Natural History and Management of Familial Paraganglioma Syndrome Type 1: Long-Term Data from a Large Family

 , , ,
, , , 
Abstract
:1. Introduction
2. Patients and Methods
3. Results
3.1. Family Case Series
3.2. Clinical Presentation and Diagnosis
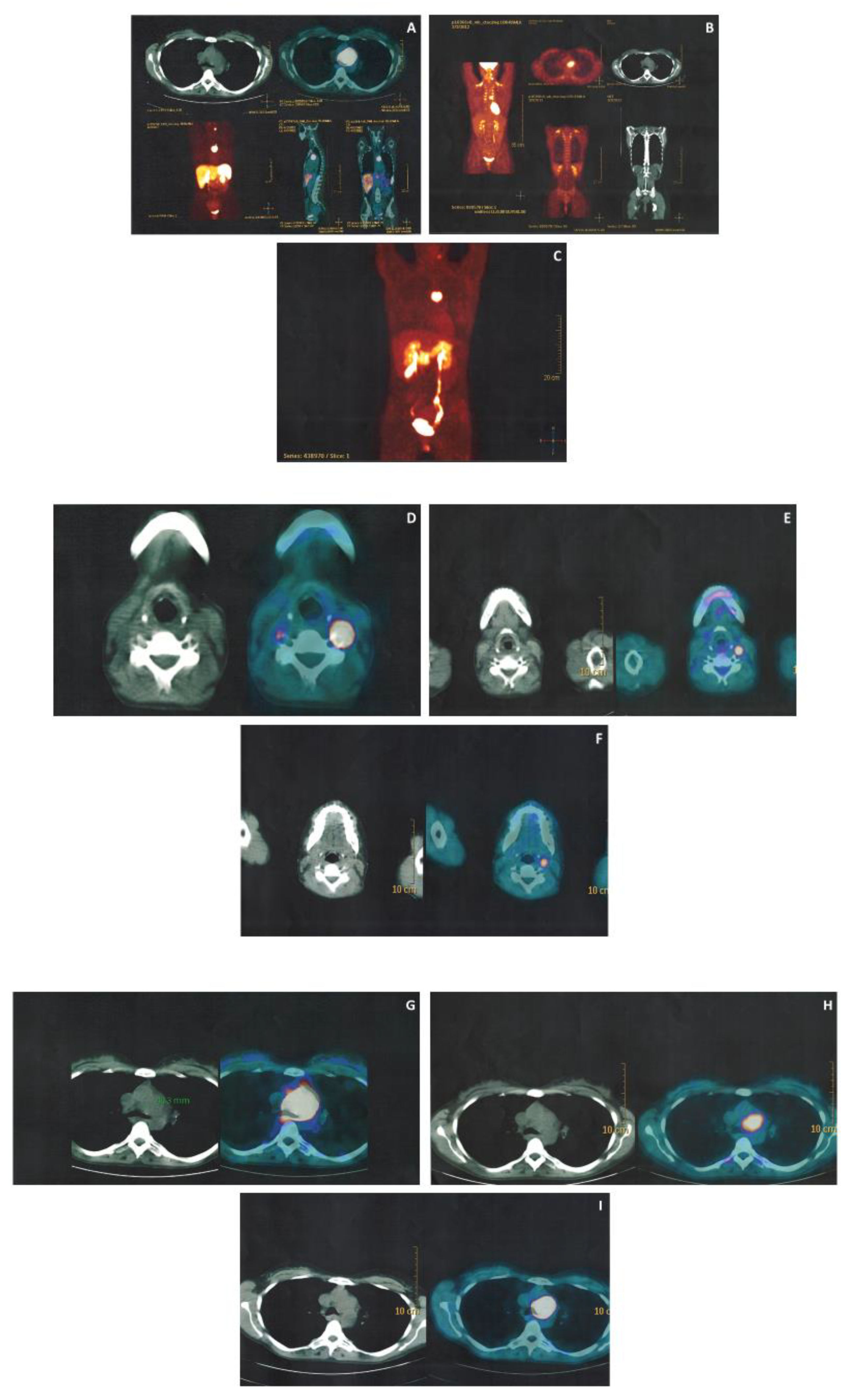
3.3. Imaging Work up
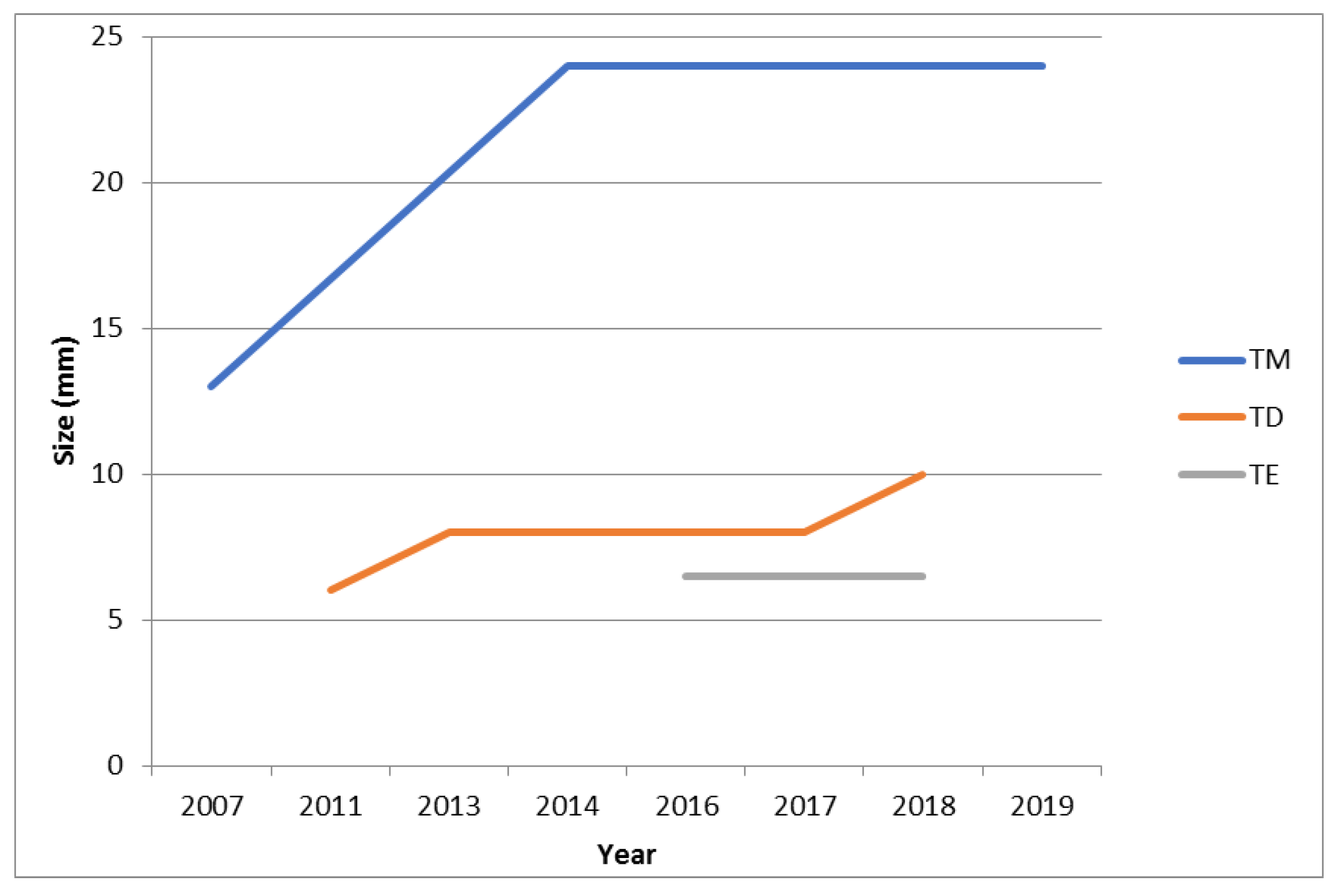
3.4. Treatment and Follow-up
4. Discussion
4.1. Natural History
4.2. Current Diagnostic Work-up
4.2.1. The Role of Serum and Urinary Markers
4.2.2. The Role of Magnetic Resonance Imaging
4.2.3. The Role of Ultrasonography
4.2.4. Functional Imaging: “Back to the Future”
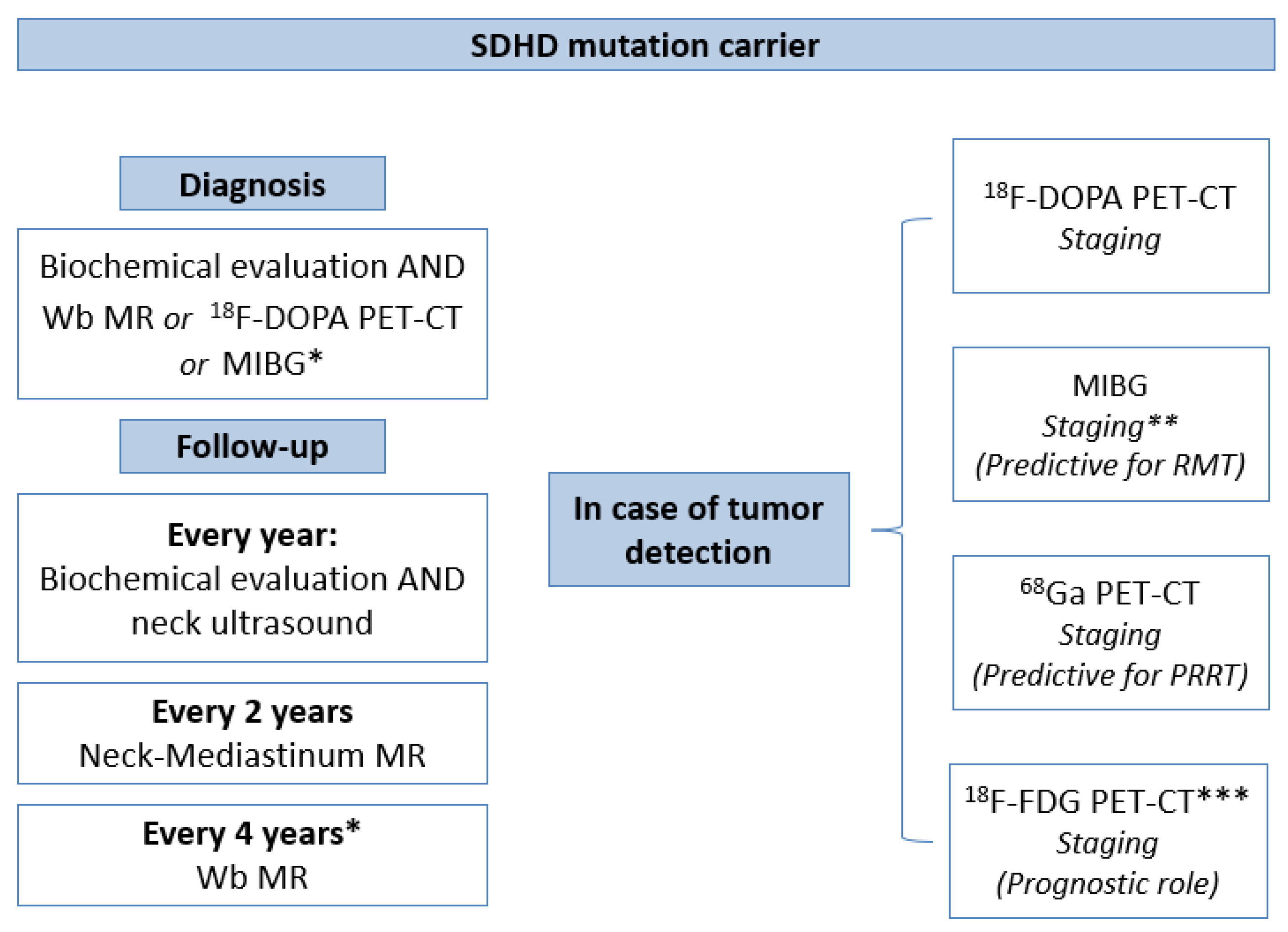
4.3. Our Novel Insight for A Precision Diagnostic Algorithm
4.4. Therapy
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Benn, D.E.; Robinson, B.G.; Clifton-Bligh, R.J. 15 Years of Paraganglioma: Clinical manifestations of paraganglioma syndromes types 1–5. Endocr. Relat. Cancer 2015, 22, T91–T103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sbardella, E.; Cranston, T.; Isidori, A.M.; Shine, B.; Pal, A.; Jafar-Mohammadi, B.; Sadler, G.; Mihai, R.; Grossman, A.B. Routine genetic screening with a multi-gene panel in patients with pheochromocytomas. Endocrine 2018, 59, 175–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baysal, B.E.; Ferrell, R.E.; Willett-Brozick, J.E.; Lawrence, E.C.; Myssiorek, D.; Bosch, A.; van der Mey, A.; Taschner, P.E.; Rubinstein, W.S.; Myers, E.N.; et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 2000, 287, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Baysal, B.E.; Maher, E.R. 15 Years of Paraganglioma: Genetics and mechanism of pheochromocytoma-paraganglioma syndromes characterized by germline SDHB and SDHD mutations. Endocr. Relat. Cancer 2015, 22, T71–T82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannelli, M.; Rapizzi, E.; Fucci, R.; Canu, L.; Ercolino, T.; Luconi, M.; Young, W.F., Jr. 15 Years of paraganglioma: Metabolism and pheochromocytoma/paraganglioma. Endocr. Relat. Cancer 2015, 22, T83–T90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenders, J.W.; Duh, Q.Y.; Eisenhofer, G.; Gimenez-Roqueplo, A.P.; Grebe, S.K.; Murad, M.H.; Naruse, M.; Pacak, K.; Young, W.F., Jr.; Endocrine, S. Pheochromocytoma and paraganglioma: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2014, 99, 1915–1942. [Google Scholar] [CrossRef] [PubMed]
- Neumann, H.P.; Pawlu, C.; Peczkowska, M.; Bausch, B.; McWhinney, S.R.; Muresan, M.; Buchta, M.; Franke, G.; Klisch, J.; Bley, T.A.; et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA 2004, 292, 943–951. [Google Scholar] [CrossRef] [Green Version]
- Bausch, B.; Wellner, U.; Bausch, D.; Schiavi, F.; Barontini, M.; Sanso, G.; Walz, M.K.; Peczkowska, M.; Weryha, G.; Dall’igna, P.; et al. Long-term prognosis of patients with pediatric pheochromocytoma. Endocr. Relat. Cancer 2014, 21, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Crona, J.; Taieb, D.; Pacak, K. New Perspectives on Pheochromocytoma and Paraganglioma: Toward a Molecular Classification. Endocr Rev. 2017, 38, 489–515. [Google Scholar] [CrossRef]
- Van Hulsteijn, L.T.; Dekkers, O.M.; Hes, F.J.; Smit, J.W.; Corssmit, E.P. Risk of malignant paraganglioma in SDHB-mutation and SDHD-mutation carriers: A systematic review and meta-analysis. J. Med. Genet. 2012, 49, 768–776. [Google Scholar] [CrossRef]
- Casey, R.T.; Warren, A.Y.; Martin, J.E.; Challis, B.G.; Rattenberry, E.; Whitworth, J.; Andrews, K.A.; Roberts, T.; Clark, G.R.; West, H.; et al. Clinical and Molecular Features of Renal and Pheochromocytoma/Paraganglioma Tumor Association Syndrome (RAPTAS): Case Series and Literature Review. J. Clin. Endocrinol. Metab. 2017, 102, 4013–4022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill, A.J.; Chou, A.; Vilain, R.; Clarkson, A.; Lui, M.; Jin, R.; Tobias, V.; Samra, J.; Goldstein, D.; Smith, C.; et al. Immunohistochemistry for SDHB divides gastrointestinal stromal tumors (GISTs) into 2 distinct types. Am. J. Surg. Pathol. 2010, 34, 636–644. [Google Scholar] [CrossRef] [PubMed]
- Gill, A.J. Succinate dehydrogenase (SDH)-deficient neoplasia. Histopathology 2018, 72, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Denes, J.; Swords, F.; Rattenberry, E.; Stals, K.; Owens, M.; Cranston, T.; Xekouki, P.; Moran, L.; Kumar, A.; Wassif, C.; et al. Heterogeneous genetic background of the association of pheochromocytoma/paraganglioma and pituitary adenoma: Results from a large patient cohort. J. Clin. Endocrinol. Metab. 2015, 100, E531–E541. [Google Scholar] [CrossRef]
- Rednam, S.P.; Erez, A.; Druker, H.; Janeway, K.A.; Kamihara, J.; Kohlmann, W.K.; Nathanson, K.L.; States, L.J.; Tomlinson, G.E.; Villani, A.; et al. Von Hippel-Lindau and Hereditary Pheochromocytoma/Paraganglioma Syndromes: Clinical Features, Genetics, and Surveillance Recommendations in Childhood. Clin. Cancer Res. 2017, 23, e68–e75. [Google Scholar] [CrossRef] [Green Version]
- Eisenhofer, G.; Lenders, J.W.; Timmers, H.; Mannelli, M.; Grebe, S.K.; Hofbauer, L.C.; Bornstein, S.R.; Tiebel, O.; Adams, K.; Bratslavsky, G.; et al. Measurements of plasma methoxytyramine, normetanephrine, and metanephrine as discriminators of different hereditary forms of pheochromocytoma. Clin. Chem. 2011, 57, 411–420. [Google Scholar] [CrossRef]
- Rao, D.; Peitzsch, M.; Prejbisz, A.; Hanus, K.; Fassnacht, M.; Beuschlein, F.; Brugger, C.; Fliedner, S.; Langton, K.; Pamporaki, C.; et al. Plasma methoxytyramine: Clinical utility with metanephrines for diagnosis of pheochromocytoma and paraganglioma. Eur. J. Endocrinol. 2017, 177, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Thompson, M.J.; Parameswaran, V.; Burgess, J.R. Clinical utility of chromogranin A for the surveillance of succinate dehydrogenase B- and succinate dehydrogenase D-related paraganglioma. Ann. Clin. Biochem. 2019, 56, 163–169. [Google Scholar] [CrossRef]
- Heesterman, B.L.; Bayley, J.P.; Tops, C.M.; Hes, F.J.; van Brussel, B.T.; Corssmit, E.P.; Hamming, J.F.; van der Mey, A.G.; Jansen, J.C. High prevalence of occult paragangliomas in asymptomatic carriers of SDHD and SDHB gene mutations. Eur. J. Hum. Genet. 2013, 21, 469–470. [Google Scholar] [CrossRef] [Green Version]
- Else, T.; Greenberg, S.; Fishbein, L. Hereditary Paraganglioma-Pheochromocytoma Syndromes. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Dreijerink, K.M.A.; Rijken, J.A.; Compaijen, C.J.; Timmers, H.; van der Horst-Schrivers, A.N.A.; van Leeuwaarde, R.S.; van Dam, P.S.; Leemans, C.R.; van Dam, E.; Dickhoff, C.; et al. Biochemically Silent Sympathetic Paraganglioma, Pheochromocytoma, or Metastatic Disease in SDHD Mutation Carriers. J. Clin. Endocrinol. Metab. 2019, 104, 5421–5426. [Google Scholar] [CrossRef]
- Favier, J.; Amar, L.; Gimenez-Roqueplo, A.P. Paraganglioma and phaeochromocytoma: From genetics to personalized medicine. Nat. Rev. Endocrinol. 2015, 11, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Muth, A.; Crona, J.; Gimm, O.; Elmgren, A.; Filipsson, K.; Stenmark Askmalm, M.; Sandstedt, J.; Tengvar, M.; Tham, E. Genetic testing and surveillance guidelines in hereditary pheochromocytoma and paraganglioma. J. Intern. Med. 2019, 285, 187–204. [Google Scholar] [CrossRef] [PubMed]
- Jasperson, K.W.; Kohlmann, W.; Gammon, A.; Slack, H.; Buchmann, L.; Hunt, J.; Kirchhoff, A.C.; Baskin, H.; Shaaban, A.; Schiffman, J.D. Role of rapid sequence whole-body MRI screening in SDH-associated hereditary paraganglioma families. Fam. Cancer 2014, 13, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Daniel, E.; Jones, R.; Bull, M.; Newell-Price, J. Rapid-sequence MRI for long-term surveillance for paraganglioma and phaeochromocytoma in patients with succinate dehydrogenase mutations. Eur. J. Endocrinol. 2016, 175, 561–570. [Google Scholar] [CrossRef] [Green Version]
- Derchi, L.E.; Serafini, G.; Rabbia, C.; De Albertis, P.; Solbiati, L.; Candiani, F.; Musante, F.; Bertoglio, C.; Rizzatto, G. Carotid body tumors: US evaluation. Radiology 1992, 182, 457–459. [Google Scholar] [CrossRef]
- Jansen, J.C.; Baatenburg de Jong, R.J.; Schipper, J.; van der Mey, A.G.; van Gils, A.P. Color Doppler imaging of paragangliomas in the neck. J. Clin. Ultrasound 1997, 25, 481–485. [Google Scholar] [CrossRef]
- Rubenthaler, J.; Lutz, J.; Reiser, M.; Clevert, D.A. Title Page—Paraganglioma of the Head and Neck: Follow-Up of Interventional Procedures with CEUS. Ultraschall Med. 2015, 36, 541–543. [Google Scholar] [CrossRef]
- Friedrich-Rust, M.; Glasemann, T.; Polta, A.; Eichler, K.; Holzer, K.; Kriener, S.; Herrmann, E.; Nierhoff, J.; Bon, D.; Bechstein, W.O.; et al. Differentiation between benign and malignant adrenal mass using contrast-enhanced ultrasound. Ultraschall Med. 2011, 32, 460–471. [Google Scholar] [CrossRef]
- Sidhu, P.S.; Cantisani, V.; Dietrich, C.F.; Gilja, O.H.; Saftoiu, A.; Bartels, E.; Bertolotto, M.; Calliada, F.; Clevert, D.A.; Cosgrove, D.; et al. The EFSUMB Guidelines and Recommendations for the Clinical Practice of Contrast-Enhanced Ultrasound (CEUS) in Non-Hepatic Applications: Update 2017 (Long Version). Ultraschall Med. 2018, 39, e2–e44. [Google Scholar] [CrossRef] [Green Version]
- Wiseman, G.A.; Pacak, K.; O’Dorisio, M.S.; Neumann, D.R.; Waxman, A.D.; Mankoff, D.A.; Heiba, S.I.; Serafini, A.N.; Tumeh, S.S.; Khutoryansky, N.; et al. Usefulness of 123I-MIBG scintigraphy in the evaluation of patients with known or suspected primary or metastatic pheochromocytoma or paraganglioma: Results from a prospective multicenter trial. J. Nucl. Med. 2009, 50, 1448–1454. [Google Scholar] [CrossRef] [Green Version]
- Castinetti, F.; Kroiss, A.; Kumar, R.; Pacak, K.; Taieb, D. 15 Years of paraganglioma: Imaging and imaging-based treatment of pheochromocytoma and paraganglioma. Endocr. Relat. Cancer 2015, 22, T135–T145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gimenez-Roqueplo, A.P.; Caumont-Prim, A.; Houzard, C.; Hignette, C.; Hernigou, A.; Halimi, P.; Niccoli, P.; Leboulleux, S.; Amar, L.; Borson-Chazot, F.; et al. Imaging work-up for screening of paraganglioma and pheochromocytoma in SDHx mutation carriers: A multicenter prospective study from the PGL.EVA Investigators. J. Clin. Endocrinol. Metab. 2013, 98, E162–E173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charrier, N.; Deveze, A.; Fakhry, N.; Sebag, F.; Morange, I.; Gaborit, B.; Barlier, A.; Carmona, E.; De Micco, C.; Garcia, S.; et al. Comparison of [(1)(1)(1)In]pentetreotide-SPECT and [(1)(8)F]FDOPA-PET in the localization of extra-adrenal paragangliomas: The case for a patient-tailored use of nuclear imaging modalities. Clin. Endocrinol. 2011, 74, 21–29. [Google Scholar] [CrossRef] [PubMed]
- King, K.S.; Chen, C.C.; Alexopoulos, D.K.; Whatley, M.A.; Reynolds, J.C.; Patronas, N.; Ling, A.; Adams, K.T.; Xekouki, P.; Lando, H.; et al. Functional imaging of SDHx-related head and neck paragangliomas: Comparison of 18F-fluorodihydroxyphenylalanine, 18F-fluorodopamine, 18F-fluoro-2-deoxy-D-glucose PET, 123I-metaiodobenzylguanidine scintigraphy, and 111In-pentetreotide scintigraphy. J. Clin. Endocrinol. Metab. 2011, 96, 2779–2785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rischke, H.C.; Benz, M.R.; Wild, D.; Mix, M.; Dumont, R.A.; Campbell, D.; Seufert, J.; Wiech, T.; Rossler, J.; Weber, W.A.; et al. Correlation of the genotype of paragangliomas and pheochromocytomas with their metabolic phenotype on 3,4-dihydroxy-6-18F-fluoro-L-phenylalanin PET. J. Nucl. Med. 2012, 53, 1352–1358. [Google Scholar] [CrossRef] [Green Version]
- Feral, C.C.; Tissot, F.S.; Tosello, L.; Fakhry, N.; Sebag, F.; Pacak, K.; Taieb, D. (18)F-fluorodihydroxyphenylalanine PET/CT in pheochromocytoma and paraganglioma: Relation to genotype and amino acid transport system L. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 812–821. [Google Scholar] [CrossRef]
- Kong, G.; Schenberg, T.; Yates, C.J.; Trainer, A.; Sachithanandan, N.; Iravani, A.; Ravi Kumar, A.; Hofman, M.S.; Akhurst, T.; Michael, M.; et al. The Role of 68Ga-DOTA-Octreotate PET/CT in Follow-Up of SDH-Associated Pheochromocytoma and Paraganglioma. J. Clin. Endocrinol. Metab. 2019, 104, 5091–5099. [Google Scholar] [CrossRef]
- Elston, M.S.; Meyer-Rochow, G.Y.; Conaglen, H.M.; Clarkson, A.; Clifton-Bligh, R.J.; Conaglen, J.V.; Gill, A.J. Increased SSTR2A and SSTR3 expression in succinate dehydrogenase-deficient pheochromocytomas and paragangliomas. Hum. Pathol. 2015, 46, 390–396. [Google Scholar] [CrossRef]
- Janssen, I.; Chen, C.C.; Taieb, D.; Patronas, N.J.; Millo, C.M.; Adams, K.T.; Nambuba, J.; Herscovitch, P.; Sadowski, S.M.; Fojo, A.T.; et al. 68Ga-DOTATATE PET/CT in the Localization of Head and Neck Paragangliomas Compared with Other Functional Imaging Modalities and CT/MRI. J. Nucl. Med. 2016, 57, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Janssen, I.; Chen, C.C.; Millo, C.M.; Ling, A.; Taieb, D.; Lin, F.I.; Adams, K.T.; Wolf, K.I.; Herscovitch, P.; Fojo, A.T.; et al. PET/CT comparing (68)Ga-DOTATATE and other radiopharmaceuticals and in comparison with CT/MRI for the localization of sporadic metastatic pheochromocytoma and paraganglioma. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 1784–1791. [Google Scholar] [CrossRef]
- Chang, C.A.; Pattison, D.A.; Tothill, R.W.; Kong, G.; Akhurst, T.J.; Hicks, R.J.; Hofman, M.S. (68)Ga-DOTATATE and (18)F-FDG PET/CT in Paraganglioma and Pheochromocytoma: Utility, patterns and heterogeneity. Cancer Imaging 2016, 16, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofman, M.S.; Lau, W.F.; Hicks, R.J. Somatostatin receptor imaging with 68 Ga DOTATATE PET/CT: Clinical utility, normal patterns, pearls, and pitfalls in interpretation. Radiographics 2015, 35, 500–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, M.G.; Netterville, J.L.; Mendenhall, W.M.; Isaacson, B.; Nussenbaum, B. Head and Neck Paragangliomas: An Update on Evaluation and Management. Otolaryngol. Head Neck Surg. 2016, 154, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Kolasinska-Cwikla, A.; Peczkowska, M.; Cwikla, J.B.; Michalowska, I.; Palucki, J.M.; Bodei, L.; Lewczuk-Myslicka, A.; Januszewicz, A. A Clinical Efficacy of PRRT in Patients with Advanced, Nonresectable, Paraganglioma-Pheochromocytoma, Related to SDHx Gene Mutation. J. Clin. Med. 2019, 8, 952. [Google Scholar] [CrossRef] [Green Version]
- Nolting, S.; Grossman, A.; Pacak, K. Metastatic Phaeochromocytoma: Spinning Towards More Promising Treatment Options. Exp. Clin. Endocrinol. Diabetes 2019, 127, 117–128. [Google Scholar] [CrossRef] [Green Version]
- Ayala-Ramirez, M.; Chougnet, C.N.; Habra, M.A.; Palmer, J.L.; Leboulleux, S.; Cabanillas, M.E.; Caramella, C.; Anderson, P.; Al Ghuzlan, A.; Waguespack, S.G.; et al. Treatment with sunitinib for patients with progressive metastatic pheochromocytomas and sympathetic paragangliomas. J. Clin. Endocrinol. Metab. 2012, 97, 4040–4050. [Google Scholar] [CrossRef]
- Van Hulsteijn, L.T.; van Duinen, N.; Verbist, B.M.; Jansen, J.C.; van der Klaauw, A.A.; Smit, J.W.; Corssmit, E.P. Effects of octreotide therapy in progressive head and neck paragangliomas: Case series. Head Neck 2013, 35, E391–E396. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Demographic Parameters: (1) Sex (2) Age at Clinical dx (yrs) (3) Age at Genetic dx (yrs) (4) Age at Last FU (yrs) | FU from Clinical Diagnosis (Months) | Tumor Lesions: (1) Number (2) Site | Size at Diagnosis (mm) | Basal Morphological Imaging | Urinary Fractionated Metanephrines | Histology | Surgery | Medical Therapy | Follow-up |
|---|---|---|---|---|---|---|---|---|---|---|
| Case 1 (T.D.) | (1) Female (2) 40 (3) 41 (4) 48 | 91 | (1) 3 (2) a: left carotid bifurcation b: right carotid bifurcation c: mediastinum | a: 18 b: 6 c: 38 | - echography - CT angiography - MR | M: 111 µg/24 h (50–340), N: 312 µg/24 (90–445) | a: PGL NSE+, synaptophysin +, CgA+, S100+; b:- c: PGL, synaptophysin +, CgA+, Ki67 < 1%, R1 | a: yes, 2013 b: no c: yes, 2013 after embolization and subsequent radiotherapy | yes, OCT LAR 30 mg every 28 days | - lesions operated (a,c): no persistence, no relapse - lesion not operated (b): 4 mm growth in 7 yrs at CT |
| Case 2 (T.E.) | (1) M (2) 44 (3) 48 (4) 48 | 44 | (1) 4 (2) a: left laterocervical b: right carotid bifurcation c: right jugular foramen d: cervical | a: NA b: 6 c: NA d: NA | - echography - CT angiography (c and d lesion were detected exclusively by functional imaging) | M: 50 µg/24 h (50–340), N: 239 µg/24 (90–445) | NA | a: yes, 2015 b: no c: no d: no | no | - lesion operated (a): no persistence, no relapse - lesion not operated (b): echography, 6.5 mm (Nov 2018); MR angiography 20 mm (April 2019) |
| Case 3 (T.I.) | (1) F (2) 19 (3) 16 (4) 19 | 15 | (1) 2 (2) a, b: left laterocervical | a: 30 b: 15 | - echography - MR angiography | M: 34 µg/24 h (50–340), N: 163 µg/24 (90–445) | NA | a, b: yes, 2019 | no | NA |
| Case 4 (T.M.) | (1) F (2) 36 (3) 44 (4) 49 | 156 | (1) 3 (2) a: left laterocervical b: at 1.5 cm of the petrous canal between carotid and jugular c: right carotid glomus | a: NA b: 13 c: 16 | - echography - CT angiography - MR angiography | M: 57 µg/24 h (50–340), N: 104 µg/24 (90–445) | a: NA c: paraganglioma S100+; 1 mitosis/50 HPF | a: yes, 2006 b: no c: yes, 2007 | no | - lesions operated (a, c): no persistence, no relapse -lesion not operated (b): 11 mm growth in 7 yrs at CT |
| Case 5 (T.R.) | (1) M (2) 50 (3) 47 (4) 52 | 26 | (1) 1 (2) a: left carotid bifurcation | a: 20 | - echography - MR angiography - CT angiography | M: 62 µg/24 h (50–340), N: 62 µg/24 (90–445) | a: Paraganglioma S100+; Synaptophysin +; CgA +/− | a: yes, 2018 | No | - lesion operated (a): no persistence, no relapse |
| Patient | Lesions (Number) | 68Ga-DOTATOC PET-CT | 18F- FDG PET-CT | 18F-DOPA PET-CT |
|---|---|---|---|---|
| All | 13 | 9/9 (7 confirmed at morphological imaging and 2 not confirmed at morphological imaging) (4 NP) | 5/6 (mean SUV max 9.2) (7 NP) | 3/4 (confirmed at morphological imaging) (9 NP) |
| Case 1 (T.D.) | 3 | 3/3 | 2/3 | 2/3 |
| Case 2 (T.E.) | 4 | 3/3 (2 not confirmed at morphological imaging) (1 NP) * | NP | NP |
| Case 3 (T.I.) | 2 | 2/2 | 2/2 | NP |
| Case 4 (T.M.) | 3 | 1/1 (2 NP) * | 1/1 | NP |
| Case 5 (T.R.) | 1 | NP | NP | 1/1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puliani, G.; Sesti, F.; Feola, T.; Di Leo, N.; Polti, G.; Verrico, M.; Modica, R.; Colao, A.; Lenzi, A.; Isidori, A.M.; et al. Natural History and Management of Familial Paraganglioma Syndrome Type 1: Long-Term Data from a Large Family. J. Clin. Med. 2020, 9, 588. https://doi.org/10.3390/jcm9020588
Puliani G, Sesti F, Feola T, Di Leo N, Polti G, Verrico M, Modica R, Colao A, Lenzi A, Isidori AM, et al. Natural History and Management of Familial Paraganglioma Syndrome Type 1: Long-Term Data from a Large Family. Journal of Clinical Medicine. 2020; 9(2):588. https://doi.org/10.3390/jcm9020588
Chicago/Turabian StylePuliani, Giulia, Franz Sesti, Tiziana Feola, Nicola Di Leo, Giorgia Polti, Monica Verrico, Roberta Modica, Annamaria Colao, Andrea Lenzi, Andrea M. Isidori, and et al. 2020. "Natural History and Management of Familial Paraganglioma Syndrome Type 1: Long-Term Data from a Large Family" Journal of Clinical Medicine 9, no. 2: 588. https://doi.org/10.3390/jcm9020588