Rethinking Lupus Nephritis Classification on a Molecular Level

 , , ,
, , ,
Abstract
:1. Introduction
2. Experimental Section
2.1. Materials and Methods
Experimental Design and Sample Collection
2.2. Sample Handling and RNA Isolation
2.3. Microarray Processing and Statistical Analysis—Discovery Toronto Cohort
2.4. NanoString Statistical Analysis (Longitudinal Cohort)
sample/mean abundance of positive controls in sample i
2.5. Clinical Outcome
2.6. Pathway Analysis
3. Results
3.1. The Molecular Profiles of LN Kidney Biopsies
3.2. Correlates of Glomerular Inflammation
3.3. Correlates of Tubulointerstitial Fibrosis and Glomerulosclerosis
3.4. Correlates of Clinical Variables
3.5. Evolution of Transcript Abundance after Treatment
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lerang, K.; Gilboe, I.-M.; Thelle, D.S.; Gran, J.T. Mortality and years of potential life loss in systemic lupus erythematosus: A population-based cohort study. Lupus 2014, 23, 1546–1552. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.E.; Korbet, S.M.; Katz, R.S.; Schwartz, M.M.; Lewis, E.J.; for the Collaborative Study Group. Value of a Complete or Partial Remission in Severe Lupus Nephritis. Clin. J. Am. Soc. Nephrol. 2008, 3, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Weening, J.J.; D’Agati, V.D.; Schwartz, M.M.; Seshan, S.V.; Alpers, C.E.; Appel, G.B.; Balow, J.E.; Bruijn, J.A.; Cook, T.; Ferrario, F.; et al. The classification of glomerulonephritis in systemic lupus erythematosus revisited. Kidney Int. 2004, 65, 521–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morel-Maroger, L.; Méry, J.P.; Droz, D.; Godin, M.; Verroust, P.; Kourilsky, O.; Richet, G. The course of lupus nephritis: Contribution of serial renal biopsies. Adv. Nephrol. Necker Hosp. 1976, 6, 79–118. [Google Scholar] [PubMed]
- Tektonidou, M.G.; Dasgupta, A.; Ward, M.M. Risk of End-Stage Renal Disease in Patients with Lupus Nephritis, 1971–2015: A Systematic Review and Bayesian Meta-Analysis. Arthritis Rheumatol. 2016, 68, 1432–1441. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, G.; D’Agati, V. The ISN/RPS 2003 classification of lupus nephritis: An assessment at 3 years. Kidney Int. 2007, 71, 491–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banfi, G.; Bertani, T.; Boeri, V.; Faraggiana, T.; Mazzucco, G.; Monga, G.; Sacchi, G. Renal Vascular Lesions as a Marker of Poor Prognosis in Patients with Lupus Nephritis. Am. J. Kidney Dis. 1991, 18, 240–248. [Google Scholar] [CrossRef]
- Austin, H.A., 3rd; Muenz, L.R.; Joyce, K.M.; Antonovych, T.A.; Kullick, M.E.; Klippel, J.H.; Decker, J.L.; Balow, J.E. Prognostic factors in lupus nephritis. Contribution of renal histologic data. Am. J. Med. 1983, 75, 382–391. [Google Scholar] [CrossRef]
- Schwartz, M.M.; Lan, S.-P.; Bernstein, J.; Hill, G.S.; Holley, K.; Lewis, E.J. Irreproducibility of the Activity and Chronicity Indices Limits Their Utility in the Management of Lupus Nephritis. Am. J. Kidney Dis. 1993, 21, 374–377. [Google Scholar] [CrossRef]
- Bajema, I.M.; Wilhelmus, S.; Alpers, C.E.; Bruijn, J.A.; Colvin, R.B.; Cook, H.T.; D’Agati, V.D.; Ferrario, F.; Haas, M.; Jennette, J.C.; et al. Revision of the International Society of Nephrology/Renal Pathology Society classification for lupus nephritis: Clarification of definitions, and modified National Institutes of Health activity and chronicity indices. Kidney Int. 2018, 93, 789–796. [Google Scholar] [CrossRef]
- Peterson, K.S.; Huang, J.-F.; Zhu, J.; D’Agati, V.; Liu, X.; Miller, N.; Erlander, M.G.; Jackson, M.R.; Winchester, R.J. Characterization of heterogeneity in the molecular pathogenesis of lupus nephritis from transcriptional profiles of laser-captured glomeruli. J. Clin. Investig. 2004, 113, 1722–1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, M.; Loupy, A.; Lefaucheur, C.; Roufosse, C.; Glotz, D.; Seron, D.; Nankivell, B.J.; Halloran, P.F.; Colvin, R.B.; Akalin, E.; et al. The Banff 2017 Kidney Meeting Report: Revised diagnostic criteria for chronic active T cell–mediated rejection, antibody-mediated rejection, and prospects for integrative endpoints for next-generation clinical trials. Am. J. Transplant. 2018, 18, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Anders, H.-J.; Rovin, B. A pathophysiology-based approach to the diagnosis and treatment of lupus nephritis. Kidney Int. 2016, 90, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Birmingham, D.J.; Merchant, M.; Waikar, S.S.; Nagaraja, H.; Klein, J.B.; Rovin, B.H. Biomarkers of lupus nephritis histology and flare: Deciphering the relevant amidst the noise. Nephrol. Dial. Transplant. 2017, 32, i71–i79. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.V.; Malvar, A.; Song, H.; Alberton, V.; Lococo, B.; Vance, J.; Zhang, J.; Yu, L.; Rovin, B.H. Characterising the immune profile of the kidney biopsy at lupus nephritis flare differentiates early treatment responders from non-responders. Lupus Sci. Med. 2015, 2, e000112. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.V.; Malvar, A.; Song, H.; Alberton, V.; Lococo, B.; Vance, J.; Zhang, J.; Yu, L.; Birmingham, D.; Rovin, B.H. Molecular imaging of the kidney in lupus nephritis to characterize response to treatment. Transl. Res. 2017, 182, 1–13. [Google Scholar] [CrossRef]
- Austin, H.A., 3rd; Muenz, L.R.; Joyce, K.M.; Antonovych, T.T.; Balow, J.E. Diffuse proliferative lupus nephritis: Identification of specific pathologic features affecting renal outcome. Kidney Int. 1984, 25, 689–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, C.D.; Frach, K.; Schlöndorff, D.; Kretzler, M. Quantitative gene expression analysis in renal biopsies: A novel protocol for a high-throughput multicenter application. Kidney Int. 2002, 61, 133–140. [Google Scholar] [CrossRef] [Green Version]
- Schmid, H.; Henger, A.; Kretzler, M. Molecular approaches to chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2006, 15, 123–129. [Google Scholar] [CrossRef]
- Gautier, L.; Cope, L.; Bolstad, B.M.; Irizarry, R.A. Affy—Analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 2004, 20, 307–315. [Google Scholar] [CrossRef]
- Dai, M.; Wang, P.; Boyd, A.D.; Kostov, G.; Athey, B.; Jones, E.G.; Bunney, W.E.; Myers, R.M.; Speed, T.P.; Akil, H.; et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005, 33, e175. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef] [Green Version]
- P’Ng, C.; Green, J.; Chong, L.C.; Waggott, D.; Prokopec, S.D.; Shamsi, M.; Nguyen, F.; Mak, D.Y.F.; Lam, F.; Albuquerque, M.A.; et al. BPG: Seamless, automated and interactive visualization of scientific data. BMC Bioinformatics 2019, 20, 42. [Google Scholar] [CrossRef]
- Bolstad, B. preprocess Core: A collection of pre-processing functions. R package version 1.44.0. 2018. Available online: https://rdrr.io/bioc/preprocessCore/ (accessed on 23 September 2019).
- Geiss, G.K.; Bumgarner, R.E.; Birditt, B.; Dahl, T.; Dowidar, N.; Dunaway, D.L.; Fell, H.P.; Ferree, S.; George, R.D.; Grogan, T.; et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat. Biotechnol. 2008, 26, 317–325. [Google Scholar] [CrossRef]
- Rusinova, I.; Forster, S.; Yu, S.; Kannan, A.; Masse, M.; Cumming, H.; Chapman, R.; Hertzog, P.J. Interferome v2.0: An updated database of annotated interferon-regulated genes. Nucleic Acids Res. 2013, 41, D1040–D1046. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef]
- Baechler, E.C.; Batliwalla, F.M.; Karypis, G.; Gaffney, P.M.; Ortmann, W.A.; Espe, K.J.; Shark, K.B.; Grande, W.J.; Hughes, K.M.; Kapur, V.; et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. USA 2003, 100, 2610–2615. [Google Scholar] [CrossRef] [Green Version]
- Bennett, L.; Palucka, A.K.; Arce, E.; Cantrell, V.; Borvak, J.; Banchereau, J.; Pascual, V. Interferon and Granulopoiesis Signatures in Systemic Lupus Erythematosus Blood. J. Exp. Med. 2003, 197, 711–723. [Google Scholar] [CrossRef] [Green Version]
- Furie, R.; Khamashta, M.; Merrill, J.T.; Werth, V.P.; Kalunian, K.; Brohawn, P.; Illei, G.G.; Drappa, J.; Wang, L.; Yoo, S. Anifrolumab, an Anti–Interferon-α Receptor Monoclonal Antibody, in Moderate-to-Severe Systemic Lupus Erythematosus. Arthritis Rheumatol. 2017, 69, 376–386. [Google Scholar] [CrossRef]
- Rullo, O.J.; Woo, J.M.; Parsa, M.F.; Hoftman, A.D.; Maranian, P.; Elashoff, D.A.; Niewold, T.B.; Grossman, J.M.; Hahn, B.H.; McMahon, M.; et al. Plasma levels of osteopontin identify patients at risk for organ damage in systemic lupus erythematosus. Arthritis Res. Ther. 2013, 15, R18. [Google Scholar] [CrossRef]
- Sakamoto, K.; Fukushima, Y.; Ito, K.; Matsuda, M.; Nagata, S.; Minato, N.; Hattori, M. Osteopontin in Spontaneous Germinal Centers Inhibits Apoptotic Cell Engulfment and Promotes Anti-Nuclear Antibody Production in Lupus-Prone Mice. J. Immunol. 2016, 197, 2177–2186. [Google Scholar] [CrossRef] [Green Version]
- Triantafyllopoulou, A.; Franzke, C.-W.; Seshan, S.V.; Perino, G.; Kalliolias, G.D.; Ramanujam, M.; Van Rooijen, N.; Davidson, A.; Ivashkiv, L.B. Proliferative lesions and metalloproteinase activity in murine lupus nephritis mediated by type I interferons and macrophages. Proc. Natl. Acad. Sci. USA 2010, 107, 3012–3017. [Google Scholar] [CrossRef] [Green Version]
- Hudkins, K.L.; Giachelli, C.M.; Eitner, F.; Couser, W.G.; Johnson, R.J.; Alpers, C.E. Osteopontin expression in human crescentic glomerulonephritis. Kidney Int. 2000, 57, 105–116. [Google Scholar] [CrossRef] [Green Version]
- Kang, E.; Moon, K.; Lee, E.; Lee, Y.; Lee, E.; Ahn, C.; Song, Y. Renal expression of galectin-3 in systemic lupus erythematosus patients with nephritis. Lupus 2009, 18, 22–28. [Google Scholar] [CrossRef]
- Nielsen, C.T.; Rasmussen, N.S.; Heegaard, N.H.; Jacobsen, S. “Kill” the messenger: Targeting of cell-derived microparticles in lupus nephritis. Autoimmun. Rev. 2016, 15, 719–725. [Google Scholar] [CrossRef]
- Nielsen, C.T.; Lood, C.; Østergaard, O.; Iversen, L.V.; Voss, A.; Bengtsson, A.; Jacobsen, S.; Heegaard, N.H.H. Plasma levels of galectin-3-binding protein reflect type I interferon activity and are increased in patients with systemic lupus erythematosus. Lupus Sci. Med. 2014, 1, e000026. [Google Scholar] [CrossRef]
- Sanz, A.B.; Sanchez-Niño, M.D.; Ramos, A.M.; Moreno, J.A.; Santamaria, B.; Ruiz-Ortega, M.; Jesus Egido, J.; Ortiz, A. NF-κB in Renal Inflammation. J. Am. Soc. Nephrol. 2010, 21, 1254–1262. [Google Scholar] [CrossRef]
- Stambe, C. p38 Mitogen-Activated Protein Kinase Activation and Cell Localization in Human Glomerulonephritis: Correlation with Renal Injury. J. Am. Soc. Nephrol. 2004, 15, 326–336. [Google Scholar] [CrossRef] [Green Version]
- Gorelik, G.; Richardson, B. Key role of ERK pathway signaling in lupus. Autoimmunity 2010, 43, 17–22. [Google Scholar] [CrossRef]
- Liu, Y.; Deng, W.; Meng, Q.; Qiu, X.; Sun, D.; Dai, C. CD8+iTregs attenuate glomerular endothelial cell injury in lupus-prone mice through blocking the activation of p38 MAPK and NF-κB. Mol. Immunol. 2018, 103, 133–143. [Google Scholar] [CrossRef]
- Strutz, F.; Zeisberg, M.; Renziehausen, A.; Raschke, B.; Becker, V.; van Kooten, C.; Müller, G. TGF-beta 1 induces proliferation in human renal fibroblasts via induction of basic fibroblast growth factor (FGF-2). Kidney int. 2001, 59, 579–592. [Google Scholar] [CrossRef]
- Liu, F.; Wang, L.; Qi, H.; Wang, J.; Wang, Y.; Jiang, W.; Xu, L.; Liu, N.; Zhuang, S. Nintedanib, a triple tyrosine kinase inhibitor, attenuates renal fibrosis in chronic kidney disease. Clin. Sci. 2017, 131, 2125–2143. [Google Scholar] [CrossRef]
- Soda, K.; Ishibe, S. The function of endocytosis in podocytes. Curr. Opin. Nephrol. Hypertens. 2013, 22, 432–438. [Google Scholar] [CrossRef] [Green Version]
- Mellors, R.C.; Mellors, J.W. Type C RNA virus-specific antibody in human systemic lupus erythematosus demonstrated by enzymoimmunoassay. Proc. Natl. Acad. Sci. USA 1978, 75, 2463–2467. [Google Scholar] [CrossRef]
- Kazazian, H.H., Jr. GENETICS: L1 Retrotransposons Shape the Mammalian Genome. Science 2000, 289, 1152–1153. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
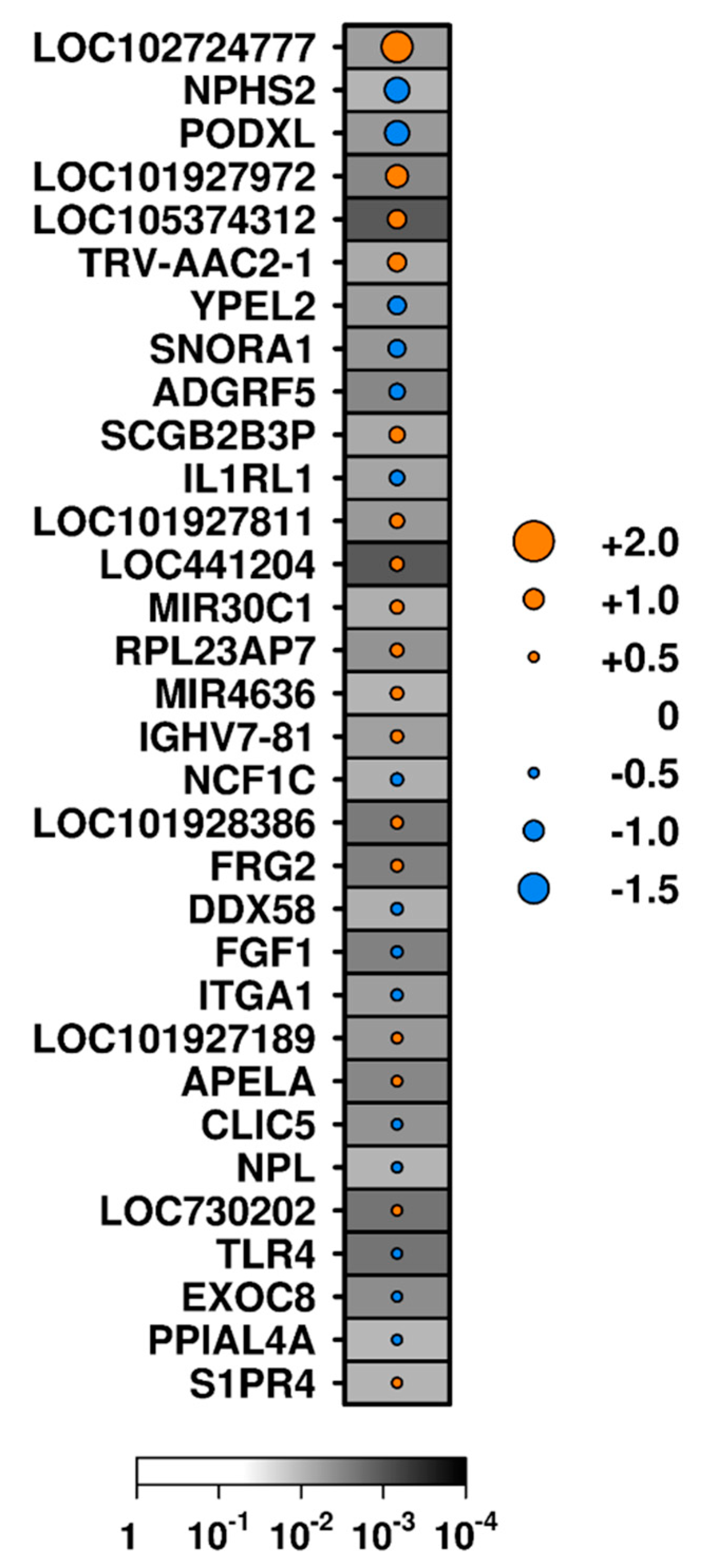{kind=link}
| Age (Years) | 33 (24–45) |
|---|---|
| Sex | n (%) |
| Female | 44 (86) |
| Male | 7 (14) |
| Creatinine (µmol/L) * | 68.5 (59.5–97.0) |
| Proteinuria (g/d) ** | 2.5 (1.5–5) |
| Mean Arterial Pressure (mm Hg) * | 98.8 ± 14.5 |
| Therapeutic Intervention | n (%) |
| Hydroxychloroquine | 30 (56) |
| RAAS Blocker | 22 (41) |
| Any Immunosuppression | 41 (76) |
| Prednisone alone | 16 (30) |
| Prednisone plus | |
| MMF | 11 (20) |
| AZT | 11 (20) |
| CNI † | 3 (6) |
| MTX † | 1 (2) |
| Histological Feature | n (%) |
|---|---|
| ISN/RPS Class | |
| I + II | 2 (4) |
| III–IV | 19 (35) |
| III–IV + V | 21 (39) |
| V | 10 (18) |
| VI | 2 (4) |
| NIH Indices | |
| Activity Index (max 24) | 3.5 (1–9) |
| Chronicity Index (max 12) | 3 (2–4) |
| Histological Lesions | |
| Cellular Proliferation | 33 (61) |
| 1 | 12 |
| 2 | 15 |
| 3 | 6 |
| Crescents | 24 (44) |
| 1 | 10 |
| 2 | 9 |
| 3 | 5 |
| Interstitial Fibrosis | 45 (83) |
| 1 | 35 |
| 2 | 6 |
| 3 | 4 |
| Any Sclerosis | 39 (72) |
| Segmental | 26 (48) |
| Global | 32 (59) |
| 1 | 20 |
| 2 | 6 |
| 3 | 6 |
| Compartment | Transcript | Toronto Cohort | Longitudinal Cohort | |
|---|---|---|---|---|
| Complete Responders | Non-Responders | |||
| Glomerular | FN1 | Positive correlation with the degree of endocapillary hypercellularity | Decreased with response | Increased |
| LGALS3 | Positive correlation with degree of crescent formation | Trend towards decrease with response | No change | |
| SPP1 | Positive correlation with the degree of endocapillary hypercellularity | Decreased with response | No change | |
| Tubulointerstitial | IL1RL1 | Decreased level in patients with interstitial fibrosis | No Change | No change |
| TLR4 | Decreased level in patients with interstitial fibrosis | No Change | No Change | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almaani, S.; Prokopec, S.D.; Zhang, J.; Yu, L.; Avila-Casado, C.; Wither, J.; Scholey, J.W.; Alberton, V.; Malvar, A.; Parikh, S.V.; et al. Rethinking Lupus Nephritis Classification on a Molecular Level. J. Clin. Med. 2019, 8, 1524. https://doi.org/10.3390/jcm8101524
Almaani S, Prokopec SD, Zhang J, Yu L, Avila-Casado C, Wither J, Scholey JW, Alberton V, Malvar A, Parikh SV, et al. Rethinking Lupus Nephritis Classification on a Molecular Level. Journal of Clinical Medicine. 2019; 8(10):1524. https://doi.org/10.3390/jcm8101524
Chicago/Turabian StyleAlmaani, Salem, Stephenie D. Prokopec, Jianying Zhang, Lianbo Yu, Carmen Avila-Casado, Joan Wither, James W. Scholey, Valeria Alberton, Ana Malvar, Samir V. Parikh, and et al. 2019. "Rethinking Lupus Nephritis Classification on a Molecular Level" Journal of Clinical Medicine 8, no. 10: 1524. https://doi.org/10.3390/jcm8101524