Scholar] [CrossRef] [Green Version]
- Gophna, U.; Konikoff, T.; Nielsen, H.B. Oscillospira and related bacteria-From metagenomic species to metabolic features. Environ. Microbiol. 2017, 19, 835–841. [Google Scholar] [CrossRef] [Green Version]
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojarvi, J.; Kootte, R.S.; Bartelsman, J.F.; Dallinga-Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012, 143, 913–916.e7. [Google Scholar] [CrossRef]
- Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Cheng, J.; Duncan, A.E.; Kau, A.L.; Griffin, N.W.; Lombard, V.; Henrissat, B.; Bain, J.R.; et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 2013, 341, 1241214. [Google Scholar] [CrossRef] [Green Version]
- Keren, N.; Konikoff, F.M.; Paitan, Y.; Gabay, G.; Reshef, L.; Naftali, T.; Gophna, U. Interactions between the intestinal microbiota and bile acids in gallstones patients. Environ. Microbiol. Rep. 2015, 7, 874–880. [Google Scholar] [CrossRef]
- Bo, T.; Shao, S.; Wu, D.; Niu, S.; Zhao, J.; Gao, L. Relative variations of gut microbiota in disordered cholesterol metabolism caused by high-cholesterol diet and host genetics. Microbiologyopen 2017, 6, e00491. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, J.S.; Ridlon, J.M.; Hylemon, P.B.; Thacker, L.R.; Heuman, D.M.; Smith, S.; Sikaroodi, M.; Gillevet, P.M. Linkage of gut microbiome with cognition in hepatic encephalopathy. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G168–G175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loubinoux, J.; Bronowicki, J.P.; Pereira, I.A.; Mougenel, J.L.; Faou, A.E. Sulfate-reducing bacteria in human feces and their association with inflammatory bowel diseases. FEMS Microbiol. Ecol. 2002, 40, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, M.D.; Fazio, S. From Lipids to Inflammation: New Approaches to Reducing Atherosclerotic Risk. Circ. Res. 2016, 118, 732–749. [Google Scholar] [CrossRef]
- Tall, A.R.; Yvan-Charvet, L. Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 2015, 15, 104–116. [Google Scholar] [CrossRef] [Green Version]
- Sniderman, A.D.; Thanassoulis, G.; Glavinovic, T.; Navar, A.M.; Pencina, M.; Catapano, A.; Ference, B.A. Apolipoprotein B Particles and Cardiovascular Disease: A Narrative Review. JAMA Cardiol. 2019, 4, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Feingold, K.R.; Grunfeld, C. The acute phase response inhibits reverse cholesterol transport. J. Lipid Res. 2010, 51, 682–684. [Google Scholar] [CrossRef] [Green Version]
- Granado-Serrano, A.B.; Martin-Gari, M.; Sanchez, V.; Riart Solans, M.; Berdun, R.; Ludwig, I.A.; Rubio, L.; Vilaprinyo, E.; Portero-Otin, M.; Serrano, J.C.E. Faecal bacterial and short-chain fatty acids signature in hypercholesterolemia. Sci. Rep. 2019, 9, 1772. [Google Scholar] [CrossRef]
- Lim, M.Y.; You, H.J.; Yoon, H.S.; Kwon, B.; Lee, J.Y.; Lee, S.; Song, Y.M.; Lee, K.; Sung, J.; Ko, G. The effect of heritability and host genetics on the gut microbiota and metabolic syndrome. Gut 2017, 66, 1031–1038. [Google Scholar] [CrossRef]
- Ghazalpour, A.; Cespedes, I.; Bennett, B.J.; Allayee, H. Expanding role of gut microbiota in lipid metabolism. Curr. Opin. Lipidol. 2016, 27, 141–147. [Google Scholar] [CrossRef]
- Zhang, Y.; Kim, B.K.; Chang, Y.; Ryu, S.; Cho, J.; Lee, W.Y.; Rhee, E.J.; Kwon, M.J.; Rampal, S.; Zhao, D.; et al. Thyroid hormones and coronary artery calcification in euthyroid men and women. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2128–2134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riviere, A.; Selak, M.; Lantin, D.; Leroy, F.; De Vuyst, L. Bifidobacteria and Butyrate-Producing Colon Bacteria: Importance and Strategies for Their Stimulation in the Human Gut. Front. Microbiol. 2016, 7, 979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Y.H.; Nishimura, Y.; Hishikawa, D.; Tsuzuki, H.; Miyahara, H.; Gotoh, C.; Choi, K.C.; Feng, D.D.; Chen, C.; Lee, H.G.; et al. Acetate and propionate short chain fatty acids stimulate adipogenesis via GPCR43. Endocrinology 2005, 146, 5092–5099. [Google Scholar] [CrossRef] [Green Version]
- Demigne, C.; Morand, C.; Levrat, M.A.; Besson, C.; Moundras, C.; Remesy, C. Effect of propionate on fatty acid and cholesterol synthesis and on acetate metabolism in isolated rat hepatocytes. Br. J. Nutr. 1995, 74, 209–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, F.; Du, Y.; Lv, Z.; Chen, S.; Zhu, J.; Sheng, H.; Guo, F. Effects of essential amino acids on lipid metabolism in mice and humans. J. Mol. Endocrinol. 2016, 57, 223–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Overall | Male | Female | p Value a | |
|---|---|---|---|---|
| No. of subjects | 1141 | 712 | 429 | |
| Age (years), mean (SD) | 45.2 (8.7) | 45.7 (8.7) | 44.4 (8.7) | 0.015 |
| BMI (kg/m2) | 23.5 (3.1) | 24.6 (2.8) | 21.8 (2.8) | <0.001 |
| Current smoker (%) | 17.2 | 26.9 | 1.17 | <0.001 |
| Alcohol intake (%) b | 38.1 | 55.2 | 9.7 | <0.001 |
| Vigorous exercise (%) c | 14.5 | 14.7 | 14.2 | 0.474 |
| SBP (mmHg), mean (SD) | 109.4 (13.5) | 114.2(12.2) | 101.5 (11.8) | <0.001 |
| DBP (mmHg), mean (SD) | 71.0 (10.1) | 74.3 (9.3) | 65.5 (9.0) | <0.001 |
| Fasting glucose (mg/dL), mean (SD) | 94.7 (14.3) | 97.3 (16.5) | 90.4 (7.7) | <0.001 |
| Total cholesterol (mg/dL), mean (SD) | 198.7 (32.4) | 202.2 (32.6) | 192.9 (31.2) | <0.001 |
| G0: <200 mg/dL (N) | 597 | 348 | 249 | |
| G1: ≥200 mg/dL (N) | 544 | 364 | 180 | |
| LDL cholesterol (mg/dL), mean (SD) | 120.7 (29.4) | 125.5 (28.7) | 112.8 (28.9) | <0.001 |
| G0: <130 mg/dL (N) | 727 | 402 | 325 | |
| G1: ≥130 mg/dL (N) | 414 | 310 | 104 | |
| HDL cholesterol (mg/dL), mean (SD) | 57.0 (14.4) | 53.1 (13.0) | 63.4 (14.4) | <0.001 |
| G0: ≥40 mg/dL in men ≥50 mg/dL in women | 979 | 622 | 357 | |
| G1: <40 mg/dL in men <50 mg/dL in women | 162 | 90 | 72 | |
| Triglyceride (mg/dL), mean (SD) | 120.4 (75.3) | 137.4 (83.5) | 92.1 (47.6) | <0.001 |
| G0: <150 mg/dL (N) | 882 | 492 | 390 | |
| G1: ≥150 mg/dL (N) | 259 | 220 | 39 | |
| ApoA1 (mg/dL), mean (SD) | 143.7 (22.4) | 140.5 (22.8) | 149.1 (20.7) | <0.001 |
| G0: ≥120 mg/dL in men ≥140 mg/dL in women | 645 | 443 | 202 | |
| G1: <120 mg/dL in men <140 mg/dL in women | 202 | 91 | 111 | |
| ApoB (mg/dL), mean (SD) | 95.5 (23.1) | 101.3 (21.9) | 85.6 (21.6) | <0.001 |
| G0: <90 mg/dL (N) | 359 | 166 | 193 | |
| G1: ≥90 mg/dL (N) | 488 | 368 | 120 | |
| hsCRP (mg/dL), p25/p50/p75 | 0.02/0.04/0.08 | 0.03/0.05/0.09 | 0.02/0.03/0.06 | <0.001 |
| Group | Total Cholesterol (n = 1141) | LDL Cholesterol (n = 1141) | HDL Cholesterol (n = 1141) | Triglyceride (n = 1141) | Apolipoprotein A1 (n = 847) | Apolipoprotein B (n = 847) |
|---|---|---|---|---|---|---|
| G0 | <200 mg/dL n = 597 | <130 mg/dL n = 727 | ≥40 mg/dL in men ≥50 mg/dL in women n = 979 | <150 mg/dL n = 882 | ≥120 mg/dL in men ≥140 mg/dL in women n = 645 | <90 mg/dL n = 359 |
| G1 | ≥200 mg/dL n = 544 | ≥130 mg/dL n = 414 | <40 mg/dL in men <50 mg/dL in women n = 162 | ≥150 mg/dL n = 259 | <120 mg/dL in men <140 mg/dL in women n = 202 | ≥90 mg/dL n = 488 |
| p-value a | p-value a | p-value a | p-value a | p-value a | p-value a | |
| Unweighted UniFrac distance | 0.199 | 0.089 | 0.395 | 0.001 ** | 0.009 ** | 0.005 ** |
| Weighted UniFrac distance | 0.152 | 0.057 | 0.610 | 0.001 ** | 0.009 ** | 0.005 ** |
| Bray–Curtis_dissimilarity | 0.753 | 0.260 | 0.432 | 0.001 ** | 0.012 ** | 0.001 ** |
| W a (Coefficients b) | ||||
|---|---|---|---|---|
| Taxa level a | Taxonomic Assignment | Triglyceride G0 vs. G1 | Apo A1 G0 vs. G1 | Apo B G0 vs. G1 |
| Phylum | p__Fusobacteria | 12 (0.015 **) | ||
| Class | p__Fusobacteria; c__Fusobacteriia | 27 (0.015 **) | ||
| p__Firmicutes; c__Bacilli | 23 (0.010 **) | |||
| Order | p__Fusobacteria; c__Fusobacteriia; o__Fusobacteriales | 45 (0.015 **) | ||
| p__Proteobacteria; c__Gammaproteobacteria; o__Enterobacteriales | 41 (0.014 *) | |||
| p__Firmicutes; c__Bacilli; o__Lactobacillales | 40 (0.008 *) | |||
| p__Tenericutes; c__Mollicutes; o__RF39 | 40 (0.008 **) | |||
| Family | p__Fusobacteria; c__Fusobacteriia; o__Fusobacteriales; f__Fusobacteriaceae | 83 (0.015 **) | ||
| p__Bacteroidetes; c__Bacteroidia; o__Bacteroidales; f__Prevotellaceae | 81 (0.024) | 84 (0.033) | ||
| p__Proteobacteria; c__Gammaproteobacteria; o__Enterobacteriales; f__Enterobacteriaceae | 71 (0.014 *) | |||
| p__Bacteroidetes; c__Bacteroidia; o__Bacteroidales; f__Odoribacteraceae | 68 (0.011 **) | |||
| p__Bacteroidetes; c__Bacteroidia; o__Bacteroidales; f__Rikenellaceae | 69 (−0.002) | |||
| Genus | p__Firmicutes; c__Clostridia; o__Clostridiales; f__Ruminococcaceae; g__Oscillospira | 223 (−0.022 **) | 201 (−0.006) | |
| p__Fusobacteria; c__Fusobacteriia; o__Fusobacteriales; f__Fusobacteriaceae; g__Fusobacterium | 218 (0.011 *) | |||
| p__Firmicutes; c__Clostridia; o__Clostridiales; f__Veillonellaceae; g__Megamonas | 217 (0.005) | |||
| p__Firmicutes; c__Clostridia; o__Clostridiales; f__Veillonellaceae; g__Megasphaera | 217 (0.004) | |||
| p__Firmicutes; c__Clostridia; o__Clostridiales; f__Veillonellaceae; g__Acidaminococcus | 216 (0.004 *) | |||
| p__Bacteroidetes; c__Bacteroidia; o__Bacteroidales; f__Prevotellaceae; g__Prevotella | 215 (0.024) | 226 (0.033) | ||
| p__Firmicutes; c__Clostridia; o__Clostridiales; f__Lachnospiraceae; g__Anaerostipes | 211 (−0.010 **) | |||
| p__Bacteroidetes; c__Bacteroidia; o__Bacteroidales; f__Odoribacteraceae; g__Odoribacter | 196 (0.010 **) | |||
| p__Firmicutes; c__Clostridia; o__Clostridiales; f__Lachnospiraceae; g__Lachnospira | 186 (0.015 **) | |||
| p__Firmicutes; c__Bacilli; o__Lactobacillales; f__Lactobacillaceae; g__Lactobacillus | 182 (0.003) | |||
| p__Firmicutes; c__Clostridia; o__Clostridiales; f__Clostridiaceae; g__Clostridium | 197 (−0.002) | |||
| Species | p__Bacteroidetes; c__Bacteroidia; o__Bacteroidales; f__Bacteroidaceae; g__Bacteroides;s__caccae | 310 (0.012 **) | ||
| p__Bacteroidetes; c__Bacteroidia; o__Bacteroidales; f__Prevotellaceae; g__Prevotella;s__copri | 339 (0.046 *) | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yun, K.E.; Kim, J.; Kim, M.-h.; Park, E.; Kim, H.-L.; Chang, Y.; Ryu, S.; Kim, H.-N. Major Lipids, Apolipoproteins, and Alterations of Gut Microbiota. J. Clin. Med. 2020, 9, 1589. https://doi.org/10.3390/jcm9051589
Yun KE, Kim J, Kim M-h, Park E, Kim H-L, Chang Y, Ryu S, Kim H-N. Major Lipids, Apolipoproteins, and Alterations of Gut Microbiota. Journal of Clinical Medicine. 2020; 9(5):1589. https://doi.org/10.3390/jcm9051589
Chicago/Turabian StyleYun, Kyung Eun, Jimin Kim, Mi-hyun Kim, Eunkyo Park, Hyung-Lae Kim, Yoosoo Chang, Seungho Ryu, and Han-Na Kim. 2020. "Major Lipids, Apolipoproteins, and Alterations of Gut Microbiota" Journal of Clinical Medicine 9, no. 5: 1589. https://doi.org/10.3390/jcm9051589