Choice of Donor Source and Conditioning Regimen for Hematopoietic Stem Cell Transplantation in Sickle Cell Disease

Abstract
:1. Introduction:
2. Myeloablative HLA-Matched Sibling HSCT Is Efficacious for Children with SCD
3. Reduced Intensity Conditioning Is Associated with Decreased Toxicity and Low Graft Failure
4. HLA-Matched Sibling HSCT Using Nonmyeloablative Conditioning Is Efficacious and Spares Morbidity
5. Alternative Donor Sources Improve Transplant Accessibility
5.1. Umbilical Cord Transplants
5.1.1. Related Umbilical Cord Transplants
5.1.2. Unrelated Umbilical Cord Transplants
5.2. Matched Unrelated Donor Transplant
5.3. Haploidentical HSCT
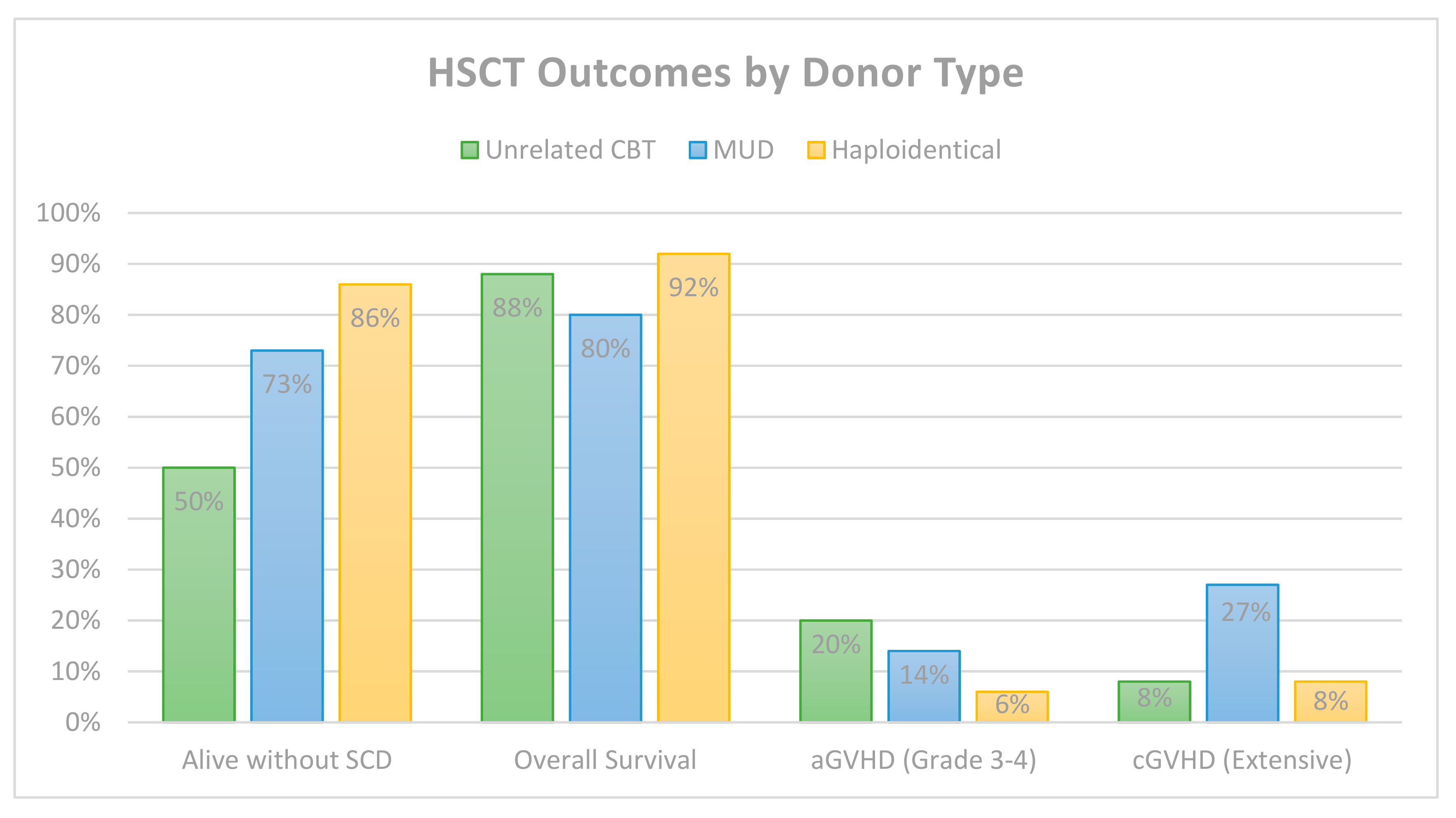
6. Comparative HSCT Outcomes by Donor Type
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Herrick, J. Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia. 1910. Yale J. Biol. Med. 2001, 74, 179–184. [Google Scholar]
- Platt, O.; Brambilla, D.; Rosse, W.; Milner, P.; Castro, O.; Steinberg, M.; Klug, P. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N. Engl. J. Med. 1994, 330, 1639–1644. [Google Scholar] [CrossRef]
- Fitzhugh, C.D.; Hsieh, M.M.; Allen, D.; Coles, W.A.; Seamon, C.; Ring, M.; Zhao, X.; Minniti, C.P.; Rodgers, G.P.; Schechter, A.N.; et al. Hydroxyurea-Increased Fetal Hemoglobin Is Associated with Less Organ Damage and Longer Survival in Adults with Sickle Cell Anemia. PLoS ONE 2015. [Google Scholar] [CrossRef]
- Walters, M.; Patience, M.; Leisenring, W.; Eckman, J.; Scott, J.; Mentzer, W. Bone Marrow Transplantation for Sickle Cell Anemia. N. Engl. J. Med. 1996, 335, 369–376. [Google Scholar] [CrossRef]
- Bernaudin, F.; Socie, G.; Kuentz, M.; Chevret, S.; Duval, M.; Bertrand, Y.; Vannier, J.P.; Yakouben, K.; Thuret, I.; Bordigoni, P.; et al. Long-term results of related myeloablative stem-cell transplantation to cure sickle cell disease. Blood 2007, 110, 2749–2756. [Google Scholar] [CrossRef]
- Gluckman, E.; Cappelli, B.; Bernaudin, F.; Labopin, M.; Volt, F.; Carreras, J. Sickle cell Disease: An international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood 2017, 129, 1548–1556. [Google Scholar] [CrossRef]
- Guilcher, G.M.T.; Truong, T.H.; Saraf, S.L.; Joseph, J.J.; Rondelli, D.; Hsieh, M.M. Curative therapies: Allogeneic hematopoietic cell transplantation from matched related donors using myeloablative, reduced intensity, and nonmyeloablative conditioning in sickle cell disease. Semin. Hematol. 2018, 55, 87–93. [Google Scholar] [CrossRef]
- Shenoy, S. Hematopoietic stem-cell transplantation for sickle cell disease: Current evidence and opinions. Ther. Adv. Hematol. 2013, 4, 335–344. [Google Scholar] [CrossRef]
- Vermylen, C.; Cornu, G.; Ferster, A.; Brichard, B.; Ninane, J.; Ferrant, A.; Zenebergh, A.; Maes, P.; Dhooge, C.; Benoit, Y.; et al. Haematopoietic stem cell transplantation for sickle cell anaemia: The first 50 patients transplanted in Belgium. Bone Marrow Transplant. 1998, 22, 1–6. [Google Scholar] [CrossRef]
- King, A.A.; Kamani, N.; Bunin, N.; Sahdev, I.; Brochstein, J.; Hayashi, R.J.; Grimley, M.; Abraham, A.; Dioguardi, J.; Chan, K.W.; et al. Successful matched sibling donor marrow transplantation following reduced intensity conditioning in children with hemoglobinopathies. Am. J. Hematol. 2015, 90, 1093–1098. [Google Scholar] [CrossRef]
- Krishnamurti, L.; Kharbanda, S.; Biernacki, M.A.; Zhang, W.; Baker, K.S.; Wagner, J.E.; Wu, C.J. Stable long-term donor engraftment following reduced-intensity hematopoietic cell transplantation for sickle cell disease. Biol. Blood Marrow Transplant. 2008, 14, 1270–1278. [Google Scholar] [CrossRef]
- Matthes-Martin, S.; Lawitschka, A.; Fritsch, G.; Lion, T.; Grimm, B.; Breuer, S.; Boztug, H.; Karlhuber, S.; Holter, W.; Peters, C.; et al. Stem cell transplantation after reduced-intensity conditioning for sickle cell disease. Eur. J. Haematol. 2013, 90, 308–312. [Google Scholar] [CrossRef]
- Bhatia, M.; Jin, Z.; Baker, C.; Geyer, M.B.; Radhakrishnan, K.; Morris, E.; Satwani, P.; George, D.; Garvin, J.; Del Toro, G.; et al. Reduced toxicity, myeloablative conditioning with BU, fludarabine, alemtuzumab and SCT from sibling donors in children with sickle cell disease. Bone Marrow Transplant. 2014, 49, 913–920. [Google Scholar] [CrossRef]
- Madden, L.M.; Hayashi, R.J.; Chan, K.W.; Pulsipher, M.A.; Douglas, D.; Hale, G.A.; Chaudhury, S.; Haut, P.; Kasow, K.A.; Gilman, A.L.; et al. Long-Term Follow-Up after Reduced-Intensity Conditioning and Stem Cell Transplantation for Childhood Nonmalignant Disorders. Biol. Blood Marrow Transplant. 2016, 22, 1467–1472. [Google Scholar] [CrossRef]
- Fitzhugh, C.; Cordes, S.; Taylor, T.; Coles, W.; Roskom, K.; Link, M. At least 20% donor myeloid chimerism is necessary to reverse the sickle phenotype after allogeneic HSCT. Blood 2017, 130, 1946–1948. [Google Scholar] [CrossRef]
- Powell, J.; Fitzhugh, C.; Kang, E.; Hsieh, M.; Schwartz, R.; Tisdale, J. Low-Dose Radiation Plus Rapamycin Promotes Long-Term Bone Marrow Chimerism. Transplantation 2005, 80, 1541–1545. [Google Scholar] [CrossRef]
- Hsieh, M.; Kang, E.; Fitzhugh, C.; Link, M.; Bolan, C.; Kurlander, R.; Childs, R.; Rodgers, G.; Powell, J.; Tisdale, J. Allogeneic Hematopoietic Stem-Cell Transplantation for Sickle Cell Disease. N. Engl. J. Med. 2009, 361, 2309–2317. [Google Scholar] [CrossRef]
- Hsieh, M.; Fitzhugh, C.; Weitzel, R.P.; Link, M.; Coles, W.; Zhao, X.; Rodgers, G.; Powell, J.; Tisdale, J. Nonmyeloablative HLA-Matched Sibling Allogeneic Hematopoietic Stem Cell Transplantation for Severe Sickle Cell Phenotype. J. Am. Med. Assoc. 2014, 312, 48–56. [Google Scholar] [CrossRef]
- Saraf, S.; Oh, A.; Patel, P.; Jalundhwala, Y.; Sweiss, K.; Koshy, M.; Campbell-Lee, S.; Gowhari, M.; Hassan, J.; Peace, D.; et al. Nonmyeloablative Stem Cell Transplantation with Alemtuzumab/Low-Dose Irradiation to Cure and Improve the Quality of Life of Adults with Sickle Cell Disease. Biol. Blood Marrow Transplant. 2016, 22, 441–448. [Google Scholar] [CrossRef]
- Alzahrani, M.; Damlaj, M.; Essa, M.; Alahmari, B.; Alaskar, A.; Hejazi, A.; Basher, E.; Abujoub, R.; Ghazi, S.; Abuelgasim, K.; et al. Outcome of Age-Adapted Approach to HLA-Identical Related Hematopoietic Stem Cell Transplantation in Severe Sickle Cell Disease: Saudi Experience. Blood 2018, 132, 3468. [Google Scholar] [CrossRef]
- Guilcher, G.M.T.; Monagel, D.A.; Nettel-Aguirre, A.; Truong, T.H.; Desai, S.J.; Bruce, A.; Shah, R.M.; Leaker, M.T.; Lewis, V.A. Nonmyeloablative Matched Sibling Donor Hematopoietic Cell Transplantation in Children and Adolescents with Sickle Cell Disease. Biol. Blood Marrow Transplant. 2019, 25, 1179–1186. [Google Scholar] [CrossRef]
- Cutler, C.; Lu, S.; Ho, V.; Koreth, J.; Alyea, E.; Soiffer, R.; Antin, J.H. Extended follow-up of methotrexate-free immunosuppression using sirolimus and tacrolimus in related and unrelated donor peripheral blood stem cell transplantation. Transplantation 2007, 109, 3108–3114. [Google Scholar] [CrossRef]
- Walters, M.C.; Patience, M.; Leisenring, W.; Eckman, J.R.; Buchanan, G.R.; Rogers, Z.R.; Olivieri, N.E.; Vichinsky, E.; Davies, S.C.; Mentzer, W.C.; et al. Barriers to bone marrow transplantation for sickle cell anemia. Biol. Blood Marrow Transplant. 1996, 2, 100–104. [Google Scholar]
- Hansbury, E.N.; Schultz, W.H.; Ware, R.E.; Aygun, B. Bone marrow transplant options and preferences in a sickle cell anemia cohort on chronic transfusions. Pediatr. Blood Cancer 2012, 58, 611–615. [Google Scholar] [CrossRef]
- Brichard, B.; Vermylen, C.; Ninane, J.; Cornu, G. Persistence of fetal hemoglobin production after successful transplantation of cord blood stm cells in a patient with sickle cell anemia. J. Pediatr. 1996, 128, 241–243. [Google Scholar] [CrossRef]
- Miniero, R.; Rocha, V.; Saracco, P.; Locatelli, F.; Brichard, B.; Nagler, A.; Roberts, I.; Yaniv, I.; Beksac, M.; Bernaudin, F.; et al. Cord blood transplantation (CBT) in hemoglobinopathies. Eurocord. Bone Marrow Transplant. 1998, 22, S78–S79. [Google Scholar]
- Gore, L.; Lane, P.; Quinones, R.; Giller, R. Successful cord blood transplantation for sickle cell anemia from a sibling who is human leukocyte antigen-identical; implications for comprehensive care. J. Pediatr. Hematol. Oncol. 2000, 22, 437–440. [Google Scholar] [CrossRef]
- Walters, M.; Quirolo, L.; Trachtenberg, E.; Edwards, S.; Hale, L.; Lee, J.; Morton-Wiley, J.; Quirolo, K.; Robertson, S.; Saba, J.; et al. Sibling Donor Cord Blood Transplantation for Thalassemia Major: Experience of the Sibling Donor Cord Blood Program. Ann. N. Y. Acad. Sci. 2005, 1054, 206–213. [Google Scholar] [CrossRef]
- Locatelli, F.; Kabbara, N.; Ruggeri, A.; Ghavamzadeh, A.; Roberts, I.; Li, C.; Bernaudin, F.; Vermylen, C.; Dalle, J.; Stein, J.; et al. Outcome of patients with hemoglobinopathies given either cord blood or bone marrow transplantation from an HLA-identical sibling. Blood 2013, 122, 1072–1078. [Google Scholar] [CrossRef]
- Soni, S.; Boulad, F.; Cowan, M.; Scaradavou, A.; Dahake, J.; Edwards, S.; Walters, M. Combined umbilical cord blood and bone marrow from HLA-identical sibling donors for hematopoietic stem cell transplantation in children with hemoglobinopathies. Pediatr. Blood Cancer 2014, 61, 1690–1694. [Google Scholar] [CrossRef]
- Bradley, M.B.; Cairo, M.S. Cord blood immunology and stem cell transplantation. Hum. Immunol. 2005, 66, 431–446. [Google Scholar] [CrossRef]
- Bauer, D.E.; Brendel, C.; Fitzhugh, C.D. Curative approaches for sickle cell disease: A review of allogeneic and autologous strategies. Blood Cells Mol. Dis. 2017, 67, 155–168. [Google Scholar] [CrossRef]
- Kamani, N.; Walters, M.; Carter, S.; Aquino, V.; Brochstein, J.A.; Chaudhury, S.; Eapen, M.; Freed, B.; Grimley, M.; Levine, J.; et al. Unrelated donor cord blood transplantation for children with severe sickle cell disease: Results of one cohort from the phase II study from the Blood and Marrow Transplant Clinical Trials Network (BMT CTN). Biol. Blood Marrow Transplant. 2012, 18, 1265–1272. [Google Scholar] [CrossRef]
- Ruggeri, A.; Eapen, M.; Scaradavou, A.; Cairo, M.; Bhatia, M.; Kurtzberg, J.; Wingard, J.; Fasth, A.; Lo Nigro, L.; Ayas, M.; et al. Umbilical cord blood transplantation for children with thalassemia and sickle cell disease. Biol. Marrow Transplant. 2011, 17, 1375–1382. [Google Scholar] [CrossRef] [Green Version]
- Abraham, A.; Cluster, A.; Jacobsohn, D.; Delgado, D.; Hulbert, M.; Kukadiya, D.; Murray, L.; Shenoy, S. Unrelated Umbilical Cord Blood Transplantation for Sickle Cell Disease Following Reduced-Intensity Conditioning: Results of a Phase I Trial. Biol. Blood Marrow Transplantat. 2017, 23, 1587–1592. [Google Scholar] [CrossRef] [Green Version]
- Adamkiewicz, T.; Szabolcs, P.; Haight, A.; Baker, K.; Staba, S.; Kedar, A.; Chiang, K.; Krishnamurti, L.; Boyer, M.; Kurtzberg, J.; et al. Unrelated cord blood transplantation in children with sickle cell disease: Review of four-center experience. Pediatr. Transplant. 2007, 11, 641–644. [Google Scholar] [CrossRef]
- Radhakrishnan, K.; Bhatia, M.; Geyer, M.; Del Toro, G.; Jin, Z.; Baker, C.; Harrison, L.; Morris, E.; Baxter-Lowe, L.; Cairo, M. Busulfan, fludarabine, and alemtuzumab conditioning and unrelated cord blood transplantation in children with sickle cell disease. Biol. Blood Marrow Transplantat. 2013, 19, 676–677. [Google Scholar] [CrossRef] [Green Version]
- Kharbanda, S.; Smith, A.; Hutchinson, S.; McKenna, D.; Ball, J.; Lamb, L.J.; Agarwal, R.; KI, W.; Wagner, J.J. Unrelated donor allogeneic hematopoieic stem cell transplantation in pateints with hemoglobinopathies using a reduced-intensity conditioning regimen and third-party mesenchymal stromal cells. Biol. Blood Marrow Transplant. 2014, 20, 581–586. [Google Scholar] [CrossRef] [Green Version]
- Gragert, L.; Eapen, M.; Williams, E.; Freeman, J.; Spellman, S.; Baitty, R.; Hartzman, R.; Rizzo, J.D.; Horowitz, M.; Confer, D.; et al. HLA match likelihoods for hematopoietic stem-cell grafts in the U.S. registry. N. Engl. J. Med. 2014, 371, 339–348. [Google Scholar] [CrossRef] [Green Version]
- Shenoy, S.; Eapen, M.; Panepinto, J.; Logan, B.; Wu, J.; Abraham, A.; Brochstein, J.A.; Chaudhury, S.; Godder, K.; Haight, A.; et al. A trial of unrelated donor marrow tranplantation for children with severe sickle cell disease. Blood 2016, 128, 2561–2567. [Google Scholar] [CrossRef]
- Krishnamurti, L.; Neuberg, D.; Sullivan, K.; Kamani, N.; Abraham, A.; Campigotto, F.; Zhang, W.; Dahdoul, T.; De Castro, L.; Parikh, S.; et al. Bone marrow transplantation for adolescents and young adults with sickle cell disease: Results of a prospective multicenter pilot study. Am. J. Hematol. 2019, 94, 446–454. [Google Scholar] [CrossRef] [Green Version]
- Strocchio, L.; Zecca, M.; Comoli, P.; Mina, T.; Giorgiani, G.; Giraldi, E.; Vinti, L.; Merli, P.; Regazzi, M.; Locatelli, F. Treosulfan-based conditioning regimen for allogeneic haematopoietic stem cell transplantation in children with sickle cell disease. Br. J. Haematol. 2015, 169, 726–736. [Google Scholar] [CrossRef]
- Marzollo, A.; Calore, E.; Tumino, M.; Pillon, M.; Gazzola, M.; Destro, R.; Colombatti, R.; Marson, P.; Tison, T.; Colpo, A.; et al. Treosulfan-based Conditioning Regimen in Sibling and Alternative Donor Hematopoietic Stem Cell Transplantation for Children with Sickle Cell Disease. Mediterr. J. Hematol. Infect. Dis. 2017. [Google Scholar] [CrossRef] [Green Version]
- Gilman, A.; Eckrich, M.; Epstein, S.; Barnhart, C.; Cannon, M.; Fukes, T.; Hyland, M.; Shah, K.; Grochowski, D.; Champion, E.; et al. Alternative donor hematopoietic stem cell tranplantation for sickle cell disease. Blood Adv. 2017, 1, 1215–1223. [Google Scholar] [CrossRef] [Green Version]
- Dallas, M.H.; Triplett, B.; Shook, D.R.; Hartford, C.; Srinivasan, A.; Laver, J.; Ware, R.; Leung, W. Long-term outcome and evaluation of organ function in pediatric patients undergoing haploidentical and matched related hematopoietic cell transplantation for sickle cell disease. Biol. Blood Marrow Transplant. 2013, 19, 820–830. [Google Scholar] [CrossRef] [Green Version]
- Luznik, L.; Jalla, S.; Engstrom, L.W.; Iannone, R.; Fuchs, E.J. Durable engraftment of major histocompatibility complex-incompatible cells after nonmyeloablative conditioning with fludarabine, low-dose total body irradiation, and posttransplantation cyclophosphamide. Blood 2001, 98, 3456–3464. [Google Scholar] [CrossRef] [Green Version]
- Kanakry, C.; Fuchs, E.; Luznik, L. Modern approaches to HLA-haploidentical blood or marrow transplantation. Nat. Rev. Clin. Oncol. 2016, 13, 10–24. [Google Scholar] [CrossRef] [Green Version]
- Luznik, L.; O’Donnell, P.V.; Fuchs, E.J. Post-transplantation cyclophosphamide for tolerance induction in HLA-haploidentical bone marrow transplantation. Semin. Oncol. 2012, 39, 683–693. [Google Scholar] [CrossRef] [Green Version]
- Klein, O.; Buddenbaum, J.; Tucker, N.; Chen, A.; CJ, G.; Loeb, D.; Zambidis, E.; Llosa, N.; Huo, J.; Robey, N.; et al. Nonmyeloablative Haploidentical Bone Marrow Transplantation with Post-Transplantation Cyclophosphamide for Pediatric and Young Adult Patients with High-Risk Hematologic Malignancies. Biol. Blood Marrow Transplantat. 2017, 23, 325–332. [Google Scholar] [CrossRef] [Green Version]
- Kanakry, C.G.; Ganguly, S.; Zahurak, M.; Bolanos-Meade, J.; Thoburn, C.; Perkins, B.; Fuchs, E.J.; Jones, R.J.; Hess, A.D.; Luznik, L. Aldehyde dehydrogenase expression drives human regulatory T cell resistance to posttransplantation cyclophosphamide. Sci. Transl. Med. 2013. [Google Scholar] [CrossRef] [Green Version]
- Wachsmuth, L.P.; Patterson, M.T.; Eckhaus, M.A.; Venzon, D.J.; Gress, R.E.; Kanakry, C.G. Post-transplantation cyclophosphamide prevents graft-versus-host disease by inducing alloreactive T cell dysfunction and suppression. J. Clin. Invest. 2019, 129, 2357–2373. [Google Scholar] [CrossRef]
- Bolanos-Meade, J.; Fuchs, E.J.; Luznik, L.; Lanzkron, S.M.; Gamper, C.J.; Jones, R.J.; Brodsky, R.A. HLA-haploidentical bone marrow transplantation with posttransplant cyclophosphamide expands the donor pool for patients with sickle cell disease. Blood 2012, 120, 4285–4291. [Google Scholar] [CrossRef] [Green Version]
- Fitzhugh, C.D.; Hsieh, M.M.; Taylor, T.; Coles, W.; Roskom, K.; Wilson, D.; Wright, E.; Jeffries, N.; Gamper, C.J.; Powell, J.; et al. Cyclophosphamide improves engraftment in patients with SCD and severe organ damage who undergo haploidentical PBSCT. Blood Adv. 2017, 1, 652–661. [Google Scholar] [CrossRef]
- Foell, J.; Pfirstinger, B.; Rehe, K.; Wolff, D.; Holler, E.; Corbacioglu, S. Haploidentical stem cell transplantation with CD3(+)-/CD19(+)- depleted peripheral stem cells for patients with advanced stage sickle cell disease and no alternative donor: Results of a pilot study. Bone Marrow Transplant. 2017, 52, 938–940. [Google Scholar] [CrossRef]
- Wiebking, V.; Hutker, S.; Schmid, I.; Immler, S.; Feuchtinger, T.; Albert, M.H. Reduced toxicity, myeloablative HLA-haploidentical hematopoietic stem cell transplantation with post-transplantation cyclophosphamide for sickle cell disease. Ann. Hematol. 2017, 96, 1373–1377. [Google Scholar] [CrossRef]
- Frangoul, H.; Evans, M.; Isbell, J.; Bruce, K.; Domm, J. Haploidentical hematopoietic stem cell transplant for patients with sickle cell disease using thiotepa, fludarabine, thymoglobulin, low dose cyclophosphamide, 200 cGy tbi and post transplant cyclophosphamide. Bone Marrow Transplant. 2018, 53, 647–650. [Google Scholar] [CrossRef]
- Saraf, S.L.; Oh, A.L.; Patel, P.R.; Sweiss, K.; Koshy, M.; Campbell-Lee, S.; Gowhari, M.; Jain, S.; Peace, D.; Quigley, J.G.; et al. Haploidentical Peripheral Blood Stem Cell Transplantation Demonstrates Stable Engraftment in Adults with Sickle Cell Disease. Biol. Blood Marrow Transplant. 2018, 24, 1759–1765. [Google Scholar] [CrossRef] [Green Version]
- Pawlowska, A.B.; Cheng, J.C.; Karras, N.A.; Sun, W.; Wang, L.D.; Bell, A.D.; Gutierrez, L.; Rosenthal, J. HLA Haploidentical Stem Cell Transplant with Pretransplant Immunosuppression for Patients with Sickle Cell Disease. Biol. Blood Marrow Transplant. 2018, 24, 185–189. [Google Scholar] [CrossRef] [Green Version]
- Gaziev, J.; Isgro, A.; Sodani, P.; Paciaroni, K.; De Angelis, G.; Marziali, M.; Ribersani, M.; Alfieri, C.; Lanti, A.; Galluccio, T.; et al. Haploidentical HSCT for hemoglobinopathies: Improved outcomes with TCRalphabeta(+)/CD19(+)-depleted grafts. Blood Adv. 2018, 2, 263–270. [Google Scholar] [CrossRef] [Green Version]
- De la Fuente, J.; Dhedin, N.; Koyama, T.; Bernaudin, F.; Kuentz, M.; Karnik, L.; Socie, G.; Culos, K.A.; Brodsky, R.A.; DeBaun, M.R.; et al. Haploidentical Bone Marrow Transplantation with Post-Transplantation Cyclophosphamide Plus Thiotepa Improves Donor Engraftment in Patients with Sickle Cell Anemia: Results of an International Learning Collaborative. Biol. Blood Marrow Transplant. 2019, 25, 1197–1209. [Google Scholar] [CrossRef]
- Bolanos-Meade, J.; Cooke, K.R.; Gamper, C.J.; Ali, S.A.; Ambinder, R.F.; Borrello, I.M.; Fuchs, E.J.; Gladstone, D.E.; Gocke, C.B.; Huff, C.A.; et al. Effect of increased dose of total body irradiation on graft failure associated with HLA-haploidentical transplantation in patients with severe haemoglobinopathies: A prospective clinical trial. Lancet Haematol. 2019, 6, e183–e193. [Google Scholar] [CrossRef]
- Foell, J.; Schulte, J.H.; Pfirstinger, B.; Troeger, A.; Wolff, D.; Edinger, M.; Hofmann, P.; Aslanidis, C.; Lang, P.; Holler, E.; et al. Haploidentical CD3 or alpha/beta T-cell depleted HSCT in advanced stage sickle cell disease. Bone Marrow Transplant. 2019. [Google Scholar] [CrossRef]
- Jaiswal, S.; Bhakuni, P.; Zaman, S.; Bansal, S.; Bharadwaj, P.; Bhargava, S.; Chakrabarti, S. T cell costimulation blockade promotes transplantation tolerance in combination with sirolimus and post-transplantation cyclophosphamide for haploidentical transplantation in children with severe aplastic anemia. Transplant. Immunol. 2017, 43, 54–59. [Google Scholar] [CrossRef]
- Fitzhugh, C.D.; Weitzel, R.P.; Hsieh, M.M.; Phang, O.A.; Madison, C.; Luznik, L.; Powell, J.D.; Tisdale, J.F. Sirolimus and post transplant Cy synergistically maintain mixed chimerism in a mismatched murine model. Bone Marrow Transplant. 2013, 48, 1335–1341. [Google Scholar] [CrossRef] [Green Version]
- Joseph, J.J.; Abraham, A.A.; Fitzhugh, C.D. When there is no match, the game is not over: Alternative donor options for hematopoietic stem cell transplantation in sickle cell disease. Semin. Hematol. 2018, 55, 94–101. [Google Scholar] [CrossRef]
- Eapen, M.; Brazauskas, R.; Walters, M.C.; Bernaudin, F.; Bo-Subait, K.; Fitzhugh, C.D.; Hankins, J.S.; Kanter, J.; Meerpohl, J.J.; Bolanos-Meade, J.; et al. Effect of donor type and conditioning regimen intensity on allogeneic transplantation outcomes in patients with sickle cell disease: A retrospective multicentre, cohort study. Lancet Haematol. 2019. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Reference | Transplant Regimen | Number of Patients (Median Age) | Alive without SCD (%) | Median Time to Neutrophil/Platelet Recovery (Days) | Grade 3–4 Acute GvHD (% of Total) | Extensive Chronic (% of Total) | Mortality (Cause) |
|---|---|---|---|---|---|---|---|
| Brichard, 1996 [25] | Bu 16 mg/kg, Cy 200 mg/kg, ATG, CSA | 1 (5) | 1 (100%) | 32/48 | 0 | 0 | 0 |
| Miniero, 1998 [26] | Bu 16 mg/kg, Cy 200 mg/kg, CSA ± MTX | 3 (3–11) | 2 (67%) | 31/40 | 0 | 0 | 0 |
| Gore, 2000 [27] | Bu 726 mg/m2, Cy 200 mg/kg, ATG, CSA | 1 (9) | 1 (100%) | 23/49 | 0 | 0 | 0 |
| Walters, 2005 † [28] | NR | 8 (NR) | 6 (75%) | 23/45 | NR | NR | 1 (intractable seizures) |
| Matthes-Martin, 2013 [12] | TBI (2 Gy), Flu 160 mg/m2, Mel 140 mg/m2, Alem 1mg/kg, CSA, MMF | 1 (11.1) | 1 (100%) | 19/NR | 0 | 0 | 0 |
| Locatelli, 2013 [29] | Bu ± Flu ± Cy ± ATG/ALG ± TT, CSA ± MTX | 30 (5.9) | 27 (90%) | 23/38 | 11 (11%) (Grade 2–3 only) * | 0 | 3 (2 hemorrhage, 1 organ failure) * |
| Soni, 2014 [30] | Bu 14–16 mg/kg, Cy 200 mg/kg, ATG, CSA ± MMF, ± MTX, ± Pred | 22 (5.2) * | 19* | 25/48 | 1* | 0 | 3 (infection, pulmonary complications, seizure) * |
| Total | -- | 66 | 57 (86%) | 25/45 | 12 (18%) * | 0 | 7 (11%) * |
| Reference | Transplant Regimen | HLA Match (n) | Number of Patients (Median Age) | Alive without SCD (%) | Median Time to Neutrophil/Platelet Recovery (Days) | Grade 3–4 Acute GvHD (% of Total) | Extensive Chronic GvHD (% of Total) | Mortality (Cause) |
|---|---|---|---|---|---|---|---|---|
| Adamkiewicz, 2007 [36] | Mixed, 4 pts myeloablative, 3 pts reduced intensity | 5/6 (2) 4/6 (5) | 7 (2.4) | 3 (43%) | 22/70 | 2 (28%) | 1 (14%) | 1 (multi-organ failure) |
| Ruggeri, 2011 [34] | Mixed, 9 pts myeloablative, 7 pts reduced intensity | 6/6 (2) 5/6 (4) 4/6 (10) | 16 (6) | 8 (50%) | 22/40 | 3 (19%) * | 2 (12%) * | 1 (aGvHD) |
| Kamani, 2012 [33] | Alem 48 mg, Flu 150 mg/m2, Mel 140 mg/m2, CSA or Tac + MMF | 6/6 (1) 5/6 (7) | 8 (13.7) | 3 (38%) | 22/41 | 0 | 1 (12%) | 1 (cGvHD) |
| Radhakrishnan, 2013 [37] | Bu 12.8–16 mg/kg, Flu 180 mg/m2, Alem 54 mg, MMF, Tac | NR | 8 (3.6) | 4 (50%) | 34/54 | 2 (25%) | 0 | 3 (infection) |
| Kharbanda, 2014 [38] | Flu 150 mg/m2, Mel 140 mg/m2, Alem 60 mg, CSA, MMF | 4/6 (2) | 2 (8) | 0 | 24/46 | 0 | 0 | 0 |
| Abraham, 2017 [35] | Alem, Flu 30 mg/m2, TT 4 mg/kg, Mel 140 mg/m2, CSA or Tac | 6/6 (2) 5/6 (7) | 9 (4) | 7 (78%) | 21/52 | 3 (43%) | 0 | 0 |
| Total | -- | 6/6 (5) 5/6 (20) 4/6 (17) | 50 | 25 (50%) | 24/50 | 10 (20%) | 4 (8%) | 6 (12%) |
| Reference | Transplant Regimen | Graft Type | Number of Patients (Median Age) | Alive without SCD (%) | Median Time to Neutrophil/Platelet Recovery (Days) | Grade 3–4 Acute GvHD (% of Total) | Extensive Chronic GvHD (% of Total) | Mortality (Cause) |
|---|---|---|---|---|---|---|---|---|
| Strocchio, 2015 [42] | TT 10 mg/kg, Treo 42 g/m2, Flu 160 mg/m2, ATG 15–30 mg/kg, CSA, MTX | BM (5) PBSCs (1) | 6 (8.4) | 5 (83%) | 21/26 | 0 | 0 | 0 |
| Shenoy, 2016 [40] | Alem 45 mg, Flu 150 mg/m2, Mel 140 mg/m2, CSA or Tac, MTX, Methylpred | BM | 29 (14) | 20 (69%) | 12/24 | 5 (17%) | 11 (38%) | 7 (GVHD) 1 (following 2nd transplant) |
| Marzollo, 2017 [43] | TT 8–10 mg/kg, Treo 42 g/m2, Flu 160 mg/m2, ATG 60 mg/kg, ± MTX, CSA | BM or T-cell depleted PBSCs * | 2 (8.5) | 2 (100%) | 20/22 | 0 | 0 | 0 |
| Gilman, 2017 [44] | Mel 140 mg/m2, TT 10mg/kg, Flu 200 mg/m2, ATG 10 mg/kg ± Ritux 150-375 mg/m2, MTX | PBSCs | 2 (9) | 2 (100%) | 14/19 | 0 | 0 | 0 |
| Krishnamur-ti, 2019 [41] | Bu 13.2 mg/kg, Flu 175 mg/m2, ATG, CSA or Tac, MTX | BM | 5 (22.5) | 3 (60%) | 17/21 | 1 (20%) | 1 (20%) | 1 (cGvHD) |
| Total | Mixed | BM or PBSCs | 44 | 32 (73%) | 17/22 | 6 (14%) | 12 (27%) | 9 (20%) |
| Reference | Transplant Regimen | Graft Type | Number of Patients (Median Age) | Alive without SCD (%) | Median Time to Neutrophil/Platelet Recovery (Days) | Grade 3–4 Acute GvHD (% of Total) | Extensive Chronic GvHD (% of Total) | Mortality (Cause) |
|---|---|---|---|---|---|---|---|---|
| Fitzhugh, 2017 [53] | Alem 1mg/kg, 400 cGy TBI, PT-Cy 100 mg/kg, Sir | PBSC | 12 (36) | 6 (50%) | 27/31 | 0 | 0 | 1 (PH and CHF) |
| Foell, 2017 [54] | TT 10 mg/kg, Flu 160 mg/m2, Treo 42 g/m2, ATG 45 mg/kg, CSA, MMF | CD3/ CD19 depleted PBSC | 9 (16) | 8 (89%) | 18/11 | 0 | 1 (moderate) (12.5%) | 1 (CMV) |
| Marzollo, 2017 [43] | TT 8–10 mg/kg, Treo 42 g/m2, Flu 160 mg/m2, ATG 20 mg/kg ± Ritux 200 mg/m2 | TCRab/ CD19 depleted PBSC | 2 (15) | 2 (100%) | 20/22 | 0 | 0 | 0 |
| Wiebking, 2017 [55] | Alem 0.4 mg/kg, Flu 150 mg/m2, Treo 42 g/m2, TT 10 mg/kg, PT-Cy 100 mg/kg, MMF, Tac | BM | 3 (8) | 3 (100%) | 17/25 | 0 | 0 | 0 |
| Gilman, 2017 [44] | Mel 140 mg/m2, TT 10 mg/kg, Flu 200 mg/m2, ATG 10 mg/kg ± Ritux 375 mg/m2, MTX | PBSC | 8 (16) | 7 (88%) | 14/19 | 1 (12.5%) | 1 (12.5%) | 1 (GvHD) |
| Frangoul, 2018 [56] | Cy 29 mg/kg, Flu 150 mg/m2, 200 cGy TBI, ATG 4.5 mg/kg, TT 10 mg/kg, PT-Cy 100 mg/kg, Sir + MMF | Primed BM or PBSC | 4 (14.3) | 4 (100%) | 22/26 | 0 | 0 | 0 |
| Saraf, 2018 [57] | Cy 29 mg/kg, Flu 150 mg/m2, 300 cGy TBI, ATG 4.5 mg/kg, PT-Cy 100 mg/kg, Tac or Sir + MMF | PBSC | 8 (28) | 6 (75%) | 22/NR | 1 (12.5%) | 1 (moderate) (12.5%) | 1 (unknown) |
| Pawlowska, 2018 [58] | Flu/Dex Pre-Conditioning, ATG 4.5 mg/kg, Flu 210 mg/m2, Bu 520 mg/m2, PT-Cy 100 mg/kg, Tac, MMF | BM or PBSC | 4 (19) | 4 (100%) | 16/19 | 0 | 0 | 0 |
| Gaziev, 2018 [59] | HU/Aza/Flu Pre-Conditioning, Bu 14 mg/kg, TT 10 mg/kg, Cy 200 mg/kg, ATG 12.5 mg/kg, CSA, MMF or methylpred | TCRαβ/CD19 depleted PBSCs | 3 (7) | 3 (100%) | 13/15 | 1 (33%) * | 3 (21%) * | 2 * |
| de la Fuente, 2019 [60] | Cy 29 mg/kg, Flu 150 mg/m2, 200 cGy TBI, ATG 4.5 mg/kg, TT 10 mg/kg PT-Cy 100 mg/kg, Sir + MMF | Primed BM | 15 (20.4) | 14 (93%) | 22/28 | 2 (13%) | 1 (moderate) (6.7%) | 0 |
| Bolaños-Meade, 2019 [61] | ATG, Flu 30 mg/m2, Cy 14.5 mg/kg, TBI 400cGy, PT-Cy 100 mg/kg, MMF, Sir | BM | 12 (16) | 11 (92%) | 28/26 | 1 (8.3%) * | 1 (moderate) (8.3%) * | 0 |
| Foell, 2019 [62] | TT 10 mg/kg, Flu 160 mg/m2, Treo 42 g/m2, ATG 45 mg/kg, CSA, MMF | CD3/CD19- or TCRαβ/CD19-depleted PBSC | 20 (14.5) | 18 (90%) | 19/10 | 0 | 0 | 2 (CMV, MAS) |
| Total | Mixed | Mixed | 100 | 86 (86%) | 20/31 | 6 (6%) | 8 (8%) | 8 (8%) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Limerick, E.; Fitzhugh, C. Choice of Donor Source and Conditioning Regimen for Hematopoietic Stem Cell Transplantation in Sickle Cell Disease. J. Clin. Med. 2019, 8, 1997. https://doi.org/10.3390/jcm8111997
Limerick E, Fitzhugh C. Choice of Donor Source and Conditioning Regimen for Hematopoietic Stem Cell Transplantation in Sickle Cell Disease. Journal of Clinical Medicine. 2019; 8(11):1997. https://doi.org/10.3390/jcm8111997
Chicago/Turabian StyleLimerick, Emily, and Courtney Fitzhugh. 2019. "Choice of Donor Source and Conditioning Regimen for Hematopoietic Stem Cell Transplantation in Sickle Cell Disease" Journal of Clinical Medicine 8, no. 11: 1997. https://doi.org/10.3390/jcm8111997