Rheumatoid Arthritis-Associated Mechanisms of Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans


Abstract
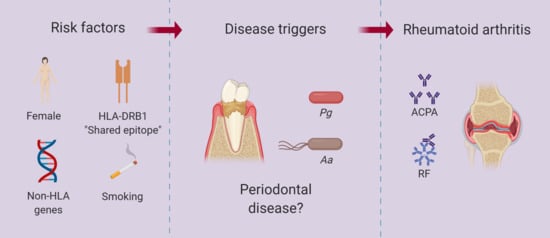
:
{kind=link}
1. Introduction
2. The Focal Infection Theory, Oral Sepsis, and RA
3. Periodontitis and RA
4. Citrullination and RA
5. P. gingivalis in RA Pathogenesis
5.1. P. gingivalis in Experimental Models of Arthritis
5.2. P. gingivalis PAD (PPAD)
5.3. PPAD-Mediated Citrullination of Bacterial and Host Proteins
5.4. Self-Endocitrullination of Pro-PPAD
5.5. PPAD in Experimental Models of Arthritis
6. Aa in RA Pathogenesis
6.1. Infection Due to Aa
6.2. Aa-Induced Hypercitrullination and the Production Citrullinated RA Autoantigens
6.3. Aa Exposure and RA Pathogenesis
7. P. gingivalis and Aa in the Mechanistic Model of RA: Causal Agents, Risk Factors, Disease Modulators, or Research Distractors
8. Therapeutic Implications
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Darrah, E.; Andrade, F. Rheumatoid arthritis and citrullination. Curr. Opin. Rheumatol. 2018, 30, 72–78. [Google Scholar] [CrossRef]
- Carmona, L.; Cross, M.; Williams, B.; Lassere, M.; March, L. Rheumatoid arthritis. Best Pract. Res. Clin. Rheumatol. 2010, 24, 733–745. [Google Scholar] [CrossRef] [PubMed]
- Mantle, A. The etiology of rheumatism considered from a bacterial point of view. Br. Med. J. 1887, 1, 1381–1384. [Google Scholar] [CrossRef] [PubMed]
- Bannatyne, G.A.; Wohlmann, A.S.; Blaxall, F.R. Rheumatoid arthritis: Its clinical history, etiology, and pathology. Lancet 1896, 147, 1120–1125. [Google Scholar] [CrossRef]
- Schuller, M. The relation of chronic villous polyarthritis to the dumb-bell shaped bacilli. Am. J. Med. Sci. 1906, 132, 231–239. [Google Scholar] [CrossRef]
- Schuller, M. Chirurgische Mittheilungen über die Chronisch-Rheumatischen Glenkentzündungen. In Archiv Fur Klinische Chirurgie; Bergman, E.V., Billroth, T., Gurlt, E., Eds.; Verlag Von August Hirschwald: Berlin, Germany, 1893; pp. 153–185. [Google Scholar]
- McInnes, I.B.; Schett, G. The Pathogenesis of Rheumatoid Arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef] [PubMed]
- Deane, K.D.; Demoruelle, M.K.; Kelmenson, L.B.; Kuhn, K.A.; Norris, J.M.; Holers, V.M. Genetic and environmental risk factors for rheumatoid arthritis. Best Pract. Res. Clin. Rheumatol. 2017, 31, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Svendsen, A.J.; Kyvik, K.O.; Houen, G.; Junker, P.; Christensen, K.; Christiansen, L.; Nielsen, C.; Skytthe, A.; Hjelmborg, J.V. On the Origin of Rheumatoid Arthritis: The Impact of Environment and Genes—A Population Based Twin Study. PLoS ONE 2013, 8, e57304. [Google Scholar] [CrossRef] [PubMed]
- Frisell, T.; Saevarsdottir, S.; Askling, J.; Frisell, T. Family history of rheumatoid arthritis: An old concept with new developments. Nat. Rev. Rheumatol. 2016, 12, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Silman, A.J.; MacGregor, A.J.; Thomson, W.; Holligan, S.; Carthy, D.; Farhan, A.; Ollier, W.E.R. Twin concordance rates for rheumatoid arthritis: Results from a nationwide study. Br. J. Rheumatol. 1993, 32, 903–907. [Google Scholar] [CrossRef] [PubMed]
- Svendsen, A.J.; Holm, N.V.; Kyvik, K.; Petersen, P.H.; Junker, P. Relative importance of genetic effects in rheumatoid arthritis: Historical cohort study of Danish nationwide twin population. BMJ 2002, 324, 264. [Google Scholar] [CrossRef] [PubMed]
- Caminer, A.C.; Haberman, R.; Scher, J.U. Human microbiome, infections, and rheumatic disease. Clin. Rheumatol. 2017, 36, 2645–2653. [Google Scholar] [CrossRef] [PubMed]
- Potempa, J.; Mydel, P.; Koziel, J. The case for periodontitis in the pathogenesis of rheumatoid arthritis. Nat. Rev. Rheumatol. 2017, 13, 606–620. [Google Scholar] [CrossRef] [PubMed]
- Scher, J.U.; Littman, D.R.; Abramson, S.B. Microbiome in Inflammatory Arthritis and Human Rheumatic Diseases. Arthritis Rheumatol. 2016, 68, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Dunbar, L.L. Oral manifestations in arthritic and gouty conditions. JAMA 1895, 24, 75–77. [Google Scholar] [CrossRef]
- Landré-Beauvais, A.J. The first description of rheumatoid arthritis. Unabridged text of the doctoral dissertation presented in 1800. Jt. Bone Spine 2001, 68, 130–143. [Google Scholar]
- Garrod, A.B. Rheumatic Gout. In The Nature and Treatment of Gout and Rheumatic Gout; Garrod, A.B., Ed.; Walton and Maberly: London, UK, 1859; pp. 526–556. [Google Scholar]
- Richards, J.H. Bacteriologic Studies in Chronic Arthritis and Chorea. J. Bacteriol. 1920, 5, 511–525. [Google Scholar] [PubMed]
- Swett, P.P. Synovectomy in chronic infectious arthritis. J. Bone Jt. Surg. 1923, 5, 110–121. [Google Scholar]
- Kinsella, R.A. Chronic infectious arthritis. J. Am. Med. Assoc. 1923, 80, 0671–0674. [Google Scholar] [CrossRef]
- Margolis, M.H.; Dorsey, A.H.E. Chronic arthritis—Bacteriology of affected tissues. Arch. Intern. Med. 1930, 46, 121–136. [Google Scholar] [CrossRef]
- Goadby, K.W. The Hunterian Lecture on the Association of Disease of the Mouth with Rheumatoid Arthritis and Certain other Forms of Rheumatism. Delivered at the Royal College of Surgeons of England on March 6th, 1911. Lancet 1911, 1, 639–649. [Google Scholar]
- Lane, S.A. A Lecture on Tertiary Syphilis, or Syphilitic Cachexia. Br. Med. J. 1873, 2, 421–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billings, I.K. Focal infection—Its broader application in the etiology of general disease. J. Am. Med. Assoc. 1914, 63, 899–903. [Google Scholar] [CrossRef]
- Lambert, J. A report of some points in the etiology and onset of 195 cases of rheumatoid arthritis. Bull. Comm. Study Spec. Dis. 1908, 2, 83–94. [Google Scholar]
- Lindsay, J. The relation of infective foci to rheumatoid arthritis. Bull. Comm. Study Spec. Dis. 1908, 2, 106–116. [Google Scholar]
- Billings, F. Chronic focal infections and their etiologic relations to arthritis and nephritis. Arch. Intern. Med. 1912, 9, 484–498. [Google Scholar] [CrossRef]
- Billings, F. Chronic focal infection as a causative factor in chronic arthritis. J. Am. Med. Assoc. 1913, 61, 819–822. [Google Scholar] [CrossRef]
- Billings, F. Mouth infection as a source of systemic disease. J. Am. Med. Assoc. 1914, 63, 2024–2025. [Google Scholar] [CrossRef]
- Bywaters, E.G.L. Historical Aspects of the Aetiology of Rheumatoid Arthritis. Br. J. Rheumatol. 1988, 110–115. [Google Scholar] [CrossRef]
- Hughes, R.A. Focal infection revisited. Br. J. Rheumatol. 1994, 33, 370–377. [Google Scholar] [CrossRef]
- Miller, W.D. The Human Mouth as a Focus of Infection. Dent. Cosmos. 1891, 33, 689–713. [Google Scholar] [CrossRef]
- Miller, W.D. Diseases of the Human Body Which Have Been Traced to the Action of Mouth-Bacteria. Am. J. Dent. Sci. 1891, 25, 311–319. [Google Scholar] [PubMed]
- Hunter, W. Oral Sepsis as a Cause of Disease. Br. Med. J. 1900, 2, 215–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, W. A Case of Pernicious Anæmia; with Observations regarding Mode of Onset, Clinical Features, Infective Nature, Prognosis, and Antiseptic and Serum Treatment of the Disease. J. R. Soc. Med. 1901, 84, 205–249. [Google Scholar]
- Hunter, W. Further observations on pernicious anaemia (seven cases): A chronic infective disease; its relation to infection from the mouth and stomach; suggested serum treatment. Lancet 1900, 155, 296–299. [Google Scholar] [CrossRef]
- Hunter, W. Further investigations regarding the infective nature and etiology of pernicius anaemia. Lancet 1903, 161, 367–371. [Google Scholar] [CrossRef]
- Roberts, H.L. Focal infection. Br. J. Derm. Syph. 1921, 33, 319–334. [Google Scholar] [CrossRef]
- Roberts, H.L. Focal infection. Br. J. Derm. Syph. 1921, 33, 353–373. [Google Scholar] [CrossRef]
- Rush, B. An Account of the Cure of Several Diseases by the Extraction of Decayed Teeth. In Medical Inquiries and Observations, 5th ed.; Rush, B., Ed.; Printed by M. Carey & Son; B. Warner; A. Finley; S. W. Conrad; T. & W. Bradford; B. & T. Kite, and Bennett and Walton; National Library of Medicine: Philadelphia, PA, USA, 1818; Volume 1, pp. 197–201. Available online: https://collections.nlm.nih.gov/catalog/nlm:nlmuid-2569006RX1-mvpart (accessed on 26 August 2019).
- Miltner, L.J.; Kulowski, J. The effect of treatment and erradication of foci on infection in chronic rheumatoid arthritis. J. Bone Jt. Surg. 1933, 15, 383–396. [Google Scholar]
- Cecil, R.; Angevine, A. Clinical and experimental observations on focal infection. Ann. Intern. Med. 1938, 12, 577–584. [Google Scholar]
- Vaizey, J.M.; Clark-Kennedy, A.E. Dental Sepsis: Anaemia, Dyspepsia, and Rheumatism. Br. Med. J. 1939, 1, 1269–1273. [Google Scholar] [CrossRef] [Green Version]
- Riggs, J.W. Suppurative Inflammation of the Gums and Absorption of the Gums and the Alveolar Process. Am. J. Dent. Sci. 1999, 32, 401–407. [Google Scholar]
- Hajishengallis, G. Immunomicrobial pathogenesis of periodontitis: Keystones, pathobionts, and host response. Trends Immunol. 2014, 35, 3–11. [Google Scholar] [CrossRef]
- Brandtzaeg, P.; Kraus, F.W. Autoimmunity and periodontal disease. Odontol. Tidskr. 1965, 73, 281. [Google Scholar]
- Snyderman, R.; McCarty, G.A. Analogous Mechanisms of Tissue Destruction in Rheumatoid Arthritis and Periodontal Disease. In Host-Parasite Interactions in Periodontal Diseases; Genco, R.J., Mergenhagen, S.E., Eds.; American Society of Microbiology: Washington, DC, USA, 1982; pp. 354–362. [Google Scholar]
- Seymour, G.J.; Powell, R.N.; Davies, W.I.R. The immunopathogenesis of progressive chronic inflammatory periodontal disease. J. Oral. Pathol. Med. 1979, 8, 249–265. [Google Scholar] [CrossRef]
- Greenwald, R.A.; Kirkwood, K. Adult periodontitis as a model for rheumatoid arthritis (with emphasis on treatment strategies). J. Rheumatol. 1999, 26, 1650–1653. [Google Scholar]
- Katz, J.; Goultschin, J.; Benoliel, R.; Brautbar, C. Human leukocyte antigen (HLA) DR4. Positive association with rapidly progressing periodontitis. J. Periodontol. 1987, 58, 607–610. [Google Scholar] [CrossRef]
- Bonfil, J.J.; Dillier, F.L.; Mercier, P.; Reviron, D.; Foti, B.; Sambuc, R.; Brodeur, J.M.; Sedarat, C. A “case control” study on the role of HLA DR4 in severe periodontitis and rapidly progressive periodontitis. Identification of types and subtypes using molecular biology (PCR.SSO). J. Clin. Periodontol. 1999, 26, 77–84. [Google Scholar] [CrossRef]
- Rizzo, A.A.; Mitchell, C.T. Chronic allergic inflammation induced by repeated deposition of antigen in rabbit gingival pockets. Periodontics 1966, 4, 5. [Google Scholar]
- McHugh, W.D. Some aspects of the development of gingival epithelium. Periodontics 1963, 1, 239–244. [Google Scholar]
- Schroeder, H. Quantitative parameters of early human gingival inflammation. Arch. Oral Biol. 1970, 15, 383–IN4. [Google Scholar] [CrossRef]
- Sugawara, M.; Yamashita, K.; Yoshie, H.; Hara, K. Detection of, and anti-collagen antibody produced by, CD5-positive B cells in inflamed gingival tissues. J. Periodontal Res. 1992, 27, 489–498. [Google Scholar] [CrossRef]
- Kristoffersen, T.; Tonder, O. Anti-immunoglobulin activity in inflamed human gingiva. J. Dent. Res. 1973, 52, 991. [Google Scholar]
- Gargiulo, A.V.; Robinson, J.; Toto, P.D.; Gargiulo, A.W. Identification of Rheumatoid Factor in Periodontal Disease. J. Periodontol. 1982, 53, 568–577. [Google Scholar] [CrossRef]
- Gargiulo, A.V., Jr.; Toto, P.D.; Robinson, J.A.; Gargiulo, A.W. Latex slide agglutination vs. ELISA system: Rheumatoid factor detection in inflamed human gingiva. J. Periodontal Res. 1985, 20, 31–34. [Google Scholar] [CrossRef]
- Malmström, M.; Natvig, J.B. IgG Rheumatoid Factor in Dental Periapical Lesions of Patients with Rheumatoid Disease. Scand. J. Rheumatol. 1975, 4, 177–185. [Google Scholar] [CrossRef]
- McGraw, W.T.; Potempa, J.; Farley, D.; Travis, J. Purification, Characterization, and Sequence Analysis of a Potential Virulence Factor from Porphyromonas gingivalis, Peptidylarginine Deiminase. Infect. Immun. 1999, 67, 3248–3256. [Google Scholar]
- Rosenstein, E.D.; Greenwald, R.A.; Kushner, L.J.; Weissmann, G. Hypothesis: The Humoral Immune Response to Oral Bacteria Provides a Stimulus for the Development of Rheumatoid Arthritis. Inflammation 2004, 28, 311–318. [Google Scholar] [CrossRef]
- Schellekens, G.A.; De Jong, B.A.; Hoogen, F.H.V.D.; Van De Putte, L.B.; Van Venrooij, W.J. Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis-specific autoantibodies. J. Clin. Investig. 1998, 101, 273–281. [Google Scholar] [CrossRef]
- Kaur, S.; White, S.; Bartold, P.M. Periodontal disease and rheumatoid arthritis: A systematic review. J. Dent. Res. 2013, 92, 399–408. [Google Scholar] [CrossRef]
- Fuggle, N.R.; Smith, T.O.; Kaul, A.; Sofat, N. Hand to Mouth: A Systematic Review and Meta-Analysis of the Association between Rheumatoid Arthritis and Periodontitis. Front. Immunol. 2016, 7, 80. [Google Scholar] [CrossRef] [Green Version]
- Araujo, V.M.; Melo, I.M.; Lima, V. Relationship between Periodontitis and Rheumatoid Arthritis: Review of the Literature. Med.iat. Inflamm. 2015, 2015, 259074. [Google Scholar] [CrossRef]
- Kindstedt, E.; Johansson, L.; Palmqvist, P.; Holm, C.K.; Kokkonen, H.; Johansson, I.; Dahlqvist, S.R.; Lundberg, P. Association Between Marginal Jawbone Loss and Onset of Rheumatoid Arthritis and Relationship to Plasma Levels of RANKL. Arthritis Rheumatol. 2018, 70, 508–515. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, K.; Nise, L.; Kats, A.; Luttropp, E.; Catrina, A.I.; Askling, J.; Jansson, L.; Alfredsson, L.; Klareskog, L.; Lundberg, K.; et al. Prevalence of Periodontitis in Patients with Established Rheumatoid Arthritis: A Swedish Population Based Case-Control Study. PLoS ONE 2016, 11, e0155956. [Google Scholar] [CrossRef]
- Sjöström, L.; Laurell, L.; Hugoson, A.; Håkansson, J.P. Periodontal conditions in adults with rheumatoid arthritis. Community Dent. Oral Epidemiol. 1989, 17, 234–236. [Google Scholar] [CrossRef]
- Eke, P.I.; Dye, B.A.; Wei, L.; Thornton-Evans, G.O.; Genco, R.J. CDC Periodontal Disease Surveillance workgroup: James Beck GDRP. Prevalence of periodontitis in adults in the United States: 2009 and 2010. J. Dent. Res. 2012, 91, 914–920. [Google Scholar] [CrossRef]
- Kassebaum, N.J.; Bernabe, E.; Dahiya, M.; Bhandari, B.; Murray, C.J.; Marcenes, W. Global burden of severe periodontitis in 1990-2010, a systematic review and meta-regression. J. Dent. Res. 2014, 93, 1045–1053. [Google Scholar] [CrossRef]
- dos Anjos, L.M.; Pereira, I.A.; ’Orsi, E.; Seaman, A.P.; Burlingame, R.W.; Morato, E.F. A comparative study of IgG second- and third-generation anti-cyclic citrullinated peptide (CCP) ELISAs and their combination with IgA third-generation CCP ELISA for the diagnosis of rheumatoid arthritis. Clin. Rheumatol. 2009, 28, 153–158. [Google Scholar] [CrossRef]
- Lutteri, L.; Malaise, M.; Chapelle, J.P. Comparison of second- and third-generation anti-cyclic citrullinated peptide antibodies assays for detecting rheumatoid arthritis. Clin. Chim. Acta 2007, 386, 76–81. [Google Scholar] [CrossRef]
- Sugawara, K.; Fujisaki, M. Properties of Peptidylarginine Deiminase from the Epidermis of Newborn Rats. J. Biochem. 1981, 89, 257–263. [Google Scholar]
- Raijmakers, R.; Zendman, A.J.; Egberts, W.V.; Vossenaar, E.R.; Raats, J.; Soede-Huijbregts, C.; Rutjes, F.P.; Van Veelen, P.A.; Drijfhout, J.W.; Pruijn, G.J. Methylation of Arginine Residues Interferes with Citrullination by Peptidylarginine Deiminases in vitro. J. Mol. Biol. 2007, 367, 1118–1129. [Google Scholar] [CrossRef]
- Kinloch, A.; Lundberg, K.; Wait, R.; Wegner, N.; Lim, N.H.; Zendman, A.J.W.; Saxne, T.; Malmstr, V.; Venables, P.J.; Kinloch, A. Synovial fluid is a site of citrullination of autoantigens in inflammatory arthritis. Arthritis Rheum. 2008, 58, 2287–2295. [Google Scholar] [CrossRef]
- Foulquier, C.; Sebbag, M.; Clavel, C.; Chapuy-Regaud, S.; Al Badine, R.; Méchin, M.C.; Vincent, C.; Nachat, R.; Yamada, M.; Takahara, H.; et al. Peptidyl arginine deiminase type 2 (PAD-2) and PAD-4 but not PAD-1, PAD-3, and PAD-6 are expressed in rheumatoid arthritis synovium in close association with tissue inflammation. Arthritis Rheum. 2007, 56, 3541–3553. [Google Scholar] [CrossRef]
- Chang, X.; Yamada, R.; Suzuki, A.; Sawada, T.; Yoshino, S.; Tokuhiro, S.; Yamamoto, K. Localization of peptidylarginine deiminase 4 (PADI4) and citrullinated protein in synovial tissue of rheumatoid arthritis. Rheumatology (Oxf.) 2005, 44, 40–50. [Google Scholar] [CrossRef]
- Arita, K.; Hashimoto, H.; Shimizu, T.; Nakashima, K.; Yamada, M.; Sato, M. Structural basis for Ca2+-induced activation of human PAD4. Nat. Struct. Mol. Biol. 2004, 11, 777–783. [Google Scholar] [CrossRef]
- Slade, D.J.; Fang, P.; Dreyton, C.J.; Zhang, Y.; Fuhrmann, J.; Rempel, D.; Bax, B.D.; Coonrod, S.A.; Lewis, H.D.; Guo, M.; et al. Protein Arginine Deiminase 2 Binds Calcium in an Ordered Fashion: Implications for Inhibitor Design. ACS Chem. Biol. 2015, 10, 1043–1053. [Google Scholar] [CrossRef] [Green Version]
- Saijo, S.; Nagai, A.; Kinjo, S.; Mashimo, R.; Akimoto, M.; Kizawa, K.; Yabe-Wada, T.; Shimizu, N.; Takahara, H.; Unno, M. Monomeric Form of Peptidylarginine Deiminase Type I Revealed by X-ray Crystallography and Small-Angle X-ray Scattering. J. Mol. Biol. 2016, 428, 3058–3073. [Google Scholar] [CrossRef]
- Kearney, P.L.; Bhatia, M.; Jones, N.G.; Yuan, L.; Glascock, M.C.; Catchings, K.L.; Yamada, M.; Thompson, P.R. Kinetic Characterization of Protein Arginine Deiminase 4: A Transcriptional Corepressor Implicated in the Onset and Progression of Rheumatoid Arthritis. Biochemistry 2005, 44, 10570–10582. [Google Scholar] [CrossRef]
- Vossenaar, E.R.; Zendman, A.J.; Van Venrooij, W.J.; Pruijn, G.J. PAD, a growing family of citrullinating enzymes: Genes, features and involvement in disease. BioEssays 2003, 25, 1106–1118. [Google Scholar] [CrossRef]
- Witalison, E.E.; Thompson, P.R.; Hofseth, L.J. Protein Arginine Deiminases and Associated Citrullination: Physiological Functions and Diseases Associated with Dysregulation. Curr. Drug Targets 2015, 16, 700–710. [Google Scholar] [CrossRef]
- Lee, C.Y.; Wang, D.; Wilhelm, M.; Zolg, D.P.; Schmidt, T.; Schnatbaum, K.; Reimer, U.; Ponten, F.; Uhlen, M.; Hahne, H.; et al. Mining the Human Tissue Proteome for Protein Citrullination. Mol. Cell. Proteom. 2018, 17, 1378–1391. [Google Scholar] [CrossRef] [Green Version]
- van Beers, J.J.; Schwarte, C.M.; Stammen-Vogelzangs, J.; Oosterink, E.; Bozic, B.; Pruijn, G.J. The rheumatoid arthritis synovial fluid citrullinome reveals novel citrullinated epitopes in apolipoprotein E, myeloid nuclear differentiation antigen, and beta-actin. Arthritis Rheum. 2013, 65, 69–80. [Google Scholar] [CrossRef]
- Romero, V.; Fert-Bober, J.; Nigrovic, P.A.; Darrah, E.; Haque, U.J.; Lee, D.M.; Van Eyk, J.; Rosen, A.; Andrade, F. Immune-Mediated Pore-Forming Pathways Induce Cellular Hypercitrullination and Generate Citrullinated Autoantigens in Rheumatoid Arthritis. Sci. Transl. Med. 2013, 5, 209ra150. [Google Scholar] [CrossRef]
- Tutturen, A.E.V.; Fleckenstein, B.; De Souza, G.A. Assessing the Citrullinome in Rheumatoid Arthritis Synovial Fluid with and without Enrichment of Citrullinated Peptides. J. Proteome Res. 2014, 13, 2867–2873. [Google Scholar] [CrossRef]
- Wang, F.; Chen, F.F.; Gao, W.B.; Wang, H.Y.; Zhao, N.W.; Xu, M.; Gao, D.Y.; Yu, W.; Yan, X.L.; Zhao, J.N.; et al. Identification of citrullinated peptides in the synovial fluid of patients with rheumatoid arthritis using LC-MALDI-TOF/TOF. Clin. Rheumatol. 2016, 35, 2185–2194. [Google Scholar] [CrossRef] [Green Version]
- Tilvawala, R.; Nguyen, S.H.; Maurais, A.J.; Nemmara, V.V.; Nagar, M.; Salinger, A.J.; Nagpal, S.; Weerapana, E.; Thompson, P.R. The Rheumatoid Arthritis-Associated Citrullinome. Cell Chem. Biol. 2018, 25, 691–704.e6. [Google Scholar] [CrossRef] [Green Version]
- Bennike, T.B.; Ellingsen, T.; Glerup, H.; Bonderup, O.K.; Carlsen, T.G.; Meyer, M.K.; Bøgsted, M.; Christiansen, G.; Birkelund, S.; Andersen, V.; et al. Proteome Analysis of Rheumatoid Arthritis Gut Mucosa. J. Proteome Res. 2017, 16, 346–354. [Google Scholar] [CrossRef]
- Konig, M.F.; Abusleme, L.; Reinholdt, J.; Palmer, R.J.; Teles, R.P.; Sampson, K.; Rosen, A.; Nigrovic, P.A.; Sokolove, J.; Giles, J.T.; et al. Aggregatibacter actinomycetemcomitans-induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis. Sci. Transl. Med. 2016, 8, 369ra176. [Google Scholar] [CrossRef]
- Schwenzer, A.; Quirke, A.M.; Marzeda, A.M.; Wong, A.; Montgomery, A.B.; Sayles, H.R.; Eick, S.; Gawron, K.; Chomyszyn-Gajewska, M.; et al. Association of Distinct Fine Specificities of Anti-Citrullinated Peptide Antibodies with Elevated Immune Responses to Prevotella intermedia in a Subgroup of Patients With Rheumatoid Arthritis and Periodontitis. Arthritis Rheumatol. 2017, 69, 2303–2313. [Google Scholar] [CrossRef]
- Macdonald, J.B.; Gibbons, R.J.; Socransky, S.S. Bacterial mechanisms in periodontal disease. Ann. N. Y. Acad. Sci. 1960, 85, 467–478. [Google Scholar] [CrossRef]
- Finegold, S.M.; Barnes, E.M. Report of Icsb Taxonomic Subcommittee on Gram-Negative Anaerobic Rods. Int. J. Syst. Bacteriol. 1977, 27, 388–391. [Google Scholar] [CrossRef]
- Reed, M.J.; Slots, J.; Mouton, C.; Genco, R.J. Antigenic Studies of Oral and Non-Oral Black-Pigmented Bacteroides Strains. Infect. Immun. 1980, 29, 564–574. [Google Scholar]
- Kaczmarek, F.S.; Coykendall, A.L. Production of phenylacetic acid by strains of Bacteroides asaccharolyticus and Bacteroides gingivalis (sp. nov). J. Clin. Microbiol. 1980, 12, 288–290. [Google Scholar]
- Shah, H.N.; Collins, M.D. Proposal for Reclassification of Bacteroides asaccharolyticus, Bacteroides gingivalis, and Bacteroides endodontalis in a New Genus, Porphyromonas. Int. J. Syst. Bacteriol. 1988, 38, 128–131. [Google Scholar] [CrossRef] [Green Version]
- Lamont, R.J.; Koo, H.; Hajishengallis, G. The oral microbiota: Dynamic communities and host interactions. Nat. Rev. Genet. 2018, 16, 745–759. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Darveau, R.P.; Curtis, M.A. The keystone-pathogen hypothesis. Nat. Rev. Microbiol. 2012, 10, 717–725. [Google Scholar] [CrossRef]
- de Smit, M.; Westra, J.; Vissink, A.; Doornbos-van der Meer, B.; Brouwer, E.; van Winkelhoff, A.J. Periodontitis in established rheumatoid arthritis patients: A cross-sectional clinical, microbiological and serological study. Arthritis Res. Ther. 2012, 14, R222. [Google Scholar] [CrossRef]
- Engström, M.; Eriksson, K.; Lee, L.; Hermansson, M.; Johansson, A.; Nicholas, A.P.; Gerasimcik, N.; Lundberg, K.; Klareskog, L.; Catrina, A.I.; et al. Increased citrullination and expression of peptidylarginine deiminases independently of P. gingivalis and A. actinomycetemcomitans in gingival tissue of patients with periodontitis. J. Transl. Med. 2018, 16, 214. [Google Scholar] [CrossRef]
- Eriksson, K.; Fei, G.; Lundmark, A.; Benchimol, D.; Lee, L.; Hu, Y.O.O.; Kats, A.; Saevarsdottir, S.; Catrina, A.I.; Klinge, B.; et al. Periodontal Health and Oral Microbiota in Patients with Rheumatoid Arthritis. J. Clin. Med. 2019, 8, 630. [Google Scholar] [CrossRef]
- Schmickler, J.; Rupprecht, A.; Patschan, S.; Patschan, D.; Müller, G.A.; Haak, R.; Mausberg, R.F.; Schmalz, G.; Kottmann, T.; Ziebolz, D.; et al. Cross-Sectional Evaluation of Periodontal Status and Microbiologic and Rheumatoid Parameters in a Large Cohort of Patients with Rheumatoid Arthritis. J. Periodontol. 2017, 88, 368–379. [Google Scholar] [CrossRef]
- Ziebolz, D.; Pabel, S.O.; Lange, K.; Krohn-Grimberghe, B.; Hornecker, E.; Mausberg, R.F. Clinical Periodontal and Microbiologic Parameters in Patients with Rheumatoid Arthritis. J. Periodontol. 2011, 82, 1424–1432. [Google Scholar] [CrossRef]
- Beyer, K.; Zaura, E.; Brandt, B.W.; Buijs, M.J.; Brun, J.G.; Crielaard, W.; Bolstad, A.I. Subgingival microbiome of rheumatoid arthritis patients in relation to their disease status and periodontal health. PLoS ONE 2018, 13, e0202278. [Google Scholar] [CrossRef]
- Laugisch, O.; Wong, A.; Sroka, A.; Kantyka, T.; Koziel, J.; Neuhaus, K.; Sculean, A.; Venables, P.J.; Potempa, J.; Möller, B.; et al. Citrullination in the periodontium—A possible link between periodontitis and rheumatoid arthritis. Clin. Oral Investig. 2015, 20, 675–683. [Google Scholar] [CrossRef]
- Lopez-Oliva, I.; Paropkari, A.D.; Saraswat, S.; Serban, S.; Yonel, Z.; Sharma, P.; De Pablo, P.; Raza, K.; Filer, A.; Chapple, I.; et al. Dysbiotic Subgingival Microbial Communities in Periodontally Healthy Patients with Rheumatoid Arthritis. Arthritis Rheumatol. 2018, 70, 1008–1013. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, D.; Jia, H.; Feng, Q.; Wang, D.; Liang, D.; Wu, X.; Li, J.; Tang, L.; Li, Y.; et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat. Med. 2015, 21, 895–905. [Google Scholar] [CrossRef]
- Mikuls, T.R.; Walker, C.; Qiu, F.; Yu, F.; Thiele, G.M.; Alfant, B.; Li, E.C.; Zhao, L.Y.; Wang, G.P.; Datta, S.; et al. The subgingival microbiome in patients with established rheumatoid arthritis. Rheumatology 2018, 57, 1162–1172. [Google Scholar] [CrossRef] [Green Version]
- Scher, J.U.; Ubeda, C.; Equinda, M.; Khanin, R.; Buischi, Y.; Viale, A.; Lipuma, L.; Attur, M.; Pillinger, M.H.; Weissmann, G.; et al. Periodontal Disease and the Oral Microbiota in New-Onset Rheumatoid Arthritis. Arthritis Rheum. 2012, 64, 3083–3094. [Google Scholar] [CrossRef]
- Mankia, K.; Cheng, Z.; Do, T.; Hunt, L.; Meade, J.; Kang, J.; Clerehugh, V.; Speirs, A.; Tugnait, A.; Hensor, E.M.A.; et al. Prevalence of Periodontal Disease and Periodontopathic Bacteria in Anti–Cyclic Citrullinated Protein Antibody–Positive At-Risk Adults Without Arthritis. JAMA Netw. Open 2019, 2, e195394. [Google Scholar] [CrossRef]
- Arvikar, S.L.; Collier, D.S.; Fisher, M.C.; Unizony, S.; Cohen, G.L.; McHugh, G.; Kawai, T.; Strle, K.; Steere, A.C. Clinical correlations with Porphyromonas gingivalis antibody responses in patients with early rheumatoid arthritis. Arthritis Res. Ther. 2013, 15, R109. [Google Scholar] [CrossRef]
- Okada, M.; Kobayashi, T.; Ito, S.; Yokoyama, T.; Komatsu, Y.; Abe, A.; Murasawa, A.; Yoshie, H. Antibody Responses to Periodontopathic Bacteria in Relation to Rheumatoid Arthritis in Japanese Adults. J. Periodontol. 2011, 82, 1433–1441. [Google Scholar] [CrossRef]
- Mikuls, T.R.; Payne, J.B.; Reinhardt, R.A.; Thiele, G.M.; Maziarz, E.; Cannella, A.C.; Holers, V.M.; Kuhn, K.A.; O’Dell, J.R. Antibody responses to Porphyromonas gingivalis (P. gingivalis) in subjects with rheumatoid arthritis and periodontitis. Int. Immunopharmacol. 2009, 9, 38–42. [Google Scholar] [CrossRef] [Green Version]
- Ogrendik, M.; Kokino, S.; Ozdemir, F.; Bird, P.S.; Hamlet, S. Serum Antibodies to Oral Anaerobic Bacteria in Patients with Rheumatoid Arthritis. MedGenMed Medscape Gen. Med. 2005, 7, 2. [Google Scholar]
- Yusof, Z.R.; Porter, S.; Greenman, J.; Scully, C. Levels of Serum IgG against Porphyromonas gingivalis in Patients with Rapidly Progressive Periodontitis, Rheumatoid Arthritis and Adult Periodontitis. J. Nihon Univ. Sch. Dent. 1995, 37, 197–200. [Google Scholar] [CrossRef]
- Moen, K.; Brun, J.G.; Madland, T.M.; Tynning, T.; Jonsson, R. Immunoglobulin G and a Antibody Responses to Bacteroides forsythus and Prevotella intermedia in Sera and Synovial Fluids of Arthritis Patients. Clin. Diagn. Lab. Immunol. 2003, 10, 1043–1050. [Google Scholar] [CrossRef]
- de Smit, M.; van de Stadt, L.A.; Janssen, K.M.; Doornbos-van der Meer, B.; Vissink, A.; van Winkelhoff, A.J.; Brouwer, E.; Westra, J.; van Schaardenburg, D. Antibodies against Porphyromonas gingivalis in seropositive arthralgia patients do not predict development of rheumatoid arthritis. Ann. Rheum. Dis. 2014, 73, 1277–1279. [Google Scholar] [CrossRef]
- Konig, M.F.; Paracha, A.S.; Moni, M.; Bingham, C.O., III; Andrade, F. Defining the role of Porphyromonas gingivalis peptidylarginine deiminase (PPAD) in rheumatoid arthritis through the study of PPAD biology. Ann. Rheum. Dis. 2015, 74, 2054–2061. [Google Scholar] [CrossRef]
- Quirke, A.M.; Lugli, E.B.; Wegner, N.; Hamilton, B.C.; Charles, P.; Chowdhury, M.; Ytterberg, A.J.; Zubarev, R.A.; Potempa, J.; Culshaw, S.; et al. Heightened immune response to autocitrullinated Porphyromonas gingivalis peptidylarginine deiminase: A potential mechanism for breaching immunologic tolerance in rheumatoid arthritis. Ann. Rheum. Dis. 2014, 73, 263–269. [Google Scholar] [CrossRef]
- Fisher, B.A.; Cartwright, A.J.; Quirke, A.M.; de Pablo, P.; Romaguera, D.; Panico, S.; Mattiello, A.; Gavrila, D.; Navarro, C.; Sacerdote, C.; et al. Smoking, Porphyromonas gingivalis and the immune response to citrullinated autoantigens before the clinical onset of rheumatoid arthritis in a Southern European nested case-control study. BMC Musculoskelet. Disord. 2015, 16, 331. [Google Scholar] [CrossRef]
- Johansson, L.; Sherina, N.; Kharlamova, N.; Potempa, B.; Larsson, B.; Israelsson, L.; Potempa, J.; Rantapää-Dahlqvist, S.; Lundberg, K. Concentration of antibodies against Porphyromonas gingivalis is increased before the onset of symptoms of rheumatoid arthritis. Arthritis Res. Ther. 2016, 18, 201. [Google Scholar] [CrossRef]
- Kharlamova, N.; Jiang, X.; Sherina, N.; Potempa, B.; Israelsson, L.; Quirke, A.-M.; Eriksson, K.; Yucel-Lindberg, T.; Venables, P.J.; Potempa, J.; et al. Antibodies to Porphyromonas gingivalis indicate interaction between oral infection, smoking and risk genes in rheumatoid arthritis etiology. Arthritis Rheumatol. 2016, 68, 604–613. [Google Scholar] [CrossRef]
- Mikuls, T.R.; Payne, J.B.; Yu, F.; Thiele, G.M.; Reynolds, R.J.; Cannon, G.W.; Markt, J.; McGowan, D.; Kerr, G.S.; Redman, R.S.; et al. Periodontitis and Porphyromonas gingivalis in Patients with Rheumatoid Arthritis. Arthritis Rheumatol. 2014, 66, 1090–1100. [Google Scholar] [CrossRef]
- Okada, M.; Kobayashi, T.; Ito, S.; Yokoyama, T.; Abe, A.; Murasawa, A.; Yoshie, H. Periodontal Treatment Decreases Levels of Antibodies to Porphyromonas gingivalis and Citrulline in Patients with Rheumatoid Arthritis and Periodontitis. J. Periodontol. 2013, 84, e74–e84. [Google Scholar] [CrossRef]
- Rinaudo-Gaujous, M.; Blasco-Baque, V.; Miossec, P.; Gaudin, P.; Farge, P.; Roblin, X.; Thomas, T.; Paul, S.; Marotte, H. Infliximab Induced a Dissociated Response of Severe Periodontal Biomarkers in Rheumatoid Arthritis Patients. J. Clin. Med. 2019, 8, 751. [Google Scholar] [CrossRef]
- Stobernack, T.; Glasner, C.; Junker, S.; Gabarrini, G.; De Smit, M.; Van Winkelhoff, A.J.; De Jong, A.; Otto, A.; Becher, D.; Van Dijl, J.M. Extracellular Proteome and Citrullinome of the Oral Pathogen Porphyromonas gingivalis. J. Proteome Res. 2016, 15, 4532–4543. [Google Scholar] [CrossRef]
- Bae, S.C.; Lee, Y.H. Association between anti-Porphyromonas gingivalis antibody, anti-citrullinated protein antibodies, and rheumatoid arthritis a meta-analysis. Z. Für Rheumatol. 2018, 77, 522–532. [Google Scholar] [CrossRef]
- Bender, P.; Burgin, W.B.; Sculean, A.; Eick, S. Serum antibody levels against Porphyromonas gingivalis in patients with and without rheumatoid arthritis - a systematic review and meta-analysis. Clin. Oral Investig. 2017, 21, 33–42. [Google Scholar] [CrossRef]
- Maresz, K.J.; Hellvard, A.; Sroka, A.; Adamowicz, K.; Bielecka, E.; Koziel, J.; Gawron, K.; Mizgalska, D.; Marcinska, K.A.; Benedyk, M.; et al. Porphyromonas gingivalis Facilitates the Development and Progression of Destructive Arthritis through Its Unique Bacterial Peptidylarginine Deiminase (PAD). PLoS Pathog. 2013, 9, e1003627. [Google Scholar] [CrossRef]
- de Aquino, S.G.; Abdollahi-Roodsaz, S.; Koenders, M.I.; van de Loo, F.A.; Pruijn, G.J.; Marijnissen, R.J.; Walgreen, B.; Helsen, M.M.; van den Bersselaar, L.A.; de Molon, R.S.; et al. Periodontal pathogens directly promote autoimmune experimental arthritis by inducing a TLR2- and IL-1-driven Th17 response. J. Immunol. 2014, 192, 4103–4111. [Google Scholar] [CrossRef]
- de Aquino, S.G.; Talbot, J.; Sônego, F.; Turato, W.M.; Grespan, R.; Avila-Campos, M.J.; Cunha, F.Q.; Cirelli, J.A. The aggravation of arthritis by periodontitis is dependent of IL-17 receptor a activation. J. Clin. Periodontol. 2017, 44, 881–891. [Google Scholar] [CrossRef]
- Marchesan, J.T.; Gerow, E.A.; Schaff, R.; Taut, A.D.; Shin, S.Y.; Sugai, J.; Brand, D.; Burberry, A.; Jorns, J.; Lundy, S.K.; et al. Porphyromonas gingivalis oral infection exacerbates the development and severity of collagen-induced arthritis. Arthritis Res. Ther. 2013, 15, R186. [Google Scholar] [CrossRef]
- Jung, H.; Jung, S.M.; Rim, Y.A.; Park, N.; Nam, Y.; Lee, J.; Park, S.H.; Ju, J.H. Arthritic role of Porphyromonas gingivalis in collagen-induced arthritis mice. PLoS ONE 2017, 12, e0188698. [Google Scholar] [CrossRef]
- Chukkapalli, S.; Rivera-Kweh, M.; Gehlot, P.; Velsko, I.; Bhattacharyya, I.; Calise, S.J.; Satoh, M.; Chan, E.K.L.; Holoshitz, J.; Kesavalu, L. Periodontal bacterial colonization in synovial tissues exacerbates collagen-induced arthritis in B10.RIII mice. Arthritis Res. Ther. 2016, 18, 161. [Google Scholar] [CrossRef]
- Sato, K.; Takahashi, N.; Kato, T.; Matsuda, Y.; Yokoji, M.; Yamada, M.; Nakajima, T.; Kondo, N.; Endo, N.; Yamamoto, R.; et al. Aggravation of collagen-induced arthritis by orally administered Porphyromonas gingivalis through modulation of the gut microbiota and gut immune system. Sci. Rep. 2017, 7, 6955. [Google Scholar] [CrossRef]
- Yamakawa, M.; Ouhara, K.; Kajiya, M.; Munenaga, S.; Kittaka, M.; Yamasaki, S.; Takeda, K.; Takeshita, K.; Mizuno, N.; Fujita, T.; et al. Porphyromonas gingivalis infection exacerbates the onset of rheumatoid arthritis in SKG mice. Clin. Exp. Immunol. 2016, 186, 177–189. [Google Scholar] [CrossRef]
- Sandal, I.; Karydis, A.; Luo, J.; Prislovsky, A.; Whittington, K.B.; Rosloniec, E.F.; Dong, C.; Novack, D.V.; Mydel, P.; Zheng, S.G.; et al. Bone loss and aggravated autoimmune arthritis in HLA-DR beta 1-bearing humanized mice following oral challenge with Porphyromonas gingivalis. Arthritis Res. Ther. 2016, 18, 249. [Google Scholar] [CrossRef]
- Gully, N.; Bright, R.; Marino, V.; Marchant, C.; Cantley, M.; Haynes, D.; Butler, C.; Dashper, S.; Reynolds, E.; Bartold, M. Porphyromonas gingivalis Peptidylarginine Deiminase, a Key Contributor in the Pathogenesis of Experimental Periodontal Disease and Experimental Arthritis. PLoS ONE 2014, 9, e100838. [Google Scholar] [CrossRef]
- Munenaga, S.; Ouhara, K.; Hamamoto, Y.; Kajiya, M.; Takeda, K.; Yamasaki, S.; Kawai, T.; Mizuno, N.; Fujita, T.; Sugiyama, E.; et al. The involvement of C5a in the progression of experimental arthritis with Porphyromonas gingivalis infection in SKG mice. Arthritis Res. Ther. 2018, 20, 247. [Google Scholar] [CrossRef] [Green Version]
- Ebbers, M.; Lübcke, P.M.; Volzke, J.; Kriebel, K.; Hieke, C.; Engelmann, R.; Lang, H.; Kreikemeyer, B.; Müller-Hilke, B.; et al. Interplay between P. gingivalis, F. nucleatum and A. actinomycetemcomitans in murine alveolar bone loss, arthritis onset and progression. Sci. Rep. 2018, 8, 15129. [Google Scholar] [CrossRef]
- Eriksson, K.; Lönnblom, E.; Tour, G.; Kats, A.; Mydel, P.; Georgsson, P.; Hultgren, C.; Kharlamova, N.; Norin, U.; Jönsson, J.; et al. Effects by periodontitis on pristane-induced arthritis in rats. J. Transl. Med. 2016, 14, 311. [Google Scholar] [CrossRef]
- Courbon, G.; Rinaudo-Gaujous, M.; Blasco-Baque, V.; Auger, I.; Caire, R.; Mijola, L.; Vico, L.; Paul, S.; Marotte, H. Porphyromonas gingivalis experimentally induces periodontis and an anti-CCP2-associated arthritis in the rat. Ann. Rheum. Dis. 2019, 78, 594–599. [Google Scholar] [CrossRef]
- Kinloch, A.J.; Alzabin, S.; Brintnell, W.; Wilson, E.; Barra, L.; Wegner, N.; Bell, D.A.; Cairns, E.; Venables, P.J. Immunization with Porphyromonas gingivalis enolase induces autoimmunity to mammalian alpha-enolase and arthritis in DR4-IE-transgenic mice. Arthritis Rheum. 2011, 63, 3818–3823. [Google Scholar] [CrossRef]
- Mangat, P.; Wegner, N.; Venables, P.J.; Potempa, J. Bacterial and human peptidylarginine deiminases: Targets for inhibiting the autoimmune response in rheumatoid arthritis? Arthritis Res. Ther. 2010, 12, 209. [Google Scholar] [CrossRef]
- Hayashi, H.; Morioka, M.; Ichimiya, S.; Yamato, K.; Hinode, D.; Nagata, A.; Nakamura, R. Participation of an arginyl residue of insulin chain B in the inhibition of hemagglutination by Porphyromonas gingivalis. Oral Microbiol. Immunol. 1993, 8, 386–389. [Google Scholar] [CrossRef]
- Goulas, T.; Mizgalska, D.; Garcia-Ferrer, I.; Kantyka, T.; Guevara, T.; Szmigielski, B.; Sroka, A.; Millán, C.; Usón, I.; Veillard, F.; et al. Structure and mechanism of a bacterial host-protein citrullinating virulence factor, Porphyromonas gingivalis peptidylarginine deiminase. Sci. Rep. 2015, 5, 11969. [Google Scholar] [CrossRef]
- Montgomery, A.B.; Kopec, J.; Shrestha, L.; Thezenas, M.L.; Burgess-Brown, N.A.; Fischer, R.; Yue, W.W.; Venables, P.J. Crystal structure of Porphyromonas gingivalis peptidylarginine deiminase: Implications for autoimmunity in rheumatoid arthritis. Ann. Rheum. Dis. 2016, 75, 1255–1261. [Google Scholar] [CrossRef]
- Bereta, G.; Goulas, T.; Madej, M.; Bielecka, E.; Solà, M.; Potempa, J.; Gomis-Rüth, F.X. Structure, function, and inhibition of a genomic/clinical variant of Porphyromonas gingivalis peptidylarginine deiminase. Protein Sci. 2019, 28, 478–486. [Google Scholar] [CrossRef]
- Gabarrini, G.; Heida, R.; Van Ieperen, N.; Curtis, M.A.; Van Winkelhoff, A.J.; Van Dijl, J.M. Dropping anchor: Attachment of peptidylarginine deiminase via A-LPS to secreted outer membrane vesicles of Porphyromonas gingivalis. Sci. Rep. 2018, 8, 8949. [Google Scholar] [CrossRef]
- Gabarrini, G.; Medina, L.M.P.; Stobernack, T.; Prins, R.C.; Espina, M.D.T.; Kuipers, J.; Chlebowicz, M.A.; Rossen, J.W.A.; Van Winkelhoff, A.J.; Van Dijl, J.M. There’s no place like OM: Vesicular sorting and secretion of the peptidylarginine deiminase of Porphyromonas gingivalis. Virulence 2018, 9, 456–464. [Google Scholar] [CrossRef]
- Casiano-Colón, A.; Marquis, R.E. Role of the arginine deiminase system in protecting oral bacteria and an enzymatic basis for acid tolerance. Appl. Environ. Microbiol. 1988, 54, 1318–1324. [Google Scholar]
- Niederman, R.; Brunkhorst, B.; Smith, S.; Weinreb, R.; Ryder, M. Ammonia as a potential mediator of adult human periodontal infection: Inhibition of neutrophil function. Arch. Oral Biol. 1990, 35, S205–S209. [Google Scholar] [CrossRef]
- Shawcross, D.L.; Wright, G.A.K.; Stadlbauer, V.; Hodges, S.J.; Davies, N.A.; Wheeler-Jones, C.; Pitsillides, A.A.; Jalan, R. Ammonia impairs neutrophil phagocytic function in liver disease. Hepatology 2008, 48, 1202–1212. [Google Scholar] [CrossRef]
- Wegner, N.; Wait, R.; Sroka, A.; Eick, S.; Nguyen, K.A.; Lundberg, K.; Kinloch, A.; Culshaw, S.; Potempa, J.; Venables, P.J. Peptidylarginine deiminase from Porphyromonas gingivalis citrullinates human fibrinogen and alpha-enolase: Implications for autoimmunity in rheumatoid arthritis. Arthritis Rheum. 2010, 62, 2662–2672. [Google Scholar] [CrossRef]
- Li, S.; Yu, Y.; Yue, Y.; Liao, H.; Xie, W.; Thai, J.; Mikuls, T.R.; Thiele, G.M.; Duryee, M.J.; Sayles, H.; et al. Autoantibodies from Single Circulating Plasmablasts React With Citrullinated Antigens and Porphyromonas gingivalis in Rheumatoid Arthritis. Arthritis Rheumatol. 2016, 68, 614–626. [Google Scholar] [CrossRef]
- Rodríguez, S.B.; Stitt, B.L.; Ash, D.E. Expression of Peptidylarginine Deiminase from Porphyromonas gingivalis in Escherichia coli: Enzyme Purification and Characterization. Arch. Biochem. Biophys. 2009, 488, 14–22. [Google Scholar] [CrossRef]
- Sato, K.; Yukitake, H.; Narita, Y.; Shoji, M.; Naito, M.; Nakayama, K. Identification of Porphyromonas gingivalis proteins secreted by the Por secretion system. FEMS Microbiol. Lett. 2013, 338, 68–76. [Google Scholar] [CrossRef]
- Shoji, M.; Sato, K.; Yukitake, H.; Kondo, Y.; Narita, Y.; Kadowaki, T.; Naito, M.; Nakayama, K. Por Secretion System-Dependent Secretion and Glycosylation of Porphyromonas gingivalis Hemin-Binding Protein 35. PLoS ONE 2011, 6, e21372. [Google Scholar] [CrossRef]
- Konig, M.F.; Bingham, C.O., III; Andrade, F. PPAD is not targeted as a citrullinated protein in rheumatoid arthritis, but remains a candidate for inducing autoimmunity. Ann. Rheum. Dis. 2015, 74, e8. [Google Scholar] [CrossRef]
- Quirke, A.-M.; Lundberg, K.; Potempa, J.; Mikuls, T.R.; Venables, P.J. PPAD remains a credible candidate for inducing autoimmunity in rheumatoid arthritis: Comment on the article by Konig et al. Ann. Rheum. Dis. 2014, 74, e7. [Google Scholar] [CrossRef]
- Kobayashi, T.; Ito, S.; Kobayashi, D.; Shimada, A.; Narita, I.; Murasawa, A.; Nakazono, K.; Yoshie, H. Serum Immunoglobulin G Levels to Porphyromonas gingivalis Peptidylarginine Deiminase Affect Clinical Response to Biological Disease-Modifying Antirheumatic Drug in Rheumatoid Arthritis. PLoS ONE 2016, 11, e0154182. [Google Scholar] [CrossRef]
- Shimada, A.; Kobayashi, T.; Ito, S.; Okada, M.; Murasawa, A.; Nakazono, K.; Yoshie, H. Expression of anti-Porphyromonas gingivalis peptidylarginine deiminase immunoglobulin G and peptidylarginine deiminase-4 in patients with rheumatoid arthritis and periodontitis. J. Periodontal Res. 2016, 51, 103–111. [Google Scholar] [CrossRef]
- Vossenaar, E.R.; Nijenhuis, S.; Helsen, M.M.A.; Van Der Heijden, A.; Senshu, T.; Berg, W.B.V.D.; Van Venrooij, W.J.; Joosten, L.A.B. Citrullination of synovial proteins in murine models of rheumatoid arthritis. Arthritis Rheum. 2003, 48, 2489–2500. [Google Scholar] [CrossRef] [Green Version]
- Cantaert, T.; Teitsma, C.; Tak, P.P.; Baeten, D. Presence and Role of Anti-Citrullinated Protein Antibodies in Experimental Arthritis Models. Arthritis Rheum. 2013, 65, 939–948. [Google Scholar] [CrossRef]
- Konig, M.F.; Darrah, E.; Andrade, F. Insights into the significance of peptidylarginine deiminase 4 and antibodies against citrullinated antigens in the absence of “true ACPAs” in an experimental model of arthritis: Comment on the article by Shelef et al. Arthritis Rheumatol. 2014, 66, 2642–2644. [Google Scholar] [CrossRef]
- Shelef, M.A.; Sokolove, J.; Robinson, W.H.; Huttenlocher, A. Insights into the significance of peptidylarginine deiminase 4 and antibodies against citrullinated antigens in the absence of “true ACPAs” in an experimental model of arthritis: Comment on the article by Shelef et al Reply. Arthritis Rheumatol. 2014, 66, 2644–2645. [Google Scholar] [CrossRef]
- Vossenaar, E.R.; Van Boekel, M.A.M.; Van Venrooij, W.J.; López-Hoyoz, M.; Merino, J.; Merino, R.; Joosten, L.A.B. Absence of citrulline-specific autoantibodies in animal models of autoimmunity. Arthritis Rheum. 2004, 50, 2370–2372. [Google Scholar] [CrossRef]
- Kingler, R. Untersuchungen über menschliche Aktinomycose. Zent. Bakteriol 1912, 62, 191–200. [Google Scholar]
- Potts, T.V.; Zambon, J.J.; Genco, R.J. Reassignment of Actinobacillus-Actinomycetemcomitans to the Genus Hemophilus as Haemophilus-Actinomycetemcomitans Comb-Nov. Int. J. Syst. Bacteriol. 1985, 35, 337–341. [Google Scholar] [CrossRef]
- Norskov-Lauritsen, N.; Kilian, M. Reclassification of Actinobacillus actinomycetemcomitans, Haemophilus aphrophilus, Haemophilus paraphrophilus and Haemophilus segnis as Aggregatibacter actinomycetemcomitans gen. nov., comb. nov., Aggregatibacter aphrophilus comb. nov. and Aggregatibacter segnis comb. nov., and emended description of Aggregatibacter aphrophilus to include V factor-dependent and V factor-independent isolates. Int. J. Syst. Evol. Microbiol. 2006, 56, 2135–2146. [Google Scholar]
- Heinrich, S.; Pulverer, G. Zur Ätiologie und Mikrobiologie der Aktinomykose III. Die pathogene Bedeutung des Actinobacillus actinomycetem-comitans unter den “Begleitbakterien” des Actinomyces israeli. Zent. Bakteriol 1959, 176, 91–101. [Google Scholar]
- Colebrook, L. The mycelial and other micro-organisms associated with human actinomycosis. Br. J. Exp. Pathol. 1920, 1, 197–212. [Google Scholar]
- King, E.O.; Tatum, H.W. Actinobacillus Actinomycetemcomitans and Hemophilus Aphrophilus. J. Infect. Dis. 1962, 111, 85–94. [Google Scholar] [CrossRef]
- Mitchell, R.G.; Gillespie, W.A. Bacterial endocarditis due to an actinobacillus. J. Clin. Pathol. 1964, 17, 511–512. [Google Scholar] [CrossRef] [Green Version]
- Page, M.I.; King, E.O. Infection Due to Actinobacillus Actinomycetemcomitans and Haemophilus Aphrophilus. N. Engl. J. Med. 1966, 275, 181–188. [Google Scholar] [CrossRef]
- Geraci, J.E.; Wilson, W.R. Symposium on infective endocarditis. III. Endocarditis due to gram-negative bacteria. Report of 56 cases. Mayo Clin. Proc. 1982, 57, 145–148. [Google Scholar]
- Kaplan, A.H.; Weber, D.J.; Oddone, E.Z.; Perfect, J.R. Infection Due to Actinobacillus-Actinomycetemcomitans - 15 Cases and Review. Rev. Infect. Dis. 1989, 11, 46–63. [Google Scholar] [CrossRef]
- van Winkelhoff, A.J.; Slots, J. Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis in nonoral infections. Periodontol. 2000. 1999, 20, 122–135. [Google Scholar] [CrossRef]
- Tanner, A.C.R.; Haffer, C.; Bratthall, G.T.; Visconti, R.A.; Socransky, S.S. A study of the bacteria associated with advancing periodontitis in man. J. Clin. Periodontol. 1979, 6, 278–307. [Google Scholar] [CrossRef]
- Slots, J.; Reynolds, H.S.; Genco, R.J. Actinobacillus actinomycetemcomitans in Human Periodontal Disease: A Cross-Sectional Microbiological Investigation. Infect. Immun. 1980, 29, 1013–1020. [Google Scholar]
- Zambon, J.J.; Christersson, L.A.; Slots, J. Actinobacillus actinomycetemcomitans in human periodontal disease. Prevalence in patient groups and distribution of biotypes and serotypes within families. J. Periodontol. 1983, 54, 707–711. [Google Scholar] [CrossRef]
- Zambon, J.J.; Slots, J.; Genco, R.J. Serology of oral Actinobacillus actinomycetemcomitans and serotype distribution in human periodontal disease. Infect. Immun. 1983, 41, 19–27. [Google Scholar] [Green Version]
- Slots, J.; Bragd, L.; Wikström, M.; Dahlén, G. The occurrence of Actinobacillus actinomycetemcomitans, Bacteroides gingivalis and Bacteroides intermedius in destructive periodontal disease in adults. J. Clin. Periodontol. 1986, 13, 570–577. [Google Scholar] [CrossRef]
- Johansson, A. Aggregatibacter actinomycetemcomitans leukotoxin: A powerful tool with capacity to cause imbalance in the host inflammatory response. Toxins (Basel) 2011, 3, 242–259. [Google Scholar] [CrossRef]
- Claesson, R.; Höglund-Åberg, C.; Haubek, D.; Johansson, A. Age-related prevalence and characteristics of Aggregatibacter actinomycetemcomitans in periodontitis patients living in Sweden. J. Oral Microbiol. 2017, 9, 1334504. [Google Scholar] [CrossRef]
- Zambon, J.J. Actinobacillus actinomycetemcomitans in human periodontal disease. J. Clin. Periodontol. 1985, 12, 1–20. [Google Scholar] [CrossRef]
- Chen, C.; Wang, T.; Chen, W. Occurrence of Aggregatibacter actinomycetemcomitans serotypes in subgingival plaque from United States subjects. Mol. Oral Microbiol. 2010, 25, 207–214. [Google Scholar] [CrossRef]
- Pahumunto, N.; Ruangsri, P.; Wongsuwanlert, M.; Piwat, S.; Dahlén, G.; Teanpaisan, R. Aggregatibacter actinomycetemcomitans serotypes and DGGE subtypes in Thai adults with chronic periodontitis. Arch. Oral Biol. 2015, 60, 1789–1796. [Google Scholar] [CrossRef]
- Kim, T.S.; Frank, P.; Eickholz, P.; Eick, S.; Kim, C.K. Serotypes of Aggregatibacter actinomycetemcomitans in patients with different ethnic backgrounds. J. Periodontol. 2009, 80, 2020–2027. [Google Scholar] [CrossRef]
- Mínguez, M.; Pousa, X.; Herrera, D.; Blasi, A.; Sánchez, M.C.; León, R.; Sanz, M. Characterization and serotype distribution of Aggregatibacter actinomycetemcomitans isolated from a population of periodontitis patients in Spain. Arch. Oral Biol. 2014, 59, 1359–1367. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.W.; Huang, Y.F.; Chan, Y.; Chou, M.Y. Relationship of Actinobacillus actinomycetemcomitans serotypes to periodontal condition: Prevalence and proportions in subgingival plaque. Eur. J. Oral Sci. 2005, 113, 28–33. [Google Scholar] [CrossRef]
- Arenas Rodrigues, V.A.; de Avila, E.D.; Nakano, V.; Avila-Campos, M.J. Qualitative, quantitative and genotypic evaluation of Aggregatibacter actinomycetemcomitans and Fusobacterium nucleatum isolated from individuals with different periodontal clinical conditions. Anaerobe 2018, 52, 50–58. [Google Scholar] [CrossRef] [Green Version]
- Åberg, C.H.; Haubek, D.; Kwamin, F.; Johansson, A.; Claesson, R. Leukotoxic Activity of Aggregatibacter actinomycetemcomitans and Periodontal Attachment Loss. PLoS ONE 2014, 9, e104095. [Google Scholar]
- Aberg, C.H.; Kelk, P.; Johansson, A. Aggregatibacter actinomycetemcomitans: Virulence of its leukotoxin and association with aggressive periodontitis. Virulence 2015, 6, 188–195. [Google Scholar] [CrossRef]
- Linhartová, I.; Bumba, L.; Mašín, J.; Basler, M.; Osička, R.; Kamanová, J.; Procházková, K.; Adkins, I.; Hejnová-Holubová, J.; Sadílková, L.; et al. RTX proteins: A highly diverse family secreted by a common mechanism. FEMS Microbiol. Rev. 2010, 34, 1076–1112. [Google Scholar] [CrossRef]
- Reinholdt, J.; Poulsen, K.; Brinkmann, C.R.; Hoffmann, S.V.; Stapulionis, R.; Enghild, J.J.; Jensen, U.B.; Boesen, T.; Vorup-Jensen, T. Monodisperse and LPS-free Aggregatibacter actinomycetemcomitans leukotoxin: Interactions with human beta2 integrins and erythrocytes. Biochim. Biophys. Acta 2013, 1834, 546–558. [Google Scholar] [CrossRef]
- Brogan, J.M.; Lally, E.T.; Poulsen, K.; Kilian, M.; DeMuth, D.R. Regulation of Actinobacillus actinomycetemcomitans leukotoxin expression: Analysis of the promoter regions of leukotoxic and minimally leukotoxic strains. Infect. Immun. 1994, 62, 501–508. [Google Scholar]
- Zambon, J.J.; Haraszthy, V.I.; Hariharan, G.; Lally, E.T.; DeMuth, D.R. The Microbiology of early-onset periodontitis: Association of highly toxic Actinobacillus actinomycetemcomitans strains with localized juvenile periodontitis. J. Periodontol. 1996, 67, 282–290. [Google Scholar] [CrossRef]
- Sampathkumar, V.; Velusamy, S.K.; Godboley, D.; Fine, D.H. Increased leukotoxin production: Characterization of 100 base pairs within the 530 base pair leukotoxin promoter region of Aggregatibacter actinomycetemcomitans. Sci. Rep. 2017, 7, 1887. [Google Scholar] [CrossRef]
- Burgess, D.; Huang, H.; Harrison, P.; Aukhil, I.; Shaddox, L. Aggregatibacter actinomycetemcomitans in African Americans with Localized Aggressive Periodontitis. JDR Clin. Transl. Res. 2017, 2, 249–257. [Google Scholar] [CrossRef]
- Oscarsson, J.; Claesson, R.; Lindholm, M.; Höglund Åberg, C.; Johansson, A. Tools of Aggregatibacter actinomycetemcomitans to Evade the Host Response. J. Clin. Med. 2019, 8, 1079. [Google Scholar] [CrossRef]
- Harvey, G.P.; Fitzsimmons, T.R.; Dhamarpatni, A.A.; Marchant, C.; Haynes, D.R.; Bartold, P.M. Expression of peptidylarginine deiminase-2 and -4, citrullinated proteins and anti-citrullinated protein antibodies in human gingiva. J. Periodontal Res. 2013, 48, 252–261. [Google Scholar] [CrossRef]
- Nesse, W.; Westra, J.; van der Wal, J.E.; Abbas, F.; Nicholas, A.P.; Vissink, A.; Brouwer, E. The periodontium of periodontitis patients contains citrullinated proteins which may play a role in ACPA (anti-citrullinated protein antibody) formation. J. Clin. Periodontol. 2012, 39, 599–607. [Google Scholar] [CrossRef] [Green Version]
- Malinin, T.I.; Pekin, T.J.; Zvaifler, N.J. Cytology of Synovial Fluid in Rheumatoid Arthritis. Am. J. Clin. Pathol. 1967, 47, 203–208. [Google Scholar] [CrossRef] [Green Version]
- Darrah, E.; Rosen, A.; Giles, J.T.; Andrade, F. Peptidylarginine deiminase 2, 3 and 4 have distinct specificities against cellular substrates: Novel insights into autoantigen selection in rheumatoid arthritis. Ann. Rheum. Dis. 2012, 71, 92–98. [Google Scholar] [CrossRef]
- Delima, A.J.; Van Dyke, T.E. Origin and function of the cellular components in gingival crevice fluid. Periodontol 2000 2003, 31, 55–76. [Google Scholar] [CrossRef]
- Konig, M.F.; Andrade, F. A critical reappraisal of neutrophil extracellular traps (NETs) and NETosis mimics based on differential requirements for protein citrullination. Front. Immunol. 2016, 7, 461. [Google Scholar] [CrossRef]
- Volkov, M.; Dekkers, J.; Loos, B.G.; Bizzarro, S.; Huizinga, T.W.J.; Praetorius, H.A.; Toes, R.E.M.; Van Der Woude, D. Comment on Aggregatibacter actinomycetemcomitans—Induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis. Sci. Transl. Med. 2018, 10, eaan8349. [Google Scholar] [CrossRef]
- Mukherjee, A.; Jantsch, V.; Khan, R.; Hartung, W.; Fischer, R.; Jantsch, J.; Ehrenstein, B.; Konig, M.F.; Andrade, F. Rheumatoid Arthritis-Associated Autoimmunity Due to Aggregatibacter actinomycetemcomitans and Its Resolution with Antibiotic Therapy. Front. Immunol. 2018, 9, 2352. [Google Scholar] [CrossRef]
- Konig, M.F.; Giles, J.T.; Teles, R.P.; Moutsopoulos, N.M.; Andrade, F. Response to comment on “Aggregatibacter actinomycetemcomitans–induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis”. Sci. Transl. Med. 2018, 10, eaao3031. [Google Scholar] [CrossRef]
- Queiroz-Junior, C.M.; Madeira, M.F.M.; Coelho, F.M.; Oliveira, C.R.; Cândido, L.C.M.; Garlet, G.P.; Teixeira, M.M.; Souza, D.D.G.; Da Silva, T.A.; Queiroz-Junior, C.M. Experimental arthritis exacerbates Aggregatibacter actinomycetemcomitans-induced periodontitis in mice. J. Clin. Periodontol. 2012, 39, 608–616. [Google Scholar] [CrossRef]
- Taichman, N.S.; Shenker, B.J.; Tsai, C.C.; Glickman, L.T.; Baehni, P.C.; Stevens, R.; Hammond, B.F. Cytopathic effects of Actinobacillus actinomycetemcomitans on monkey blood leukocytes. J. Periodontal Res. 1984, 19, 133–145. [Google Scholar] [CrossRef]
- Rantapää-Dahlqvist, S.; De Jong, B.A.W.; Berglin, E.; Hallmans, G.; Wadell, G.; Stenlund, H.; Sundin, U.; Van Venrooij, W.J. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. 2003, 48, 2741–2749. [Google Scholar] [CrossRef]
- Nielen, M.M.J.; Van Schaardenburg, D.; Reesink, H.W.; Van De Stadt, R.J.; Van Der Horst-Bruinsma, I.E.; De Koning, M.H.M.T.; Habibuw, M.R.; Vandenbroucke, J.P.; Dijkmans, B.A.C.; Van Der Horst-Bruinsma, I.E. Specific autoantibodies precede the symptoms of rheumatoid arthritis: A study of serial measurements in blood donors. Arthritis Rheum. 2004, 50, 380–386. [Google Scholar] [CrossRef]
- Makrygiannakis, D.; Klint, E.A.; Lundberg, I.E.; Lofberg, R.; Ulfgren, A.K.; Klareskog, L.; Catrina, A.I. Citrullination is an inflammation-dependent process. Ann. Rheum. Dis. 2006, 65, 1219–1222. [Google Scholar] [CrossRef] [Green Version]
- Ramseier, C.A.; Anerud, A.; Dulac, M.; Lulic, M.; Cullinan, M.P.; Seymour, G.J.; Faddy, M.J.; Bürgin, W.; Schätzle, M.; Lang, N.P. Natural history of periodontitis: Disease progression and tooth loss over 40 years. J. Clin. Periodontol. 2017, 44, 1182–1191. [Google Scholar] [CrossRef] [Green Version]
- Dal, P.M.; van der Goot, F.G. Pore-forming toxins: Ancient, but never really out of fashion. Nat. Rev. Microbiol. 2016, 14, 77–92. [Google Scholar]
- Samuels, J.; Ng, Y.-S.; Coupillaud, C.; Paget, D.; Meffre, E. Impaired early B cell tolerance in patients with rheumatoid arthritis. J. Exp. Med. 2005, 201, 1659–1667. [Google Scholar] [CrossRef]
- Samuels, J.; Ng, Y.S.; Coupillaud, C.; Paget, D.; Meffre, E. Human B cell tolerance and its failure in rheumatoid arthritis. Ann. N. Y. Acad. Sci. 2005, 1062, 116–126. [Google Scholar] [CrossRef]
- Meffre, E.; Wardemann, H. B-cell tolerance checkpoints in health and autoimmunity. Curr. Opin. Immunol. 2008, 20, 632–638. [Google Scholar] [CrossRef]
- Menard, L.; Samuels, J.; Ng, Y.S.; Meffre, E. Inflammation-independent defective early B cell tolerance checkpoints in rheumatoid arthritis. Arthritis Rheum. 2011, 63, 1237–1245. [Google Scholar] [CrossRef]
- Borsotti, C.; Danzl, N.M.; Nauman, G.; Hölzl, M.A.; French, C.; Chavez, E.; Khosravi-Maharlooei, M.; Glauzy, S.; Delmotte, F.R.; Meffre, E.; et al. HSC extrinsic sex-related and intrinsic autoimmune disease–related human B-cell variation is recapitulated in humanized mice. Blood Adv. 2017, 1, 2007–2018. [Google Scholar] [CrossRef]
- Marotte, H. Determining the Right Time for the Right Treatment—Application to Preclinical Rheumatoid Arthritis. JAMA Netw. Open 2019, 2, e195358. [Google Scholar] [CrossRef]
- Kaur, S.; Bright, R.; Proudman, S.M.; Bartold, P.M. Does periodontal treatment influence clinical and biochemical measures for rheumatoid arthritis? A systematic review and meta-analysis. Semin. Arthritis Rheum. 2014, 44, 113–122. [Google Scholar] [CrossRef]
- Monsarrat, P.; De Grado, G.F.; Constantin, A.; Willmann, C.; Nabet, C.; Sixou, M.; Cantagrel, A.; Barnetche, T.; Mehsen-Cetre, N.; Schaeverbeke, T.; et al. The effect of periodontal treatment on patients with rheumatoid arthritis: The ESPERA randomised controlled trial. Jt. Bone Spine 2019, in press. [Google Scholar] [CrossRef]
- Bright, R.; Thiele, G.M.; Manavis, J.; Mikuls, T.R.; Payne, J.B.; Bartold, P.M. Gingival tissue, an extrasynovial source of malondialdehyde-acetaldehyde adducts, citrullinated and carbamylated proteins. J. Periodontal Res. 2018, 53, 139–143. [Google Scholar] [CrossRef]
- Darrah, E.; Andrade, F. Editorial: citrullination, and carbamylation, and malondialdehyde-acetaldehyde! Oh my! Entering the forest of autoantigen modifications in rheumatoid arthritis. Arthritis Rheumatol. 2015, 67, 604–608. [Google Scholar] [CrossRef]
- Henle, J. Von den Miasmen und Kontagien: Und Von den Miasmatisch-Kontagiösen Krankheiten (1840); Verlag von J. A. Barth: Leipzig, Germany, 1910. [Google Scholar]
- Koch, R. Über Bakteriologische Forschung. In Verhandlungen des X; Internationalen Medicinischen Congresses: Berlin, Germany, 1890; Volume 1. [Google Scholar]
- Rivers, T.M. Viruses and Koch’s postulates. J. Bacteriol. 1937, 33, 1–12. [Google Scholar]
- Correa, M.G.; Sacchetti, S.B.; Ribeiro, F.V.; Pimentel, S.P.; Casarin, R.C.V.; Cirano, F.R.; Casati, M.Z. Periodontitis increases rheumatic factor serum levels and citrullinated proteins in gingival tissues and alter cytokine balance in arthritic rats. PLoS ONE 2017, 12, e0174442. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez-Bañuelos, E.; Mukherjee, A.; Darrah, E.; Andrade, F. Rheumatoid Arthritis-Associated Mechanisms of Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans. J. Clin. Med. 2019, 8, 1309. https://doi.org/10.3390/jcm8091309
Gómez-Bañuelos E, Mukherjee A, Darrah E, Andrade F. Rheumatoid Arthritis-Associated Mechanisms of Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans. Journal of Clinical Medicine. 2019; 8(9):1309. https://doi.org/10.3390/jcm8091309
Chicago/Turabian StyleGómez-Bañuelos, Eduardo, Amarshi Mukherjee, Erika Darrah, and Felipe Andrade. 2019. "Rheumatoid Arthritis-Associated Mechanisms of Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans" Journal of Clinical Medicine 8, no. 9: 1309. https://doi.org/10.3390/jcm8091309