Energy Metabolism Disturbances in Cell Models of PARK2 CNV Carriers with ADHD

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Neuropsychiatric Assessment and Genotyping
2.2. Skin Biopsies and Fibroblast Primary Cultures
2.3. hiPSCs Generation and Pluripotency Assays
2.4. Differentiation of hiPSCs into Neurons with a Midbrain Dopamine Like Phenotype
2.5. Immunofluorescence and Mitostaining
2.6. Molecular Karyotyping
2.7. RNA Extraction, Two-Steps Reverse Transcription PCR (RT-PCR), and Quantitative RT-PCR (RT-qPCR)
2.8. Protein Concentration
2.9. Stressor Paradigms
2.10. ATP Production, Oxygen Consumption, and ROS Production
2.11. Data Analysis
3. Results
3.1. Confirmation of PARK2 CNVs
3.2. Neurological and Psychiatric Assessment
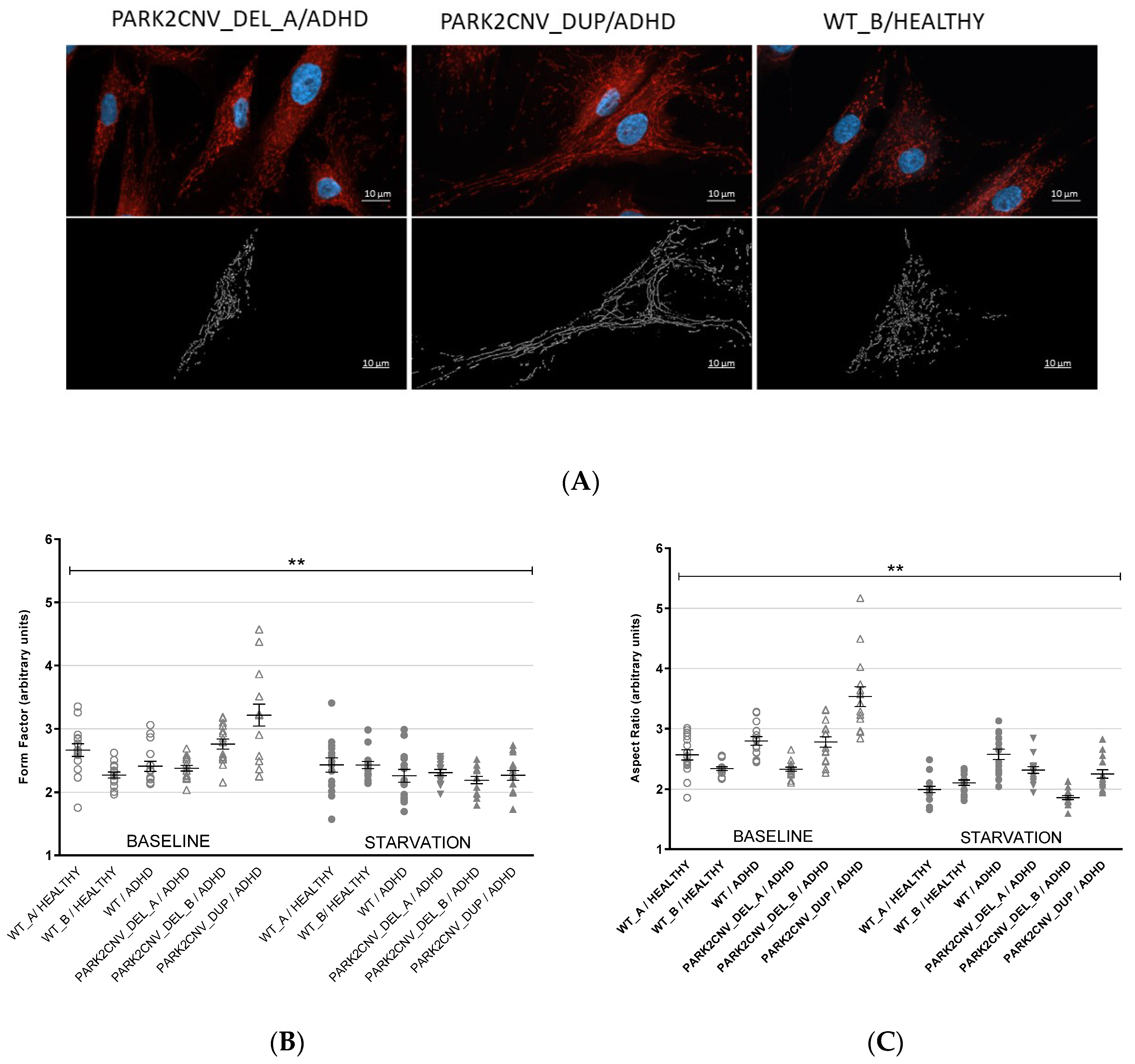
3.3. Evaluation of Mitochondrial Network Morphology
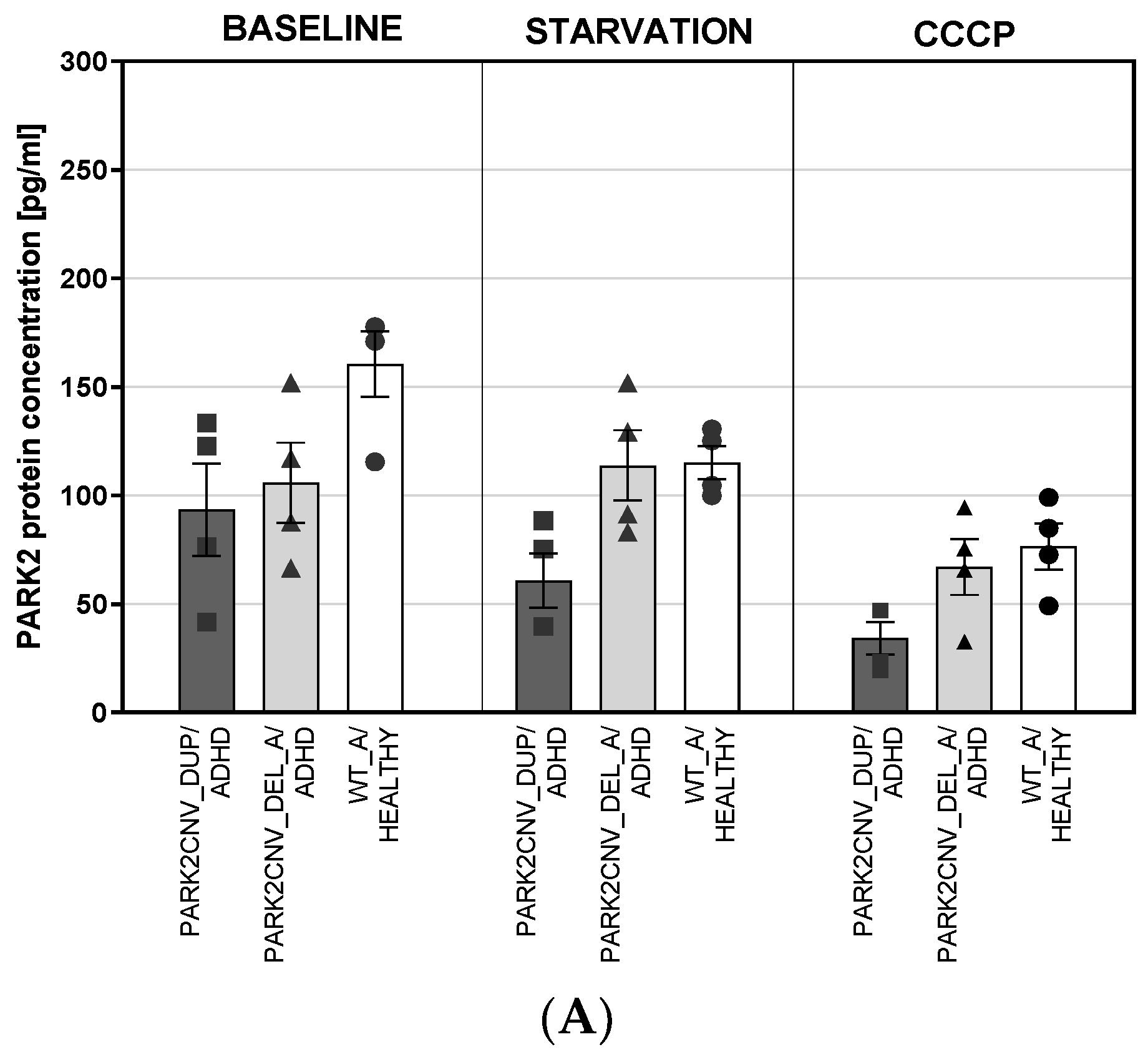
3.4. PARK2 Gene and Protein Expression
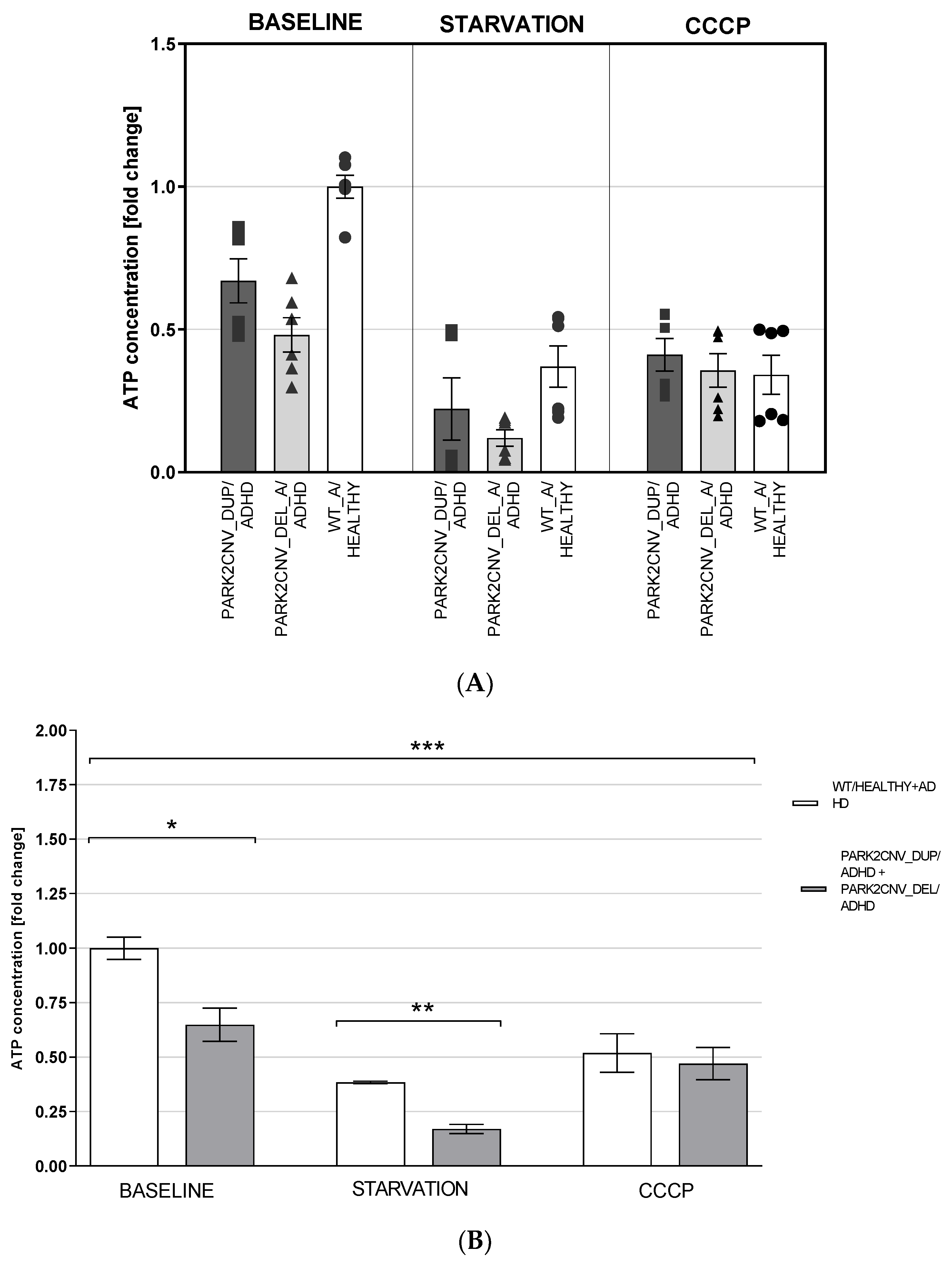
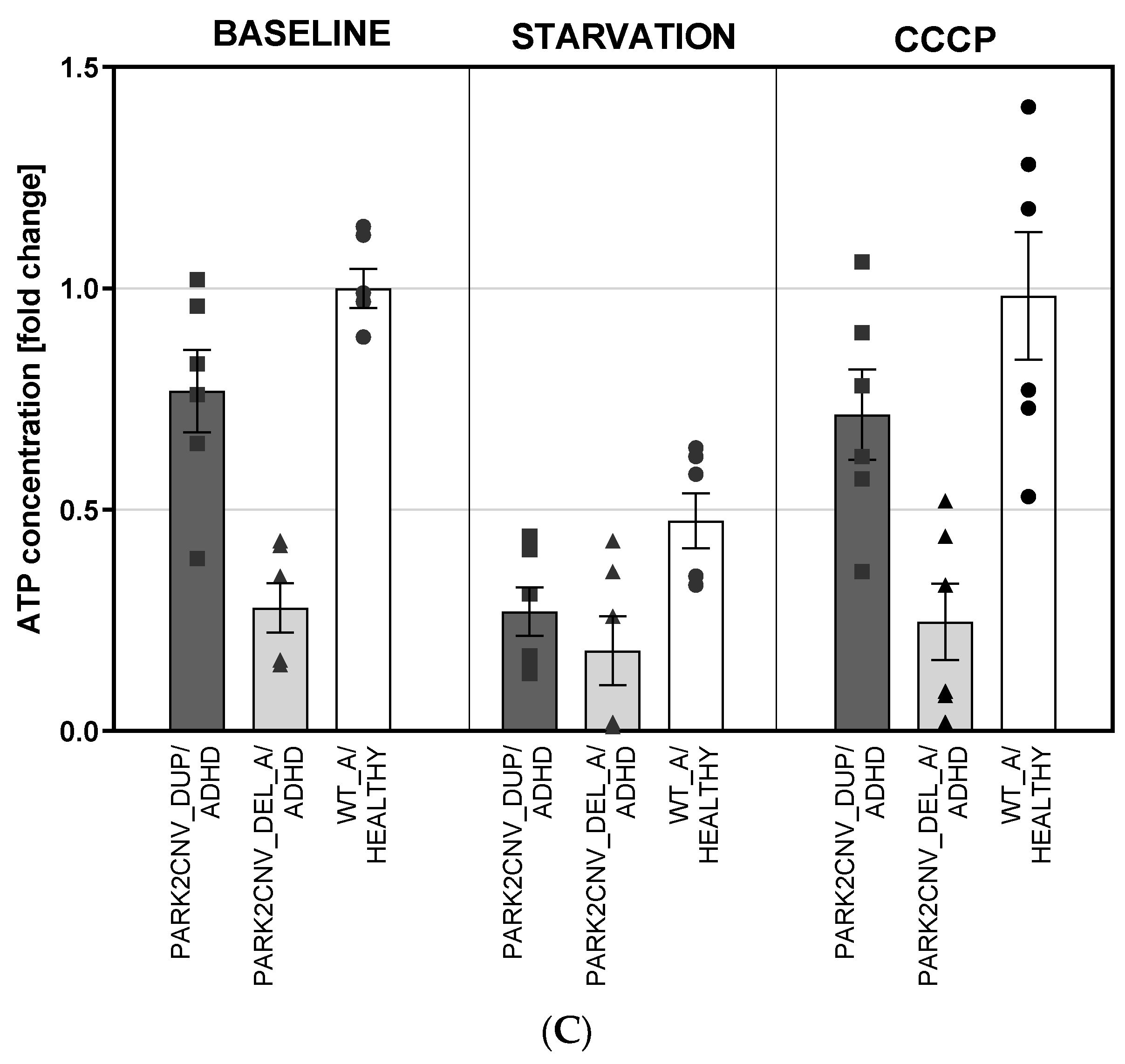
3.5. ATP Levels
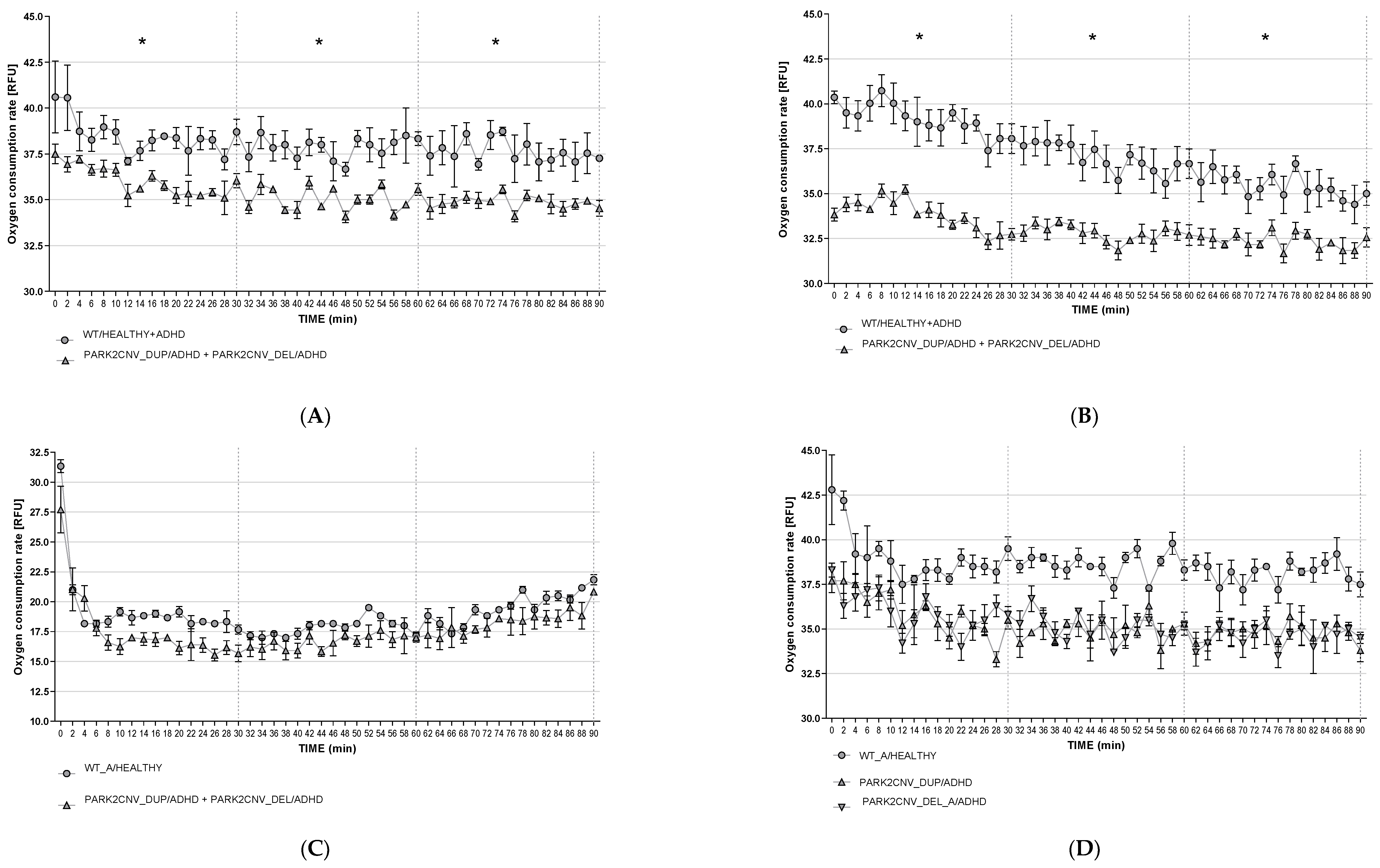
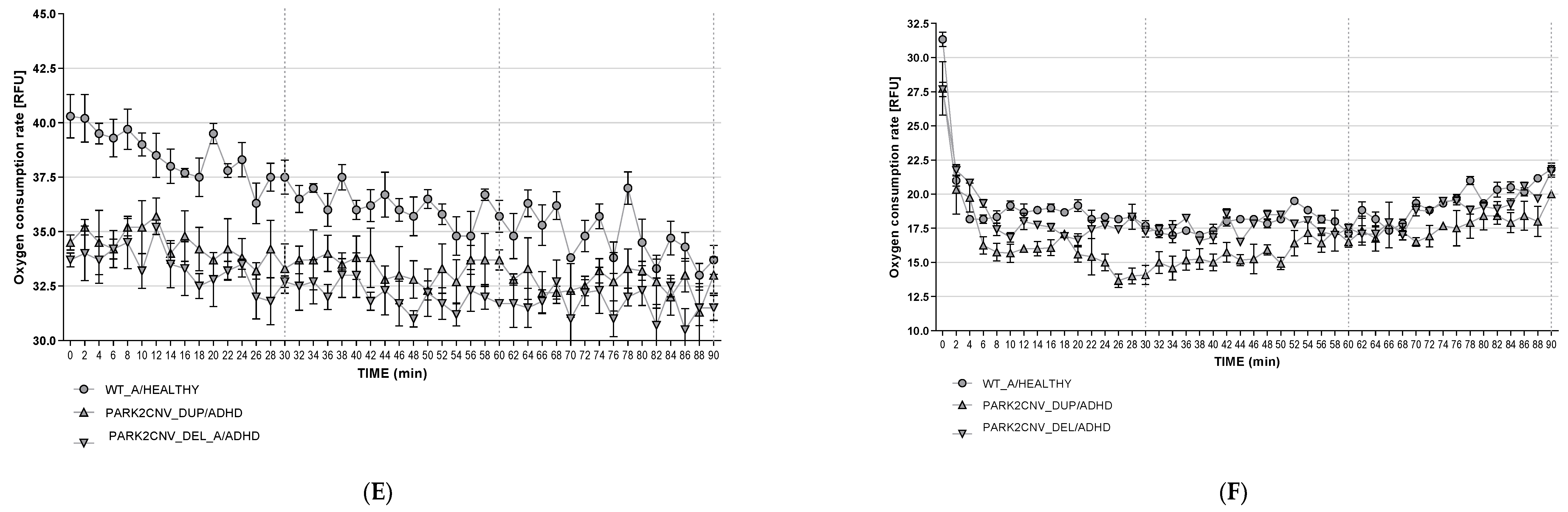
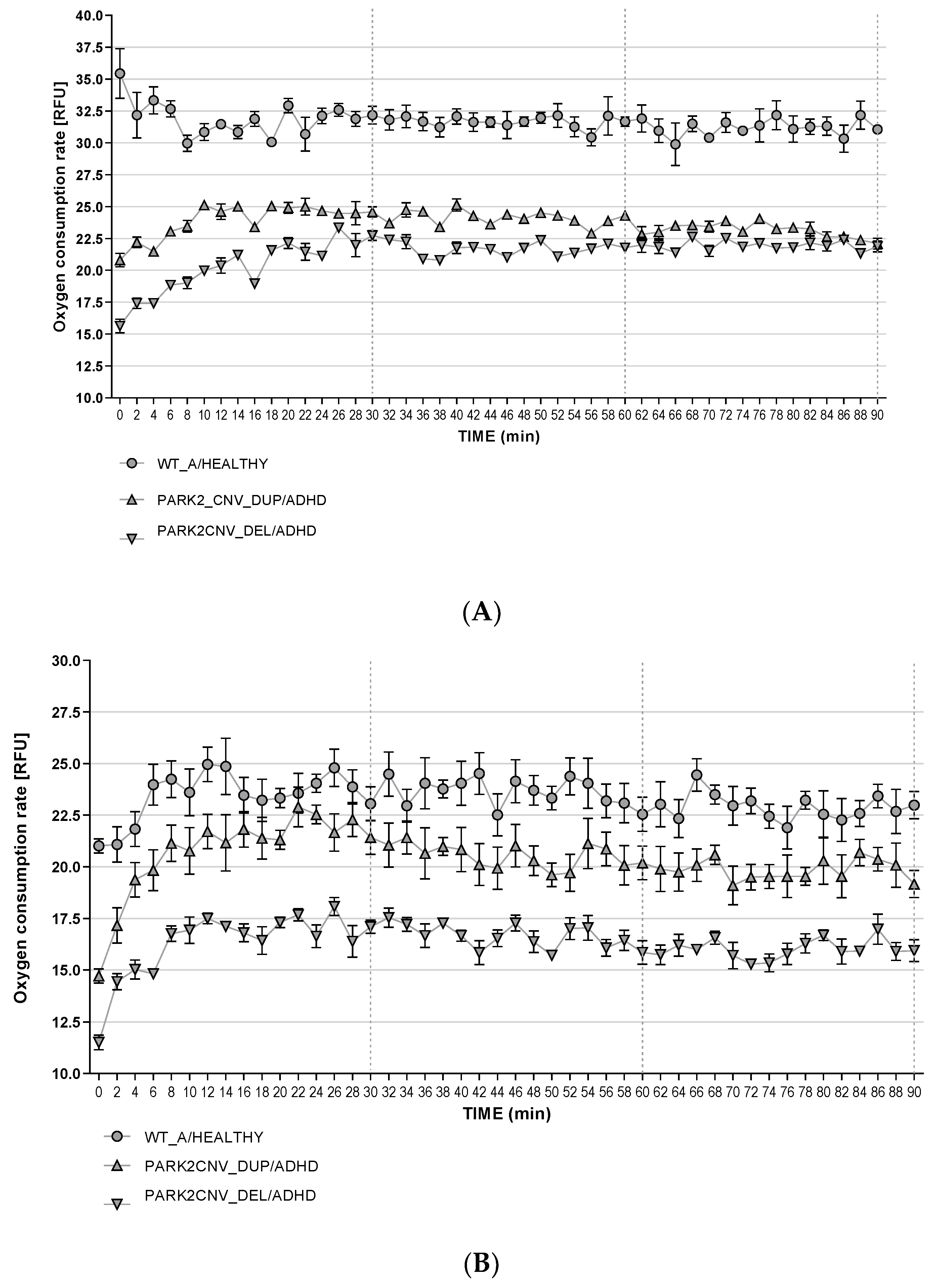
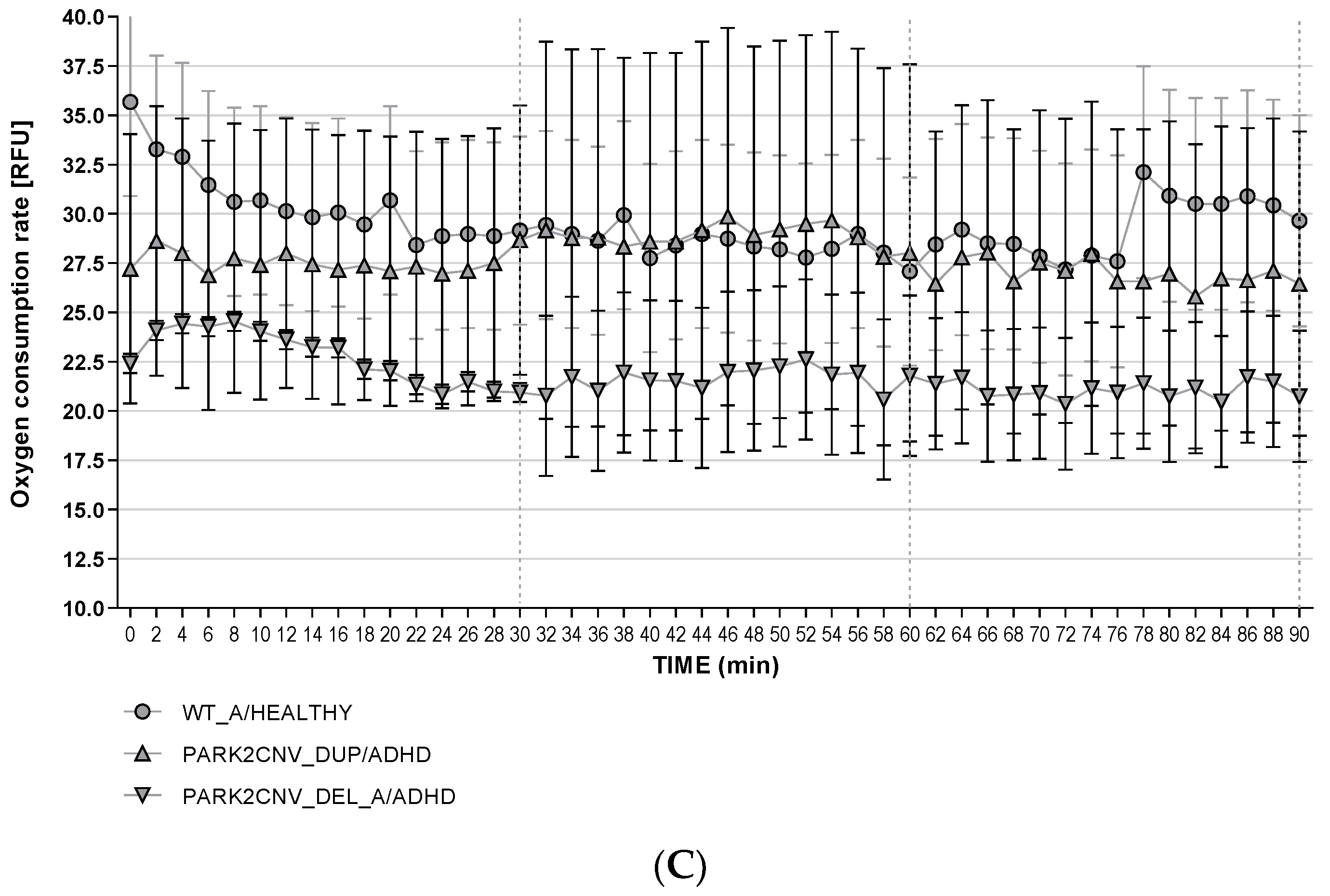
3.6. Oxygen Consumption Rates (OCR)
3.7. Reactive Oxygen Species Production (ROS)
4. Discussion
5. Conclusions
5.1. Limitations
5.2. Data Availability
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADHD | Attention-Deficit-/Hyperactivity Disorder |
| ASD | Autism Spectrum Disorder |
| ATP | Adenosine Triphosphate |
| CCCP | Carbonyl Cyanide m-Chlorophenyl Hydrazine |
| CNV | Copy Number Variation/Variants |
| HDF | Human Dermal Fibroblast |
| hiPSC | human induced Pluripotent Stem Cells |
| mDAN | mature Dopaminergic Neurons |
| MQC | Mitochondria Quality Control system |
| OCR | Oxygen Consumption Rate |
| PARK2CNV_DUP/ADHD | PARK2 duplication carrier with ADHD |
| PARK2CNV_DEL_A/ADHD | PARK2 deletion carrier with ADHD, A |
| PARK2CNV_DEL_B/ADHD | PARK2 deletion carrier with ADHD, B |
| PD NMS | Non-Motor Symptom Assessment Scale for Parkinson’s disease |
| ROS | Reactive Oxygen Species |
| UPDRS | Unified Parkinson’s Disease Rating Scale |
| WT/ADHD | Wildtype in PARK2 locus and ADHD |
| WT_A/HEALTHY | Wildtype in PARK2 locus and healthy control A |
| WT_B/HEALTHY | Wildtype in PARK2 locus and healthy control B |
References
- Bernardi, S.; Faraone, S.V.; Cortese, S.; Kerridge, B.T.; Pallanti, S.; Wang, S.; Blanco, C. The lifetime impact of attention deficit hyperactivity disorder: Results from the National Epidemiologic Survey on Alcohol and Related Conditions (NESARC). Psychol. Med. 2012, 42, 875–887. [Google Scholar] [CrossRef] [Green Version]
- Farley, M.; Cottle, K.J.; Bilder, D.; Viskochil, J.; Coon, H.; McMahon, W. Mid-life social outcomes for a population-based sample of adults with ASD. Autism. Res. 2018, 11, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Baird, G.; Simonoff, E.; Pickles, A.; Chandler, S.; Loucas, T.; Meldrum, D.; Charman, T. Prevalence of disorders of the autism spectrum in a population cohort of children in South Thames: The Special Needs and Autism Project (SNAP). Lancet 2006, 368, 210–215. [Google Scholar] [CrossRef]
- Brikell, I.; Kuja-Halkola, R.; Larsson, H. Heritability of attention-deficit hyperactivity disorder in adults. Am. J. Med. Genet. B Neuropsychiatr Genet. 2015, 168, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Waye, M.M.Y.; Cheng, H.Y. Genetics and epigenetics of autism: A Review. Psychiatry Clin. Neurosci. 2018, 72, 228–244. [Google Scholar] [CrossRef] [PubMed]
- Faraone, S.V.; Perlis, R.H.; Doyle, A.E.; Smoller, J.W.; Goralnick, J.J.; Holmgren, M.A.; Sklar, P. Molecular genetics of attention-deficit/hyperactivity disorder. Biol. Psychiatry 2005, 57, 1313–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, T.D.; Middleton, F.; Faraone, S.V. Environmental risk factors for attention-deficit hyperactivity disorder. Acta Paediatr. 2007, 96, 1269–1274. [Google Scholar] [CrossRef]
- Franke, B.; Michelini, G.; Asherson, P.; Banaschewski, T.; Bilbow, A.; Buitelaar, J.K.; Cormand, B.; Faraone, S.V.; Ginsberg, Y.; Haavik, J.; et al. Live fast, die young? A review on the developmental trajectories of ADHD across the lifespan. Eur. Neuropsychopharmacol. 2018, 28, 1059–1088. [Google Scholar]
- Nikolas, M.A.; Burt, S.A. Genetic and environmental influences on ADHD symptom dimensions of inattention and hyperactivity: A meta-analysis. J. Abnorm. Psychol. 2010, 119, 1–17. [Google Scholar] [CrossRef]
- Stankiewicz, Z.; Lupski, J.R. Structural variation in the human genome and its role in disease. Annu. Rev. Med. 2010, 61, 437–455. [Google Scholar] [CrossRef]
- Elia, J.; Gai, X.; Xie, H.; Perin, J.; Geiger, E.; Glessner, J.; D’Arcy, M.; DeBerardinis, R.; Frackelton, E.; Kim, C.; et al. Rare structural variants found in attention-deficit hyperactivity disorder are preferentially associated with neurodevelopmental genes. Mol. Psychiatry 2010, 15, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Elia, J.; Glessner, J.T.; Wang, K.; Takahashi, N.; Shtir, C.J.; Hadley, D.; A Sleiman, P.M.; Zhang, H.; E Kim, C.; Robison, R.J.; et al. Genome-wide copy number variation study associates metabotropic glutamate receptor gene networks with attention deficit hyperactivity disorder. Nat. Genet. 2012, 44, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Jacob, C.P.; Weber, H.; Retz, W.; Kittel-Schneider, S.; Heupel, J.; Renner, T.; Lesch, K.-P.; Reif, A. Acetylcholine-metabolizing butyrylcholinesterase (BCHE) copy number and single nucleotide polymorphisms and their role in attention-deficit/hyperactivity syndrome. J. Psychiatr. Res. 2013, 47, 1902–1908. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.; Odonovan, M.C.; Thapar, A.; Langley, K.; Williams, N. The relative contribution of common and rare genetic variants to ADHD. Transl. Psychiatry 2015, 5, e506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarick, I.; Volckmar, A.-L.; Pütter, C.; Pechlivanis, S.; Nguyen, T.T.; Dauvermann, M.R.; Beck, S.; Albayrak, Ö.; Scherag, S.; Gilsbach, S.; et al. Genome-wide analysis of rare copy number variations reveals PARK2 as a candidate gene for attention-deficit/hyperactivity disorder. Mol. Psychiatry 2014, 19, 115–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarffe, L.A.; Stevens, D.A.; Dawson, V.L.; Dawson, T.M. Parkin and PINK1: Much more than mitophagy. Trends Neurosci. 2014, 37, 315–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hang, L.; Thundyil, J.; Lim, K.L. Mitochondrial dysfunction and Parkinson disease: A Parkin-AMPK alliance in neuroprotection. Ann. N. Y. Acad. Sci. 2015, 1350, 37–47. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [Green Version]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef]
- Marazziti, D.; Baroni, S.; Picchetti, M.; Landi, P.; Silvestri, S.; Vatteroni, E.; Dell’Osso, M.C. Psychiatric disorders and mitochondrial dysfunctions. Eur. Rev. Med. Pharmacol. Sci. 2012, 16, 270–275. [Google Scholar]
- Verma, P.; Singh, A.; Nthenge-Ngumbau, D.N.; Rajamma, U.; Sinha, S.; Mukhopadhyay, K.; Mohanakumar, K.P. Attention deficit-hyperactivity disorder suffers from mitochondrial dysfunction. BBA Clin. 2016, 6, 153–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogetofte, H.; Jensen, P.; Okarmus, J.; Schmidt, S.I.; Agger, M.; Ryding, M.; Nørregaard, P.; Fenger, C.; Zeng, X.; Graakjær, J.; et al. Perturbations in RhoA signalling cause altered migration and impaired neuritogenesis in human iPSC-derived neural cells with PARK2 mutation. Neurobiol. Dis. 2019, 132, 104581. [Google Scholar] [CrossRef] [PubMed]
- Bogetofte, H.; Jensen, P.; Ryding, M.; Schmidt, S.I.; Okarmus, J.; Ritter, L.; Worm, C.S.; Hohnholt, M.C.; Azevedo, C.; Roybon, L.; et al. PARK2 Mutation Causes Metabolic Disturbances and Impaired Survival of Human iPSC-Derived Neurons. Front. Cell Neurosci. 2019, 13, 297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klinkenberg, M.; Gispert, S.; Dominguez-Bautista, J.A.; Braun, I.; Auburger, G.; Jendrach, M. Restriction of trophic factors and nutrients induces PARKIN expression. Neurogenetics 2012, 13, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Yamano, K.; Matsuda, N.; Tanaka, K. The ubiquitin signal and autophagy: An orchestrated dance leading to mitochondrial degradation. EMBO Rep. 2016, 17, 300–316. [Google Scholar] [CrossRef]
- Lehrl, S.; Triebig, G.; Fischer, B. Multiple choice vocabulary test MWT as a valid and short test to estimate premorbid intelligence. Acta Neurol. Scand 1995, 91, 335–345. [Google Scholar] [CrossRef]
- Reif, A.; Herterich, S.; Strobel, A.; Ehlis, A.-C.; Saur, D.; Jacob, C.P.; Wienker, T.; Töpner, T.; Fritzen, S.; Walter, U.; et al. A neuronal nitric oxide synthase (NOS-I) haplotype associated with schizophrenia modifies prefrontal cortex function. Mol. Psychiatry 2006, 11, 286–300. [Google Scholar] [CrossRef] [Green Version]
- Zanellati, M.C.; Monti, V.; Barzaghi, C.; Reale, C.; Nardocci, N.; Albanese, A.; Valente, E.M.; Ghezzi, D.; Garavaglia, B. Mitochondrial dysfunction in Parkinson disease: Evidence in mutant PARK2 fibroblasts. Front. Genet. 2015, 6, 78. [Google Scholar] [CrossRef] [Green Version]
- Chu, C.-L.; Lee, I.H.; Chi, M.H.; Chen, K.C.; Chen, P.S.; Yao, W.J.; Jou, I.-M.; Yang, Y.K. Availability of dopamine transporters and auditory P300 abnormalities in adults with attention-deficit hyperactivity disorder: Preliminary results. CNS Spectr. 2018, 23, 264–270. [Google Scholar] [CrossRef] [Green Version]
- Challman, T.D.; Lipsky, J.J. Methylphenidate: Its pharmacology and uses. Mayo Clin. Proc. 2000, 75, 711–721. [Google Scholar] [CrossRef] [Green Version]
- Kriks, S.; Shim, J.-W.; Piao, J.; Ganat, Y.M.; Wakeman, D.R.; Xie, Z.; Carrillo-Reid, L.; Auyeung, G.; Antonacci, C.; Buch, A.; et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 2011, 480, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Weykopf, B.; Haupt, S.; Jungverdorben, J.; Flitsch, L.J.; Hebisch, M.; Liu, G.H.; Suzuki, K.; Belmonte, J.C.I.; Peitz, M.; Blaess, S.; et al. Induced pluripotent stem cell-based modeling of mutant LRRK2-associated Parkinson’s disease. Eur. J. Neurosci. 2019, 49, 561–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burbulla, L.F.; Kruger, R. The use of primary human fibroblasts for monitoring mitochondrial phenotypes in the field of Parkinson’s disease. J. Vis. Exp. 2012, 68, e4228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickrell, A.M.; Huang, C.-H.; Kennedy, S.R.; Ordureau, A.; Sideris, D.P.; Hoekstra, J.G.; Harper, J.W.; Youle, R.J. Endogenous Parkin Preserves Dopaminergic Substantia Nigral Neurons following Mitochondrial DNA Mutagenic Stress. Neuron 2015, 87, 371–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flinn, L.; Mortiboys, H.; Volkmann, K.; Köster, R.W.; Ingham, P.W.; Bandmann, O. Complex I deficiency and dopaminergic neuronal cell loss in parkin-deficient zebrafish (Danio rerio). Brain 2009, 132, 1613–1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarraf, S.A.; Raman, M.; Guarani-Pereira, V.; Sowa, M.E.; Huttlin, E.L.; Gygi, S.P.; Harper, J.W. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 2013, 496, 372–376. [Google Scholar] [CrossRef]
- Haylett, W.; Swart, C.; Van Der Westhuizen, F.; Van Dyk, H.; Van Der Merwe, L.; Van Der Merwe, C.; Loos, B.; Carr, J.; Kinnear, C.; Bardien, S. Altered Mitochondrial Respiration and Other Features of Mitochondrial Function in Parkin-Mutant Fibroblasts from Parkinson’s Disease Patients. Parkinsons Dis. 2016, 2016, 1819209. [Google Scholar]
- Noda, S.; Sato, S.; Fukuda, T.; Tada, N.; Uchiyama, Y.; Tanaka, K.; Hattori, N. Loss of Parkin contributes to mitochondrial turnover and dopaminergic neuronal loss in aged mice. Neurobiol. Dis. 2020, 136, 104717. [Google Scholar] [CrossRef]
- McCann, R.F.; Ross, D.A. So Happy Together: The Storied Marriage Between Mitochondria and the Mind. Biol. Psychiatry 2018, 83, e47–e49. [Google Scholar] [CrossRef]
- Manji, H.K.; Kato, T.; Di Prospero, N.A.; Ness, S.; Beal, M.F.; Krams, M.; Chen, G. Impaired mitochondrial function in psychiatric disorders. Nat. Rev. Neurosci. 2012, 13, 293–307. [Google Scholar] [CrossRef]
- Sheng, Z.H.; Cai, Q. Mitochondrial transport in neurons: Impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 2012, 13, 77–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, R.; Faust, T.; Ramos, A.; Ishizuka, K.; Sawa, A. Dynamic Changes of the Mitochondria in Psychiatric Illnesses: New Mechanistic Insights From Human Neuronal Models. Biol. Psychiatry 2018, 83, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Bonello, F.; Hassoun, S.-M.; Mouton-Liger, F.; Shin, Y.S.; Muscat, A.; Tesson, C.; Lesage, S.; Beart, P.M.; Brice, A.; Krupp, J.; et al. LRRK2 impairs PINK1/Parkin-dependent mitophagy via its kinase activity: Pathologic insights into Parkinson’s disease. Hum. Mol. Genet. 2019, 28, 1645–1660. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Fang, H.; Groom, L.; Cheng, A.; Zhang, W.; Liu, J.; Wang, X.; Li, K.; Han, P.; Zheng, M.; et al. Superoxide flashes in single mitochondria. Cell 2008, 134, 279–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziviani, E.; Whitworth, A.J. How could Parkin-mediated ubiquitination of mitofusin promote mitophagy? Autophagy 2010, 6, 660–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gai, W.P.; Yuan, H.; Li, X.; Power, J.; Blumbergs, P.; Jensen, P. In situ and in vitro study of colocalization and segregation of alpha-synuclein, ubiquitin, and lipids in Lewy bodies. Exp. Neurol. 2000, 166, 324–333. [Google Scholar] [CrossRef]
- Gispert, S.; Ricciardi, F.; Kurz, A.; Azizov, M.; Hoepken, H.-H.; Becker, D.; Voos, W.; Leuner, K.; Müller, W.E.; Kudin, A.P.; et al. Parkinson phenotype in aged PINK1-deficient mice is accompanied by progressive mitochondrial dysfunction in absence of neurodegeneration. PLoS ONE 2009, 4, e5777. [Google Scholar] [CrossRef]
- Lopresti, A.L. Oxidative and nitrosative stress in ADHD: Possible causes and the potential of antioxidant-targeted therapies. Atten. Defic. Hyperact. Disord. 2015, 7, 237–247. [Google Scholar] [CrossRef]
- Joseph, N.; Zhang-James, Y.; Perl, A.; Faraone, S.V. Oxidative Stress and ADHD: A Meta-Analysis. J. Atten. Disord. 2015, 19, 915–924. [Google Scholar] [CrossRef]
- Li, Z.; Okamoto, K.-I.; Hayashi, Y.; Sheng, M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 2004, 119, 873–887. [Google Scholar] [CrossRef] [Green Version]
- Mattson, M.P.; Gleichmann, M.; Cheng, A. Mitochondria in neuroplasticity and neurological disorders. Neuron 2008, 60, 748–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xavier, J.M.; Rodrigues, C.M.; Sola, S. Mitochondria: Major Regulators of Neural Development. Neuroscientist 2016, 22, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.; Rho, J.M.; Masino, S.A. Metabolic Dysfunction Underlying Autism Spectrum Disorder and Potential Treatment Approaches. Front. Mol. Neurosci. 2017, 10, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palladino, V.S.; Chiocchetti, A.G.; Frank, L.; Haslinger, D.; McNeill, R.; Radtke, F.; Till, A.; Haupt, S.; Brüstle, O.; Günther, K.; et al. Energy Metabolism Disturbances in Cell Models of PARK2 CNV Carriers with ADHD. J. Clin. Med. 2020, 9, 4092. https://doi.org/10.3390/jcm9124092
Palladino VS, Chiocchetti AG, Frank L, Haslinger D, McNeill R, Radtke F, Till A, Haupt S, Brüstle O, Günther K, et al. Energy Metabolism Disturbances in Cell Models of PARK2 CNV Carriers with ADHD. Journal of Clinical Medicine. 2020; 9(12):4092. https://doi.org/10.3390/jcm9124092
Chicago/Turabian StylePalladino, Viola Stella, Andreas G. Chiocchetti, Lukas Frank, Denise Haslinger, Rhiannon McNeill, Franziska Radtke, Andreas Till, Simone Haupt, Oliver Brüstle, Katharina Günther, and et al. 2020. "Energy Metabolism Disturbances in Cell Models of PARK2 CNV Carriers with ADHD" Journal of Clinical Medicine 9, no. 12: 4092. https://doi.org/10.3390/jcm9124092