Identification of the Gene Expression Rules That Define the Subtypes in Glioma

 , , and
, , and
Abstract
:1. Introduction
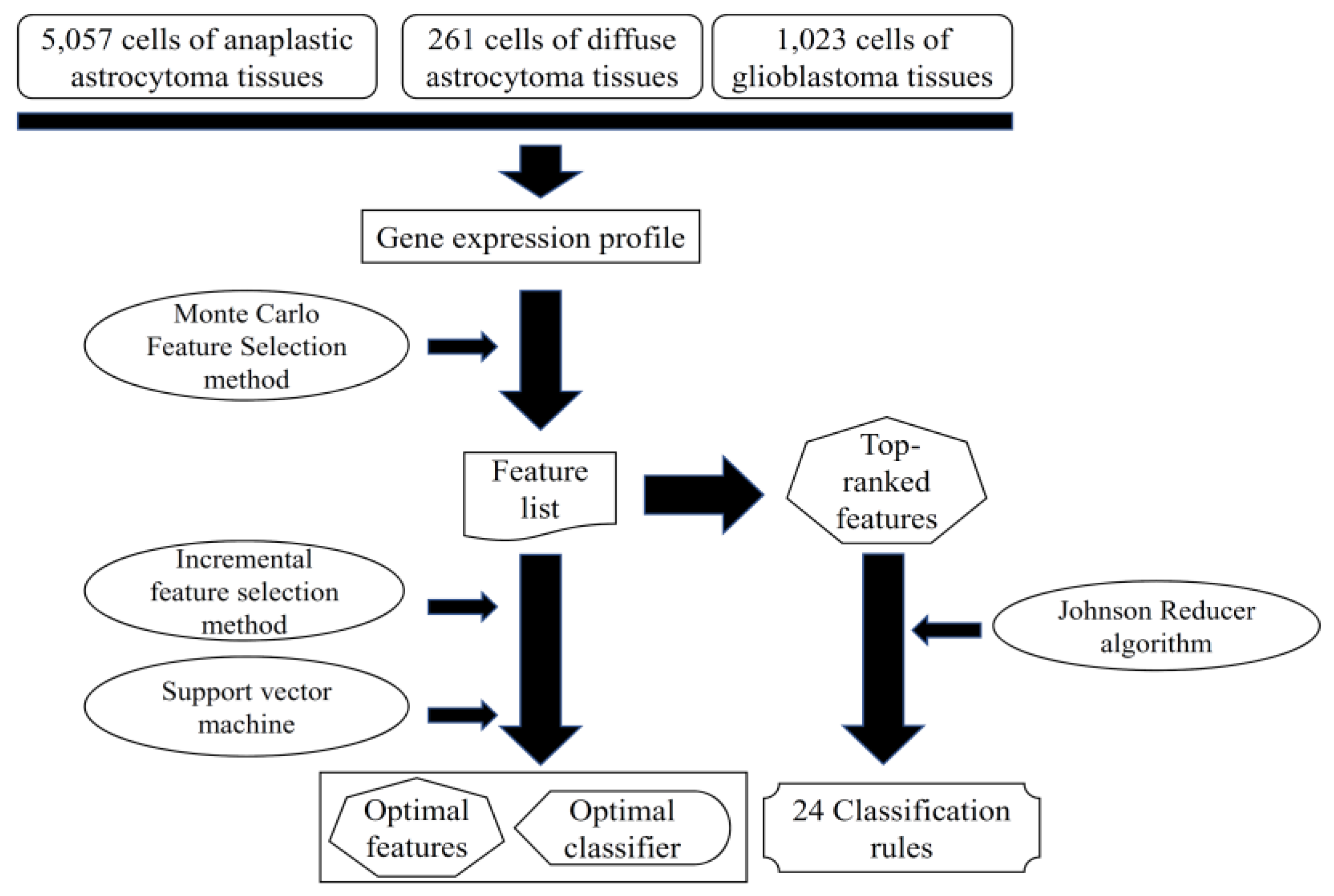
2. Materials and Methods
2.1. Dataset
2.2. Feature Selection
2.2.1. Monte Carlo Feature Selection Method
2.2.2. Rule Learning
2.2.3. Incremental Feature Selection
2.3. Support Vector Machine
2.4. Performance Measurement
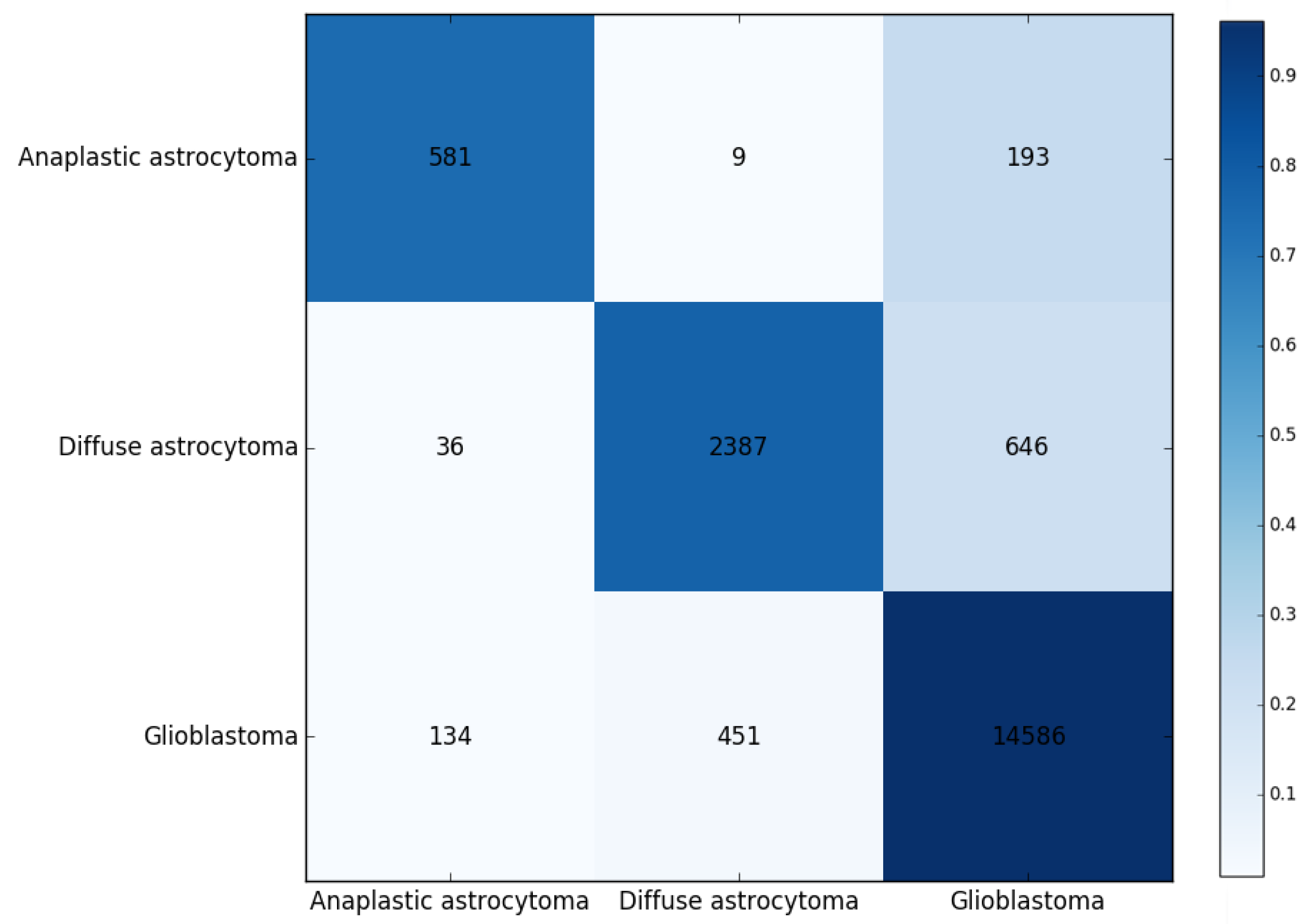
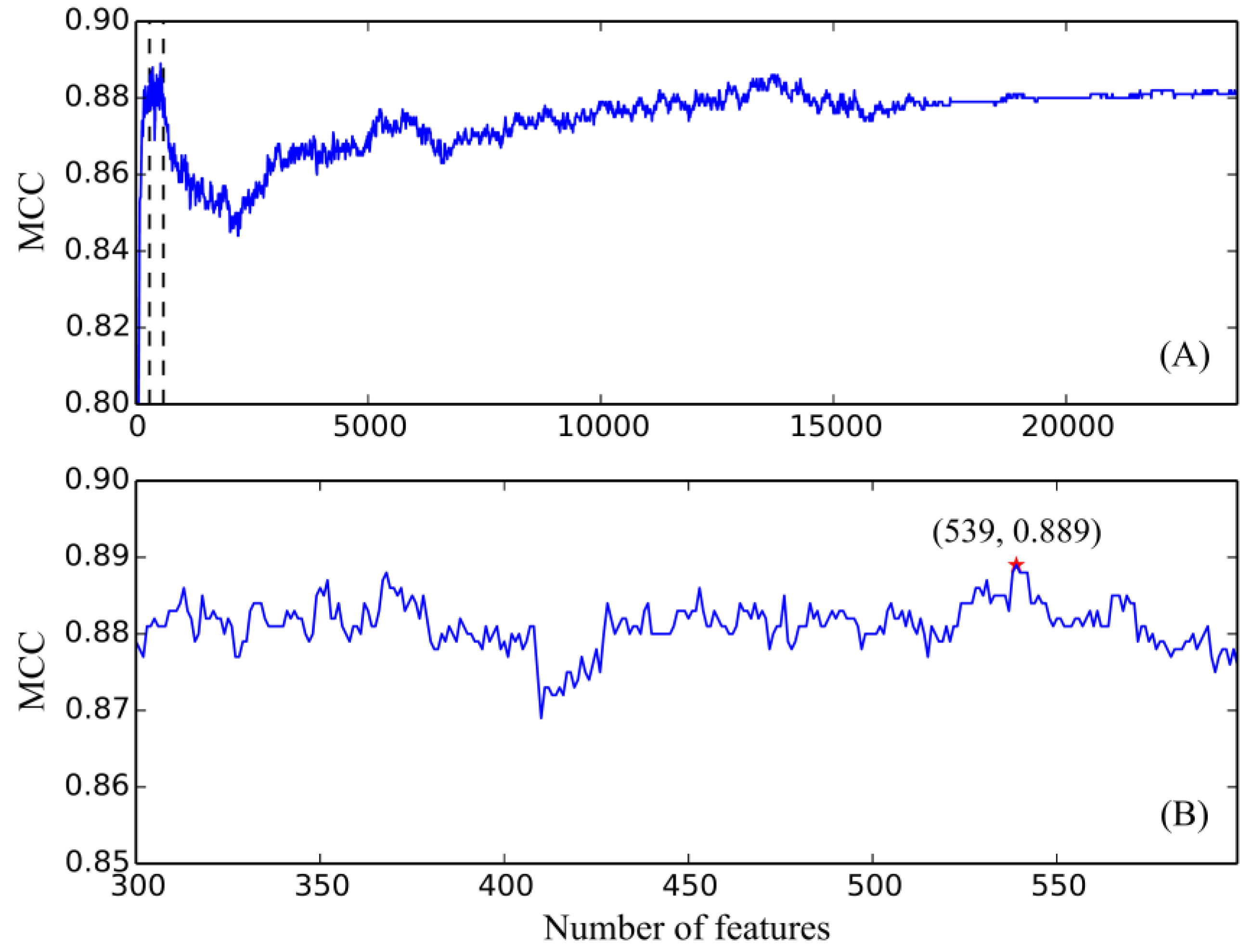
3. Results
4. Discussion
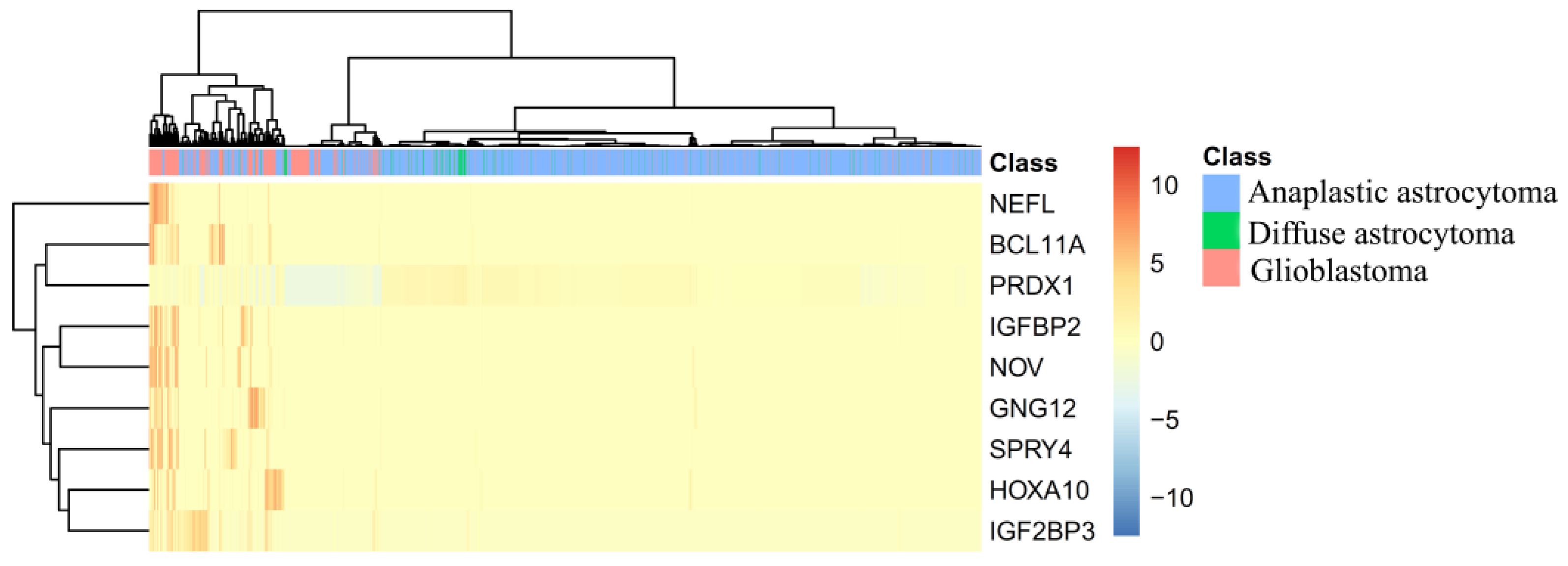
4.1. Analysis of Optimal Genes That May Contribute to the Recognition of Each Glioma Subtype
4.2. Analysis of Optimal Rules for Quantitative Identification of Each Glioma Subtype
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Stetson, L.; Virk, S.M.; Barnholtz-Sloan, J.S. Epidemiology of gliomas. Cancer Treat. Res. 2015, 163, 1–14. [Google Scholar] [PubMed]
- Lopez Juarez, A.; He, D.; Richard Lu, Q. Oligodendrocyte progenitor programming and reprogramming: Toward myelin regeneration. Brain Res. 2016, 1638, 209–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, H.; Hernandez, M.R. Heterogeneity of astrocytes in human optic nerve head. J. Comp. Neurol. 1995, 362, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Athanassakis, I.; Zarifi, I.; Evangeliou, A.; Vassiliadis, S. L-carnitine accelerates the in vitro regeneration of neural network from adult murine brain cells. Brain Res. 2002, 932, 70–78. [Google Scholar] [CrossRef]
- Wang, D.; Couture, R.; Hong, Y. Activated microglia in the spinal cord underlies diabetic neuropathic pain. Eur. J. Pharmacol. 2014, 728, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Liu, D.; Yang, Z.; Guo, N. Central and peripheral nervous systems: Master controllers in cancer metastasis. Cancer Metastasis Rev. 2013, 32, 603–621. [Google Scholar] [CrossRef] [PubMed]
- Alomar, S.A. Clinical manifestation of central nervous system tumor. Semin. Diagn. Pathol. 2010, 27, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fidler, I.J. The biology of brain metastasis: Challenges for therapy. Cancer J. 2015, 21, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Omuro, A.; DeAngelis, L.M. Glioblastoma and other malignant gliomas: A clinical review. JAMA 2013, 310, 1842–1850. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.; Murphy, B.; Miller, R.; Menon, V.; Banik, N.L.; Giglio, P.; Lindhorst, S.M.; Varma, A.K.; Vandergrift, W.A.; Patel, S.J.; et al. Mechanisms and clinical significance of histone deacetylase inhibitors: Epigenetic glioblastoma therapy. Anticancer Res. 2015, 35, 615–625. [Google Scholar] [PubMed]
- Nikolaev, S.; Santoni, F.; Garieri, M.; Makrythanasis, P.; Falconnet, E.; Guipponi, M.; Vannier, A.; Radovanovic, I.; Bena, F.; Forestier, F.; et al. Extrachromosomal driver mutations in glioblastoma and low-grade glioma. Nat. Commun. 2014, 5, 5690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faguer, R.; Tanguy, J.Y.; Rousseau, A.; Clavreul, A.; Menei, P. Early presentation of primary glioblastoma. Neurochirurgie 2014, 60, 188–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tsuda, M.; Kanno, H.; Murata, J.; Mahabir, R.; Ishida, Y.; Kimura, T.; Tanino, M.; Nishihara, H.; Nagashima, K.; et al. Differential diagnosis of small cell glioblastoma and anaplastic oligodendroglioma: A case report of an elderly man. Brain Tumor. Pathol. 2014, 31, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Yalaza, C.; Ak, H.; Cagli, M.S.; Ozgiray, E.; Atay, S.; Aydin, H.H. R132h mutation in idh1 gene is associated with increased tumor hif1-alpha and serum vegf levels in primary glioblastoma multiforme. Ann. Clin. Lab. Sci. 2017, 47, 362–364. [Google Scholar] [PubMed]
- Liu, A.; Hou, C.; Chen, H.; Zong, X.; Zong, P. Genetics and epigenetics of glioblastoma: Applications and overall incidence of idh1 mutation. Front Oncol. 2016, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Reuss, D.E.; Mamatjan, Y.; Schrimpf, D.; Capper, D.; Hovestadt, V.; Kratz, A.; Sahm, F.; Koelsche, C.; Korshunov, A.; Olar, A.; et al. Idh mutant diffuse and anaplastic astrocytomas have similar age at presentation and little difference in survival: A grading problem for who. Acta. Neuropathol. 2015, 129, 867–873. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Guo, Y.; Zhang, C.; Zhang, L.; Li, M.; Guan, P. The expression of neuroglobin in astrocytoma. Brain Tumor. Pathol. 2012, 29, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Melaragno, M.J.; Prayson, R.A.; Murphy, M.A.; Hassenbusch, S.J.; Estes, M.L. Anaplastic astrocytoma with granular cell differentiation: Case report and review of the literature. Hum. Pathol. 1993, 24, 805–808. [Google Scholar] [CrossRef]
- Tirosh, I.; Venteicher, A.S.; Hebert, C.; Escalante, L.E.; Patel, A.P.; Yizhak, K.; Fisher, J.M.; Rodman, C.; Mount, C.; Filbin, M.G.; et al. Single-cell rna-seq supports a developmental hierarchy in human oligodendroglioma. Nature 2016, 539, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Venteicher, A.S.; Tirosh, I.; Hebert, C.; Yizhak, K.; Neftel, C.; Filbin, M.G.; Hovestadt, V.; Escalante, L.E.; Shaw, M.L.; Rodman, C.; et al. Decoupling genetics, lineages, and microenvironment in idh-mutant gliomas by single-cell rna-seq. Science 2017, 355, eaai8478. [Google Scholar] [CrossRef] [PubMed]
- Draminski, M.; Rada-Iglesias, A.; Enroth, S.; Wadelius, C.; Koronacki, J.; Komorowski, J. Monte carlo feature selection for supervised classification. Bioinformatics 2008, 24, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.A.; Setiono, R. Incremental feature selection. Appl. Intell. 1998, 9, 217–230. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef] [Green Version]
- Ohrn, A. Discernibility and Rough Sets in Medicine: Tools and Applications. Ph.D. Thesis, Norwegian University of Science and Technology, Trondheim, Norway, 1999. [Google Scholar]
- Chen, L.; Li, J.; Zhang, Y.H.; Feng, K.; Wang, S.; Zhang, Y.; Huang, T.; Kong, X.; Cai, Y.D. Identification of gene expression signatures across different types of neural stem cells with the monte-carlo feature selection method. J. Cell. Biochem. 2018, 119, 3394–3403. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Cai, Y. Identification of the functional alteration signatures across different cancer types with support vector machine and feature analysis. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2218–2227. [Google Scholar] [CrossRef] [PubMed]
- MCFS-ID. Available online: http://www.ipipan.eu/staff/m.draminski/mcfs.html. (accessed on 15 April 2017).
- Pan, X.Y.; Shen, H.B. Robust prediction of b-factor profile from sequence using two-stage svr based on random forest feature selection. Protein Pept. Lett. 2009, 16, 1447–1454. [Google Scholar] [CrossRef]
- Mirza, A.H.; Berthelsen, C.H.; Seemann, S.E.; Pan, X.; Frederiksen, K.S.; Vilien, M.; Gorodkin, J.; Pociot, F. Transcriptomic landscape of lncrnas in inflammatory bowel disease. Genome Med. 2015, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, S.; Zhang, Y.-H.; Li, J.; Xing, Z.-H.; Yang, J.; Huang, T.; Cai, Y.-D. Identify key sequence features to improve crispr sgrna efficacy. IEEE Access 2017, 5, 26582–26590. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Huang, T.; Chen, L.; Xu, Y.; Hu, Y.; Hu, L.D.; Cai, Y.; Kong, X. Identifying and analyzing different cancer subtypes using rna-seq data of blood platelets. Oncotarget 2017, 8, 87494–87511. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, Y.-H.; Wang, S.; Zhang, Y.; Huang, T.; Cai, Y.-D. Prediction and analysis of essential genes using the enrichments of gene ontology and kegg pathways. PLoS ONE 2017. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chu, C.; Zhang, Y.H.; Zhu, C.; Kong, X.; Huang, T.; Cai, Y.D. Analysis of gene expression profiles in the human brain stem, cerebellum and cerebral cortex. PLoS OONE 2016, 11, e0159395. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, Q.; Lu, J.; Cai, Y.-D. Analysis and prediction of nitrated tyrosine sites with the mrmr method and support vector machine algorithm. Curr. Bioinform. 2018, 13, 3–13. [Google Scholar] [CrossRef]
- Fang, Y.; Chen, L. A binary classifier for prediction of the types of metabolic pathway of chemicals. Comb. Chem. High Throughput Screen. 2017, 20, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chu, C.; Feng, K. Predicting the types of metabolic pathway of compounds using molecular fragments and sequential minimal optimization. Chem. High Throughput Screen. 2016, 19, 136–143. [Google Scholar] [CrossRef]
- Platt, J. Sequential Minimal Optimizaton: A Fast Algorithm for Training Support Vector Machines; Technical Report MSR-TR-98-14; Microsoft Res: Redmond, WA, USA, 1998. [Google Scholar]
- Downloading and Installing Weka. Available online: https://www.cs.waikato.ac.nz/ml/weka/downloading.html. (accessed on 10 March 2017).
- Matthews, B.W. Comparison of the predicted and observed secondary structure of t4 phage lysozyme. Biochim. Biophys. Acta 1975, 405, 442–451. [Google Scholar] [CrossRef]
- Chen, L.; Chu, C.; Zhang, Y.-H.; Zheng, M.-Y.; Zhu, L.; Kong, X.; Huang, T. Identification of drug-drug interactions using chemical interactions. Curr. Bioinform. 2017, 12, 526–534. [Google Scholar] [CrossRef]
- Zhao, X.; Chen, L.; Lu, J. A similarity-based method for prediction of drug side effects with heterogeneous information. Math. Biosci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, S.; Zhang, Y.-H.; Wei, L.; Xu, X.; Huang, T.; Cai, Y.-D. Prediction of nitrated tyrosine residues in protein sequences by extreme learning machine and feature selection methods. Chem. High Throughput Screen. 2018, 21, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Gorodkin, J. Comparing two k-category assignments by a k-category correlation coefficient. Comput. Biol. Chem. 2004, 28, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Kohavi, R. A Study of Cross-Validation and Bootstrap for Accuracy Estimation and Model Selection, International joint Conference on artificial intelligence, Montreal, Quebec, Canada, 1995; Lawrence Erlbaum Associates Ltd.: Mahwah, NJ, USA, 1995; pp. 1137–1145. [Google Scholar]
- Chen, L.; Zhang, Y.-H.; Huang, T.; Cai, Y.-D. Gene expression profiling gut microbiota in different races of humans. Sci. Rep. 2016, 6, 23075. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Pan, X.; Hu, X.; Zhang, Y.-H.; Wang, S.; Huang, T.; Cai, Y.-D. Gene expression differences among different msi statuses in colorectal cancer. Int. J. Cancer 2018, 143, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Urbonaviciene, G.; Frystyk, J.; Urbonavicius, S.; Lindholt, J.S. Igf-i and igfbp2 in peripheral artery disease: Results of a prospective study. Scand. Cardiovasc. J. 2014, 48, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, D.; Hsieh, A.; Stea, B.; Ellsworth, R. Igfbp2 promotes glioma tumor stem cell expansion and survival. Biochem. Biophys. Res. Commun. 2010, 397, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.C.; Jung, T.H.; Jung, D.Y.; Park, W.K.; Cho, H. Indatraline inhibits rho- and calcium-mediated glioblastoma cell motility and angiogenesis. Biochem. Biophys. Res. Commun. 2014, 443, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Taniuchi, K.; Furihata, M.; Hanazaki, K.; Iwasaki, S.; Tanaka, K.; Shimizu, T.; Saito, M.; Saibara, T. Peroxiredoxin 1 promotes pancreatic cancer cell invasion by modulating p38 mapk activity. Pancreas 2015, 44, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Svendsen, A.; Verhoeff, J.J.; Immervoll, H.; Brogger, J.C.; Kmiecik, J.; Poli, A.; Netland, I.A.; Prestegarden, L.; Planaguma, J.; Torsvik, A.; et al. Expression of the progenitor marker ng2/cspg4 predicts poor survival and resistance to ionising radiation in glioblastoma. Acta Neuropathol. 2011, 122, 495–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiestler, B.; Claus, R.; Hartlieb, S.A.; Schliesser, M.G.; Weiss, E.K.; Hielscher, T.; Platten, M.; Dittmann, L.M.; Meisner, C.; Felsberg, J.; et al. Malignant astrocytomas of elderly patients lack favorable molecular markers: An analysis of the noa-08 study collective. Neuro-oncology 2013, 15, 1017–1026. [Google Scholar] [CrossRef] [PubMed]
- Marchal, P.O.; Kavvadas, P.; Abed, A.; Kazazian, C.; Authier, F.; Koseki, H.; Hiraoka, S.; Boffa, J.J.; Martinerie, C.; Chadjichristos, C.E. Reduced nov/ccn3 expression limits inflammation and interstitial renal fibrosis after obstructive nephropathy in mice. PLoS ONE 2015, 10, e0137876. [Google Scholar] [CrossRef] [PubMed]
- Perbal, B. Nov (nephroblastoma overexpressed) and the ccn family of genes: Structural and functional issues. Mol. Pathol. 2001, 54, 57–79. [Google Scholar] [CrossRef] [PubMed]
- Benini, S.; Perbal, B.; Zambelli, D.; Colombo, M.P.; Manara, M.C.; Serra, M.; Parenza, M.; Martinez, V.; Picci, P.; Scotlandi, K. In ewing’s sarcoma ccn3(nov) inhibits proliferation while promoting migration and invasion of the same cell type. Oncogene 2005, 24, 4349–4361. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, P.N.; Cleveland, D.W.; Griffin, J.W.; Landes, P.W.; Cowan, N.J.; Price, D.L. Neurofilament gene expression: A major determinant of axonal caliber. Proc. Natl. Acad. Sci. USA 1987, 84, 3472–3476. [Google Scholar] [CrossRef] [PubMed]
- Morosetti, R.; Servidei, T.; Mirabella, M.; Rutella, S.; Mangiola, A.; Maira, G.; Mastrangelo, R.; Koeffler, H.P. The ppargamma ligands pgj2 and rosiglitazone show a differential ability to inhibit proliferation and to induce apoptosis and differentiation of human glioblastoma cell lines. Int. J. Oncol. 2004, 25, 493–502. [Google Scholar] [PubMed]
- Fantini, S.; Salsi, V.; Vitobello, A.; Rijli, F.M.; Zappavigna, V. Microrna-196b is transcribed from an autonomous promoter and is directly regulated by cdx2 and by posterior hox proteins during embryogenesis. Biochim. Biophys. Acta 2015, 1849, 1066–1080. [Google Scholar] [CrossRef] [PubMed]
- Maurel-Zaffran, C.; Chauvet, S.; Jullien, N.; Miassod, R.; Pradel, J.; Aragnol, D. Nessy, an evolutionary conserved gene controlled by hox proteins during drosophila embryogenesis. Mech. Dev. 1999, 86, 159–163. [Google Scholar] [CrossRef]
- Kurscheid, S.; Bady, P.; Sciuscio, D.; Samarzija, I.; Shay, T.; Vassallo, I.; Criekinge, W.V.; Daniel, R.T.; van den Bent, M.J.; Marosi, C.; et al. Chromosome 7 gain and DNA hypermethylation at the hoxa10 locus are associated with expression of a stem cell related hox-signature in glioblastoma. Genome Biol. 2015, 16, 16. [Google Scholar] [CrossRef] [PubMed]
- Hale, J.S.; Otvos, B.; Sinyuk, M.; Alvarado, A.G.; Hitomi, M.; Stoltz, K.; Wu, Q.; Flavahan, W.; Levison, B.; Johansen, M.L.; et al. Cancer stem cell-specific scavenger receptor cd36 drives glioblastoma progression. Stem. Cells 2014, 32, 1746–1758. [Google Scholar] [CrossRef] [PubMed]
- Pietras, A.; Katz, A.M.; Ekstrom, E.J.; Wee, B.; Halliday, J.J.; Pitter, K.L.; Werbeck, J.L.; Amankulor, N.M.; Huse, J.T.; Holland, E.C. Osteopontin-cd44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth. Cell Stem Cell 2014, 14, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Niemczyk, M.; Ito, Y.; Huddleston, J.; Git, A.; Abu-Amero, S.; Caldas, C.; Moore, G.E.; Stojic, L.; Murrell, A. Imprinted chromatin around diras3 regulates alternative splicing of gng12-as1, a long noncoding rna. Am. J. Hum. Genet. 2013, 93, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Chen, Q.; Li, C.; Wang, L.; Qian, X.; Jiang, C.; Liu, X.; Wang, X.; Li, H.; Kang, C.; et al. Mir-124 governs glioma growth and angiogenesis and enhances chemosensitivity by targeting r-ras and n-ras. Neuro-oncology 2014, 16, 1341–1353. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhan, W.; Xie, S.; Hu, J.; Shi, Q.; Zhou, X.; Wu, Y.; Wang, S.; Fei, Z.; Yu, R. Over-expression of rap2a inhibits glioma migration and invasion by down-regulating p-akt. Cell Biol. Int. 2014, 38, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Kleihues, P. Epidemiology and etiology of gliomas. Acta Neuropathol. 2005, 109, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Lv, Z.; Guo, E. Knockdown of long noncoding rna spry4-it1 suppresses glioma cell proliferation, metastasis and epithelial-mesenchymal transition. Int. J. Clin. Exp. Pathol. 2015, 8, 9140–9146. [Google Scholar] [PubMed]
- Fu, J.; Rodova, M.; Nanta, R.; Meeker, D.; Van Veldhuizen, P.J.; Srivastava, R.K.; Shankar, S. Npv-lde-225 (erismodegib) inhibits epithelial mesenchymal transition and self-renewal of glioblastoma initiating cells by regulating mir-21, mir-128, and mir-200. Neuro-oncology 2013, 15, 691–706. [Google Scholar] [CrossRef] [PubMed]
- Joo, Y.N.; Eun, S.Y.; Park, S.W.; Lee, J.H.; Chang, K.C.; Kim, H.J. Honokiol inhibits u87mg human glioblastoma cell invasion through endothelial cells by regulating membrane permeability and the epithelial-mesenchymal transition. Int. J. Oncol. 2014, 44, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Balci, T.B.; Sawyer, S.L.; Davila, J.; Humphreys, P.; Dyment, D.A. Brain malformations in a patient with deletion 2p16.1: A refinement of the phenotype to bcl11a. Eur. J. Med. Genet. 2015, 58, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Bergerson, R.J.; Collier, L.S.; Sarver, A.L.; Been, R.A.; Lugthart, S.; Diers, M.D.; Zuber, J.; Rappaport, A.R.; Nixon, M.J.; Silverstein, K.A.; et al. An insertional mutagenesis screen identifies genes that cooperate with mll-af9 in a murine leukemogenesis model. Blood 2012, 119, 4512–4523. [Google Scholar] [CrossRef] [PubMed]
- Estruch, S.B.; Buzon, V.; Carbo, L.R.; Schorova, L.; Luders, J.; Estebanez-Perpina, E. The oncoprotein bcl11a binds to orphan nuclear receptor tlx and potentiates its transrepressive function. PLoS ONE 2012, 7, e37963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Y.; Ma, J.; Xue, Y.; Wang, P.; Li, Z.; Liu, J.; Chen, L.; Xi, Z.; Teng, H.; Wang, Z.; et al. Knockdown of long non-coding rna xist exerts tumor-suppressive functions in human glioblastoma stem cells by up-regulating mir-152. Cancer Lett. 2015, 359, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Seo, H.S.; Choi, S.J.; Park, H.S.; Lee, J.Y.; Lee, K.H.; Park, J.Y. Characterization of the two genes differentially expressed during development in human fetal astrocytes. Yonsei. Med. J. 2003, 44, 1059–1068. [Google Scholar] [CrossRef] [PubMed]
- Sakakini, N.; Turchi, L.; Bergon, A.; Holota, H.; Rekima, S.; Lopez, F.; Paquis, P.; Almairac, F.; Fontaine, D.; Baeza-Kallee, N.; et al. A positive feed-forward loop associating egr1 and pdgfa promotes proliferation and self-renewal in glioblastoma stem cells. J. Biol. Chem. 2016, 291, 10684–10699. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Wang, Z.; Wang, Y.; Liang, Z.; Zhang, X.; Zhao, Z.; Jiao, B. Eukaryotic initiation factor 3c silencing inhibits cell proliferation and promotes apoptosis in human glioma. Oncol. Rep. 2015, 33, 2954–2962. [Google Scholar] [CrossRef] [PubMed]
- Grohar, P.J.; Kim, S.; Rangel Rivera, G.O.; Sen, N.; Haddock, S.; Harlow, M.L.; Maloney, N.K.; Zhu, J.; O’Neill, M.; Jones, T.L.; et al. Functional genomic screening reveals splicing of the ews-fli1 fusion transcript as a vulnerability in ewing sarcoma. Cell Rep. 2016, 14, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.M.; Chen, Y.; Liu, L.; Zhang, C.G.; Wang, W.; Gong, K.; Huang, Z.; Guo, M.X.; Li, W.X.; Li, W. C1orf61 acts as a tumor activator in human hepatocellular carcinoma and is associated with tumorigenesis and metastasis. FASEB J. 2013, 27, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Iwase, A.; Bayasula, B.; Nagatomo, Y.; Kondo, M.; Nakahara, T.; Takikawa, S.; Goto, M.; Kotani, T.; Kiyono, T.; et al. Cyp51a1 induced by growth differentiation factor 9 and follicle-stimulating hormone in granulosa cells is a possible predictor for unfertilization. Reprod. Sci. 2015, 22, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Salemi, M.; Fraggetta, F.; Galia, A.; Pepe, P.; Cimino, L.; Condorelli, R.A.; Calogero, A.E. Cerebellar degeneration-related autoantigen 1 (cdr1) gene expression in prostate cancer cell lines. Int. J. Biol. Markers 2014, 29, e288–e290. [Google Scholar] [CrossRef] [PubMed]
- Halatsch, M.E.; Low, S.; Mursch, K.; Hielscher, T.; Schmidt, U.; Unterberg, A.; Vougioukas, V.I.; Feuerhake, F. Candidate genes for sensitivity and resistance of human glioblastoma multiforme cell lines to erlotinib. Laboratory investigation. J. Neurosurg. 2009, 111, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Jarboe, J.S.; Anderson, J.C.; Duarte, C.W.; Mehta, T.; Nowsheen, S.; Hicks, P.H.; Whitley, A.C.; Rohrbach, T.D.; McCubrey, R.O.; Chiu, S.; et al. Marcks regulates growth and radiation sensitivity and is a novel prognostic factor for glioma. Clin. Cancer Res. 2012, 18, 3030–3041. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; McDonald, J.T.; Naidu, M.; Hahnfeldt, P.; Hlatky, L. A proposed quantitative index for assessing the potential contribution of reprogramming to cancer stem cell kinetics. Stem. Cells Int. 2014, 2014, 249309. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Li, S.; Bodhinathan, K.; Meyers, C.; Chen, W.; Campbell-Thompson, M.; McIntyre, L.; Foster, T.C.; Muzyczka, N.; Kumar, A. Enhanced expression of pctk1, tcf12 and ccnd1 in hippocampus of rats: Impact on cognitive function, synaptic plasticity and pathology. Neurobiol. Learn. Mem. 2012, 97, 69–80. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rules | Criteria | Glioma Subtype | Rules | Criteria | Glioma Subtype |
|---|---|---|---|---|---|
| Rule1 | XIST ≥ 2.725 LOC100190986 ≤ 1.956 GATM ≥ 4.826 PRDX1 ≥ 6.064 | diffuse astrocytoma | Rule2 | XIST ≥ 3.588 LOC100190986 ≤ 1.609 SLC1A3 ≥ 5.404 HLA-B ≤ 7.228 | diffuse astrocytoma |
| Rule3 | XIST ≥ 3.132 RPL7 ≥ 9.478 RPL8 ≤ 7.502 EGR1 ≤ 6.442 | diffuse astrocytoma | Rule4 | XIST ≥ 2.601 EIF3C ≤ 0.477 HNRNPH1 ≥ 6.813 C1orf61 ≤ 6.456 | diffuse astrocytoma |
| Rule5 | XIST ≥ 2.395 CYP51A1 ≥ 5.810 CDR1 ≥ 6.717 | diffuse astrocytoma | Rule6 | XIST ≥ 2.395 SKP1 ≥ 6.479 SEPT7 ≥ 5.342 RPL30 ≥ 7.419 | diffuse astrocytoma |
| Rule7 | XIST ≥ 2.395 SFPQ ≥ 4.772 JAM3 ≤ 0.000 | diffuse astrocytoma | Rule8 | XIST ≥ 3.021 RPL30 ≥ 8.453 PPIA ≥ 7.077 DDX5 ≤ 6.823 | diffuse astrocytoma |
| Rule9 | PCDHB7 ≥ 3.827 HNRNPH1 ≥ 6.670 | diffuse astrocytoma | Rule10 | RHOB ≥ 6.545 HSPA1A ≥ 4.446 | diffuse astrocytoma |
| Rule11 | RPSAP58 ≤ 1.280 HSPA1B ≥ 5.291 PRDX1 ≤ 0.000 MARCKS ≥ 3.464 | glioblastoma | Rule12 | TCF12 ≤ 4.952 COL20A1 ≥ 0.800 CBR1 ≥ 0.4222 MTRNR2L2 ≥ 12.850 | glioblastoma |
| Rule13 | NRCAM ≤ 0.999 HSPA1B ≥ 4.754 XIST ≥ 1.034 HSPA1B ≥ 7.275 | glioblastoma | Rule14 | RPSAP58 ≤ 1.414 PRDX1 ≤ 1.657 MTRNR2L8 ≥ 12.074 RPL8 ≥ 7.374 | glioblastoma |
| Rule15 | NRCAM ≤ 2.392 FOS ≤ 5.642 RPL35 ≥ 6.606 C1orf61 ≥ 6.700 MARCKS ≤ 4.770 | glioblastoma | Rule16 | FAM110B ≤ 2.527 RPSAP58 ≤ 0.165 NEAT1 ≥ 5.045 ITPR2 ≥ 2.118 HLA-C ≥ 6.293 NAPSB ≥ 4.988 | glioblastoma |
| Rule17 | FAM110B ≤ 2.607 RPSAP58 ≤ 0.000 SUSD5 ≥ 0.573 SUSD5 ≥ 2.515 | glioblastoma | Rule18 | TCF12 ≤ 4.215 RHOB ≤ 0.180 TMBIM6 ≤ 4.695 RPS26 ≤ 5.572 JAM3 ≥ 1.876 | glioblastoma |
| Rule19 | RIA2 ≤ 3.045 PRDX1 ≤ 0.000 MCL1 ≤ 2.387 | glioblastoma | Rule20 | NRCAM ≤ 1.090 DDX5 ≤ 6.520 SIRPB1 ≥ 1.014 EIF1 ≤ 7.690 NDUFA4 ≥ 0.811 | glioblastoma |
| Rule21 | SMOC1 ≤ 1.959 RPSAP58 ≤ 0.000 RPS26 ≤ 4.504 APOE ≤ 0.797 RPL7A ≥ 7.267 | glioblastoma | Rule22 | NRCAM ≤ 0.548 CD97 ≥ 0.856 CYBB ≥ 5.756 RPSAP58 ≤ 0.952 ITPR2 ≥ 2.769 EIF1 ≤ 8.648 | glioblastoma |
| Rule23 | NRCAM ≤ 0.548 MT2A ≥ 8.374 PFKFB3 ≥ 4.147 | glioblastoma | Rule24 | Other conditions | anaplastic astrocytoma |
| Rank | Gene Symbol | Description | Relative importance (RI) |
|---|---|---|---|
| 1 | IGFBP2 | Insulin-Like Growth Factor Binding Protein 2 | 0.1375 |
| 2 | PRDX1 | Peroxiredoxin 1 | 0.1226 |
| 3 | NOV | Nephroblastoma Overexpressed | 0.1194 |
| 4 | NEFL | Neurofilament Light | 0.1100 |
| 5 | HOXA10 | Homeobox A10 | 0.1059 |
| 6 | GNG12 | G Protein Subunit Gamma 12 | 0.0942 |
| 7 | IGF2BP3 | Insulin Like Growth Factor 2 MRNA Binding Protein 3 | 0.0891 |
| 8 | SPRY4 | Sprouty RTK Signaling Antagonist 4 | 0.0865 |
| 9 | BCL11A | B Cell CLL/Lymphoma 11A | 0.0847 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, Y.-D.; Zhang, S.; Zhang, Y.-H.; Pan, X.; Feng, K.; Chen, L.; Huang, T.; Kong, X. Identification of the Gene Expression Rules That Define the Subtypes in Glioma. J. Clin. Med. 2018, 7, 350. https://doi.org/10.3390/jcm7100350
Cai Y-D, Zhang S, Zhang Y-H, Pan X, Feng K, Chen L, Huang T, Kong X. Identification of the Gene Expression Rules That Define the Subtypes in Glioma. Journal of Clinical Medicine. 2018; 7(10):350. https://doi.org/10.3390/jcm7100350
Chicago/Turabian StyleCai, Yu-Dong, Shiqi Zhang, Yu-Hang Zhang, Xiaoyong Pan, KaiYan Feng, Lei Chen, Tao Huang, and Xiangyin Kong. 2018. "Identification of the Gene Expression Rules That Define the Subtypes in Glioma" Journal of Clinical Medicine 7, no. 10: 350. https://doi.org/10.3390/jcm7100350