
The TGF-β/Smad4 Signaling Pathway in Pancreatic Carcinogenesis and Its Clinical Significance


Abstract
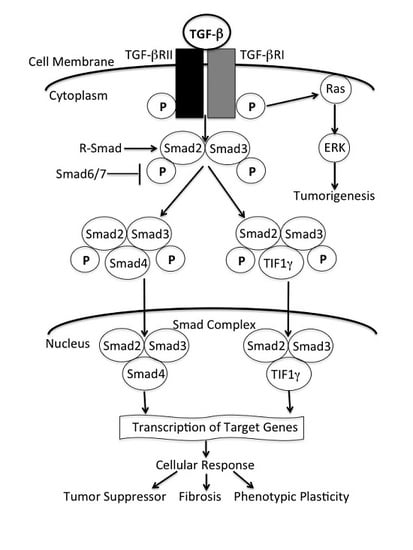
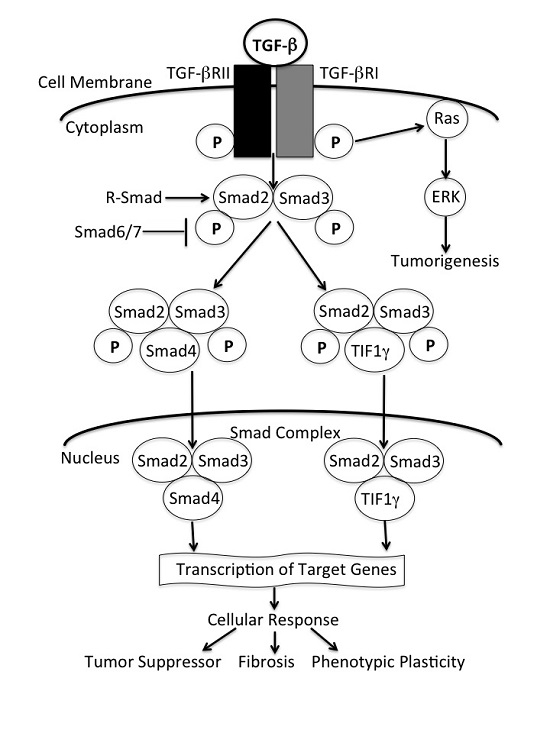
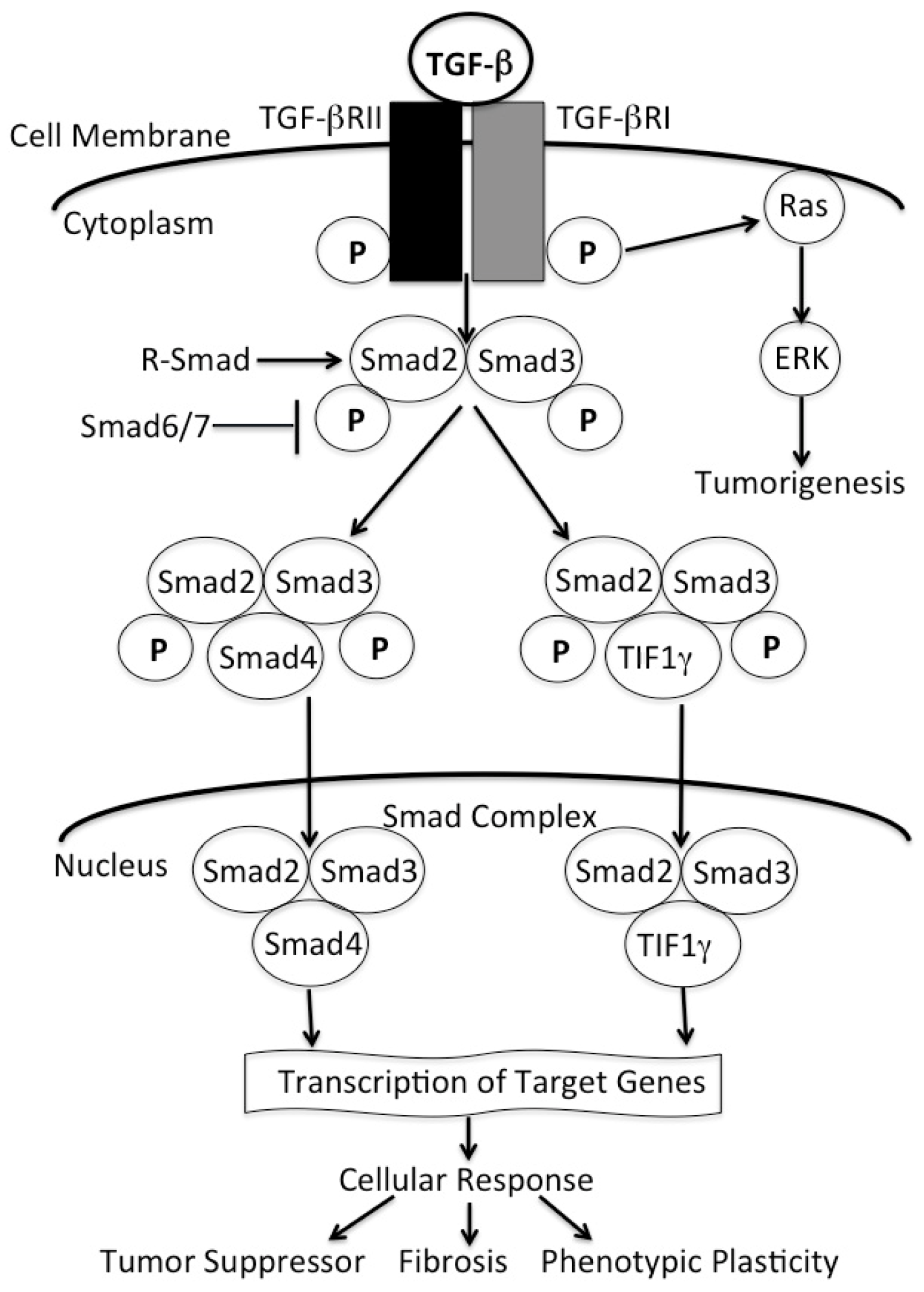
:
{kind=link}
{kind=link}
1. Introduction
2. TGF-β Signaling
2.1. Smad-Dependent Pathway
2.2. Non-Smad-Dependent Pathway
3. TGF-β/Smad4 Signaling in Pancreatic Cell as a Tumor Suppressor
4. Transcriptional Intermediary Factor 1 Gamma (Tif1γ) in the Regulation of the TGF-β Pathway
5. TGF-β/Smad4 as a Prognostic Marker of PDAC
6. TGF-β/Smad4 as a Therapeutic target for PDAC
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kato, Y.; Inoue, H.; Yoshioka, U.; Fujiyama, Y.; Bamba, T. Effects of transforming growth factor β1, interleukin-1β, tumor necrosis factor a and platelet-derived growth factor on the collagen synthesis and the proliferation of periacinal fibroblastoid cells isolated and cultured from rat pancreatic acini. Pathophysiology 1999, 3, 175–179. [Google Scholar] [CrossRef]
- Krzemień, S.; Knapczyk, P. Current review on the role of transforming growth factor beta (TGF-beta) in some pathological disorders. Wiad. Lek. 2005, 58, 536–539. [Google Scholar] [PubMed]
- Kajdaniuk, D.; Marek, B.; Borgiel-Marek, H.; Kos-Kudla, B. Transforming growth factor β1 (TGFβ1) in physiology and pathology. Endokrynol. Polska 2013, 64, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, G.; Schwarz, R.E.; Higgins, L.; McEnroe, G.; Chakravarty, S.; Dugar, S.; Reiss, M. Targeting endogenous transforming growth factor beta receptor signaling in SMAD4-deficient human pancreatic carcinoma cells inhibits their invasive phenotype1. Cancer Res. 2004, 64, 5200–5211. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Lee, K.T.; Lee, J.K.; Paik, S.W.; Rhee, J.C.; Choi, K.W. Clinical usefulness of carbohydrate antigen 19-9 as a screening test for pancreatic cancer in an asymptomatic population. J. Gastroenterol. Hepatol. 2004, 19, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.W.; Li, Y.W.; Feng, R.; Yu, J.B.; Li, J.; Zhang, Y.; Li, J.C.; Wang, Y.X. TGF-β/Smad2/3 signal pathway involves in U251 cell proliferation and apoptosis. Gene 2015, 562, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M. Pancreatic Cancer. N. Engl. J. Med. 2010, 362, 1605–1617. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Holly, E.A.; Bracci, P.M. Survival in population-based pancreatic cancer patients: San Francisco Bay area, 1995–1999. Am. J. Epidemiol. 2011, 174, 1373–1381. [Google Scholar] [CrossRef] [PubMed]
- Sultana, A.; Cox, T.; Ghaneh, P.; Neoptolemos, J.P. Adjuvant therapy for pancreatic cancer. Recent Results Cancer Res. 2012, 196, 65–88. [Google Scholar] [PubMed]
- Katz, L.H.; Li, Y.; Chen, J.S.; Muñoz, N.M.; Majumdar, A.; Chen, J.; Mishra, L. Targeting TGF-β signaling in cancer. Expert Opin. Ther. Targets 2013, 17, 743–760. [Google Scholar] [CrossRef] [PubMed]
- Todorović-Raković, N.; Milovanović, J.; Nikolić-Vukosavljević, D. TGF-β and its coreceptors in cancerogenesis: An overview. Biomark. Med. 2011, 5, 855–863. [Google Scholar] [CrossRef] [PubMed]
- López-Casillas, F.; Wrana, J.L.; Massagué, J. Betaglycan presents ligand to the TGF beta signaling receptor. Cell 1993, 73, 1435–1444. [Google Scholar] [CrossRef]
- Shi, Y.; Massagué, J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Kretzschmar, M.; Doody, J.; Massagué, J. Opposing BMP and EGF signalling pathways converge on the TGF-beta family mediator Smad1. Nature 1997, 389, 618–622. [Google Scholar] [PubMed]
- Wrana, J.L.; Attisano, L.; Wieser, R.; Ventura, F.; Massagué, J. Mechanism of activation of the TGF-beta receptor. Nature 1994, 370, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Pouponnot, C.; Massagué, J. Dual role of the Smad4/DPC4 tumor suppressor in TGFβ-inducible transcriptional complexes. Genes Dev. 1997, 11, 3157–3167. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Musci, T.; Derynck, R. The tumor suppressor Smad4/DPC 4 as a central mediator of Smad function. Curr. Biol. 1997, 7, 270–276. [Google Scholar] [CrossRef]
- Chen, Y.; Lebrun, J.J.; Vale, W. Regulation of transforming growth factor β- and activin-induced transcription by mammalian Mad proteins. Proc. Natl. Acad. Sci. USA 1996, 93, 12992–12997. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Feng, X.H.; Derynck, R. Smad3 and Smad4 cooperate with c-Jun/c-Fos to mediate TGF-beta-induced transcription. Nature 1998, 394, 909–913. [Google Scholar] [PubMed]
- Akhurst, R.J.; Hata, A. Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Akhurst, R.J.; Balmain, A. TGF-β signaling in tumor suppression and cancer progression. Nat. Genet. 2001, 29, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Mulder, K.M.; Morris, S.L. Activation of p21ras by transforming growth factor beta in epithelial cells. J. Biol. Chem. 1992, 267, 5029–5031. [Google Scholar] [PubMed]
- Yan, Z.; Winawer, S.; Friedman, E. Two different signal transduction pathways can be activated by transforming growth factor-beta-1 in epithelial cells. J. Biol. Chem. 1994, 269, 13231–13237. [Google Scholar] [PubMed]
- Lee, M.K.; Pardoux, C.; Hall, M.C.; Lee, P.S.; Warburton, D.; Qing, J.; Smith, S.M.; Derynck, R. TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007, 26, 3957–3967. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial-mesenchymal transitions in development and pathologies. Curr. Opin. Cell Biol. 2003, 15, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Dedhar, S.; Kalluri, R.; Thompson, E.W. The epithelial-mesenchymal transition: New insights in signaling, development, and disease. J. Cell Biol. 2006, 172, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [PubMed]
- Jaffe, A.B.; Hall, A. Rho GTPases: Biochemistry and biology. Annu. Rev. Cell Dev. Biol. 2005, 21, 247–269. [Google Scholar] [CrossRef] [PubMed]
- Wilkes, M.C.; Mitchell, H.; Penheiter, S.G.; Doré, J.J.; Suzuki, K.; Edens, M.; Sharma, D.K.; Pagano, R.E.; Leof, E.B. Transforming growth factor-beta activation of phosphatidylinositol 3-kinase is independent of Smad2 and Smad3 and regulates fibroblast responses via p21-activated kinase-2. Cancer Res. 2005, 65, 10431–10440. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Katuri, V.; Dillner, A.; Mishra, B.; Deng, C.X.; Mishra, L. Disruption of transforming growth factor-β signaling in ELF β-spectrin-deficient mice. Science 2003, 299, 574–577. [Google Scholar] [CrossRef] [PubMed]
- Mishra, L.; Derynck, R.; Mishra, B. Transforming growth factor-β signaling in stem cells and cancer. Science 2005, 310, 68–71. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Katuri, V.; Srinivasan, R.; Fogt, F.; Redman, R.; Anand, G.; Said, A.; Fishbein, T.; Zasloff, M.; Reddy, E.P.; et al. Transforming growth factor-beta suppresses nonmetastatic colon cancer through Smad4 and adaptor protein ELF at an early stage of tumorigenesis. Cancer Res. 2005, 65, 4228–4237. [Google Scholar] [CrossRef] [PubMed]
- Moses, H.L.; Roberts, A.B.; Derynck, R. The discovery and early days of TGF-β: A historical perspective. Cold Spring Harb. Perspect. Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.; Novitskiy, S.; Moses, H.L. The roles of TGFβ in the tumour microenvironment. Nat. Rev. Cancer 2013, 13, 788–799. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, T.; Sunamura, M.; Horii, A. Molecular mechanisms of pancreatic carcinogenesis. Cancer Sci. 2006, 97, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Duda, D.G.; Sunamura, M.; Lefter, L.P.; Furukawa, T.; Yokoyama, T.; Yatsuoka, T.; Abe, T.; Inoue, H.; Motoi, F.; Egawa, S.; et al. Restoration of SMAD4 by gene therapy reverses the invasive phenotype in pancreatic adenocarcinoma cells. Oncogene 2003, 22, 6857–6864. [Google Scholar] [CrossRef] [PubMed]
- Evan, G.I.; Vousden, K.H. Proliferation, cell cycle and apoptosis in cancer. Nature 2001, 411, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Lecanda, J.; Ganapathy, V.; D’Aquino-Ardalan, C.; Evans, B.; Cadacio, C.; Ayala, A.; Gold, L.I. TGFβ prevents proteasomal degradation of the cyclin-dependent kinase inhibitor p27kip1 for cell cycle arrest. Cell Cycle 2009, 8, 742–756. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, C.; Bass, B.L. Role of transforming growth factor-beta in growth and injury response of the pancreatic duct epithelium in vitro. J. Gastrointest. Surg. 1999, 3, 178–184. [Google Scholar] [CrossRef]
- Tachibana, I.; Imoto, M.; Adjei, P.N.; Gores, G.J.; Subramaniam, M.; Spelsberg, T.C.; Urrutia, R. Overexpression of the TGFbeta-regulated zinc finger encoding gene, TIEG, induces apoptosis in pancreatic epithelial cells. J. Clin. Investig. 1997, 99, 2365–2374. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.; Kleeff, J.; Lopez, M.E.; Bockman, I.; Massaqué, J.; Korc, M. Transfection of the type I TGF-beta receptor restores TGF-β responsiveness in pancreatic cancer. Int. J. Cancer 1998, 78, 255–260. [Google Scholar] [CrossRef]
- Moustakas, A.; Heldin, C.H. Non-Smad TGF-beta signals. J. Cell Sci. 2005, 118, 3573–3584. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Feng, X.H. Abrogation of transforming growth factor-β signaling in pancreatic cancer. World J. Surg. 2005, 29, 312–316. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Wu, W.; Huang, C.; Cen, G.; Jiang, T.; Cao, J.; Huang, K.; Qiu, Z. SMAD4 and its role in pancreatic cancer. Tumour Biol. 2015, 36, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Mancera, P.A.; Guerra, C.; Barbacid, M.; Tuveson, D.A. What we have learned about pancreatic cancer from mouse models. Gastroenterology 2012, 142, 1079–1092. [Google Scholar] [CrossRef] [PubMed]
- Bardeesy, N.; Cheng, K.H.; Berger, J.H.; Chu, G.C.; Pahler, J.; Olson, P.; Hezel, A.F.; Horner, J.; Lauwers, G.Y.; Hanahan, D.; et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev. 2006, 20, 3130–3146. [Google Scholar] [CrossRef] [PubMed]
- Ijichi, H.; Chytil, A.; Gorska, A.E.; Aakre, M.E.; Fujitani, Y.; Fujitani, S.; Wright, C.V.; Moses, H.L. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-beta signaling in cooperation with active Kras expression. Genes Dev. 2006, 20, 3147–3160. [Google Scholar] [CrossRef] [PubMed]
- Izeradjene, K.; Combs, C.; Best, M.; Gopinathan, A.; Wagner, A.; Grady, W.M.; Deng, C.X.; Hruban, R.H.; Adsay, N.V.; Tuveson, D.A.; et al. KrasG12D and Smad4/Dpc4 haploinsufficiency cooperate to induce mucinous cystic neoplasms and invasive adenocarcinoma of the pancreas. Cancer Cell. 2007, 11, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Leung, L.; Radulovich, N.; Zhu, C.Q.; Wang, D.; To, C.; Ibrahimov, E.; Tsao, M.S. Loss of canonical Smad4 signaling promotes KRAS driven malignant transformation of human pancreatic duct epithelial cells and metastasis. PLoS ONE 2013, 8, e84366. [Google Scholar] [CrossRef] [PubMed]
- Krantz, S.B.; Shields, M.A.; Dangi-Garimella, S.; Munshi, H.G.; Bentrem, D.J. Contribution of epithelial-to-mesenchymal transition and cancer stem cells to pancreatic cancer progression. J. Surg. Res. 2012, 173, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Ikushima, H.; Miyazono, K. TGFbeta signalling: A complex web in cancer progression. Nat. Rev. Cancer 2010, 10, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Rev. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Meulmeester, E.; Ten Dijke, P. The dynamic roles of TGF-β in cancer. J. Pathol. 2011, 223, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Vincent, T.; Neve, E.P.; Johnson, J.R.; Kukalev, A.; Rojo, F.; Albanell, J.; Pietras, K.; Virtanen, I.; Philipson, L.; Leopold, P.L.; et al. A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-β mediated epithelial-mesenchymal transition. Nat. Cell Biol. 2009, 11, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Bracken, C.P.; Gregory, P.A.; Kolesnikoff, N.; Bert, A.G.; Wang, J.; Shannon, M.F.; Goodall, G.J. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. 2008, 68, 7846–7854. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Akhurst, R.J. Differentiation plasticity regulated by TGF-β family proteins in development and disease. Nat. Cell Biol. 2007, 9, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.; Robinson, M.; Smith, E.; Huntley, S.; Prime, S.; Paterson, I. Induction of an epithelial to mesenchymal transition in human immortal and malignant keratinocytes by TGF-β1 involves MAPK, Smad and AP-1 signalling pathways. J. Cell. Biochem. 2005, 95, 918–931. [Google Scholar] [CrossRef] [PubMed]
- Pardali, E.; ten Dijke, P. Transforming growth factor-beta signaling and tumor angiogenesis. Front. Biosci. 2009, 14, 4848–4861. [Google Scholar] [CrossRef]
- Padua, D.; Massagué, J. Roles of TGFβ in metastasis. Cell Res. 2009, 19, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Hagedorn, H.G.; Bachmeier, B.E.; Nerlich, A.G. Synthesis and degradation of basement membranes and extracellular matrix and their regulation by TGF-β in invasive carcinomas (Review). Int. J. Oncol. 2001, 18, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Elsner, T.; Botella, L.M.; Velasco, B.; Corbí, A.; Attisano, L.; Bernabéu, C. Synergistic cooperation between hypoxia and transforming growth factor-beta pathways on human vascular endothelial growth factor gene expression. J. Biol. Chem. 2001, 276, 38527–38535. [Google Scholar] [CrossRef] [PubMed]
- Achyut, B.R.; Yang, L. Transforming growth factor-β in the gastrointestinal and hepatic tumor microenvironment. Gastroenterology 2011, 141, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, A.A.; Vimalachandran, D.; Thompson, C.C.; Jenkins, R.E.; Nedjadi, T.; Shekouh, A.; Campbell, F.; Dodson, A.; Prime, W.; Crnogorac-Jurcevic, T.; et al. The expression of S100A8 in pancreatic cancer-associated monocytes is associated with the Smad4 status of pancreatic cancer cells. Proteomics 2007, 11, 1929–1940. [Google Scholar] [CrossRef] [PubMed]
- Ang, C.W.; Nedjadi, T.; Sheikh, A.A.; Tweedle, E.M.; Tonack, S.; Honap, S.; Jenkins, R.E.; Park, B.K.; Schwarte-Waldhoff, I.; Khattak, I.; et al. Smad4 loss is associated with fewer S100A8-positive monocytes in colorectal tumors and attenuated response to S100A8 in colorectal and pancreatic cancer cells. Carcinogenesis 2010, 9, 1541–1551. [Google Scholar] [CrossRef] [PubMed]
- Basso, D.; Greco, E.; Padoan, A.; Fogar, P.; Scorzeto, M.; Fadi, E.; Bozzato, D.; Moz, S.; Navaglia, F.; Zambon, C.F.; et al. Altered intracellular calcium fluxes in pancreatic cancer induced diabetes mellitus: Relevance of the S100A8 N-terminal peptide (NT-S100A8). J. Cell. Physiol. 2011, 226, 456–468. [Google Scholar] [CrossRef] [PubMed]
- Gurumurthy, S.; Bardeesy, N. Uncapping NF-κB activity in pancreatic cancer. EMBO J. 2011, 30, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Hagopian, M.M.; Brekken, R.A. Stromal TGFβR2 signaling: A gateway to progression for pancreatic cancer. Mol. Cell. Oncol. 2014, 2, e975606. [Google Scholar] [CrossRef] [PubMed]
- Danen, E.H.; Yamada, K.M. Fibronectin, integrins, and growth control. J. Cell. Physiol. 2001, 189, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Eliceiri, B.P. Integrin and growth factor receptor crosstalk. Circ. Res. 2001, 89, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Weis, S.M.; Cheresh, D.A. Tumor angiogenesis: Molecular pathways and therapeutic targets. Nat. Med. 2011, 17, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Keller, E.T.; Garfield, D.H.; Shen, K.; Wang, J. Stromal cells in tumor microenvironment and breast cancer. Cancer Metastasis Rev. 2013, 32, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Kuang, P.P.; Joyce-Brady, M.; Zhang, X.H.; Jean, J.C.; Goldstein, R.H. Fibulin-5 gene expression in human lung fibroblasts is regulated by TGF-β and phosphatidylinositol 3-kinase activity. Am. J. Physiol. Cell Physiol. 2006, 291, C1412–C1421. [Google Scholar] [CrossRef] [PubMed]
- Ignotz, R.A.; Massagué, J. Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J. Biol. Chem. 1986, 261, 4337–4345. [Google Scholar] [PubMed]
- Kajdaniuk, D.; Marek, B.; Borgiel-Marek, H.; Kos-Kudła, B. Vascular endothelial growth factor (VEGF)—Part 1: In physiology and pathophysiology. Endokrynol. Polska 2011, 62, 444–455. [Google Scholar]
- Kajdaniuk, D.; Marek, B.; Foltyn, W.; Kos-Kudła, B. Vascular endothelial growth factor (VEGF)—Part 2: In endocrinology and oncology. Endokrynol. Polska 2011, 62, 456–464. [Google Scholar]
- Kuwahara, F.; Kai, H.; Tokuda, K.; Kai, M.; Takeshita, A.; Egashira, K.; Imaizumi, T. Transforming growth factor-beta function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats. Circulation 2002, 106, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Vincent, D.F.; Gout, J.; Chuvin, N.; Arfi, V.; Pommier, R.M.; Bertolino, P.; Jonckheere, N.; Ripoche, D.; Kaniewski, B.; Martel, S.; et al. Tif1 suppresses murine pancreatic tumoral transformation by a Smad4-independent pathway. Am. J. Pathol. 2012, 180, 2214–2221. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Zacchigna, L.; Cordenonsi, M.; Soligo, S.; Adorno, M.; Rugge, M.; Piccolo, S. Germ-layer specification and control of cell growth by Ectodermin, a Smad4 ubiquitin ligase. Cell 2005, 121, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Mamidi, A.; Cordenonsi, M.; Montagner, M.; Zacchigna, L.; Adorno, M.; Martello, G.; Stinchfield, M.J.; Soligo, S.; Morsut, L.; et al. FAM/USP9x, a deubiquitinating enzyme essential for TGFβ signaling, controls Smad4 monoubiquitination. Cell 2009, 136, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Ligr, M.; Wu, X.; Daniels, G.; Zhang, D.; Wang, H.; Hajdu, C.; Wang, J.; Pan, R.; Pei, Z.; Zhang, L.; et al. Imbalanced expression of Tif1γ inhibits pancreatic ductal epithelial cell growth. Am. J. Cancer Res. 2014, 4, 196–210. [Google Scholar] [PubMed]
- He, W.; Dorn, D.C.; Erdjument-Bromage, H.; Tempst, P.; Moore, M.A.; Massague, J. Hematopoiesis controlled by distinct TIF1gamma and Smad4 branches of the TGFbeta pathway. Cell 2006, 125, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Vincent, D.F.; Yan, K.-P.; Treilleux, I.; Gay, F.; Arfi, V.; Kaniewski, B.; Marie, J.C.; Lepinasse, F.; Martel, S.; Goddard-Leon, S.; et al. Inactivation of TIF1γ cooperates with Kras to induce cystic tumors of the pancreas. PLoS Genet. 2009, 5, e1000575. [Google Scholar] [CrossRef]
- Massagué, J. TGFβ in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.; Serrano, O.K.; Wolfgang, C.L.; Parmigiani, G.; Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Eshleman, J.R.; et al. SMAD4 gene mutations are associated with poor prognosis in pancreatic cancer. Clin. Cancer Res. 2009, 15, 4674–4679. [Google Scholar] [CrossRef] [PubMed]
- Iacobuzio-Donahue, C.A.; Fu, B.; Yachida, S.; Luo, M.; Abe, H.; Henderson, C.M.; Vilardell, F.; Wang, Z.; Keller, J.W.; Banerjee, P.; et al. DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer. J. Clin. Oncol. 2009, 27, 1806–1813. [Google Scholar] [CrossRef] [PubMed]
- Maitra, A.; Molberg, K.; Albores-Saavedra, J.; Lindberg, G. Loss of Dpc4 expression in colonic adenocarcinomas correlates with the presence of metastatic disease. Am. J. Pathol. 2000, 157, 1105–1111. [Google Scholar] [CrossRef]
- Zhao, J.; Liang, Y.; Yin, Q.; Liu, S.; Wang, Q.; Tang, Y.; Cao, C. Clinical and prognostic significance of serum transforming growth factor-beta1 levels in patients with pancreatic ductal adenocarcinoma. Braz. J. Med. Biol. Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Tascilar, M.; Skinner, H.G.; Rosty, C.; Sohn, T.; Wilentz, R.E.; Offerhaus, G.J.; Adsay, V.; Abrams, R.A.; Cameron, J.L.; Kern, S.E.; et al. The Smad4 protein and prognosis of pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2001, 12, 4115–4121. [Google Scholar]
- Du, Y.; Zhou, X.; Huang, Z.; Qiu, T.; Wang, J.; Zhu, W.; Wang, T.; Liu, P. Meta-Analysis of the Prognostic Value of Smad4 Immunohistochemistry in Various Cancers. PLoS ONE 2014, 10, e110182. [Google Scholar] [CrossRef] [PubMed]
- Xing, S.; Yang, H.; Liu, J.; Zheng, X.; Feng, J.; Li, X.; Li, W. Prognostic Value of SMAD4 in Pancreatic Cancer: A Meta-Analysis. Transl. Oncol. 2016, 1, 1–7. [Google Scholar]
- Friess, H.; Yamanaka, Y.; Büchler, M.; Berger, H.G.; Kobrin, M.S.; Baldwin, R.L.; Korc, M. Enhanced expression of the type II transforming growth factor β receptor in human pancreatic cancer cells without alteration of type III receptor expression. Cancer Res. 1993, 53, 2704–2707. [Google Scholar] [PubMed]
- Friess, H.; Yamanaka, Y.; Büchler, M.; Ebert, M.; Beger, H.G.; Gold, L.I.; Korc, M. Enhanced expression of transforming growth factor beta isoforms in pancreatic cancer correlates with decreased survival. Gastroenterology 1993, 105, 1846–1856. [Google Scholar] [CrossRef]
- Inman, G.J.; Nicolás, F.J.; Hill, C.S. Nucleocytoplasmic shuttling of Smads 2, 3, and 4 permits sensing of TGF-β receptor activity. Mol. Cell 2002, 10, 283–294. [Google Scholar] [CrossRef]
- Nicolás, F.J.; Hill, C.S. Attenuation of the TGF-beta-Smad signaling pathway in pancreatic tumor cells confers resistance to TGF-beta-induced growth arrest. Oncogene 2003, 22, 3698–3711. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Hu, W.; Yue, F.; Zou, J.; Li, W.; Chen, Q.; Yao, Q.; Sun, W.; Liu, L. Transforming Growth Factor TGFβ Increases Levels of Microtubule-Associated Protein MAP1S and Autophagy Flux in Pancreatic Ductal Adenocarcinomas. PLoS ONE 2015, 10, e0143150. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, S.; Bradshaw, A.-D.; Gera, S.; Dewan, M.Z.; Xu, R. The TGF-β/Smad4 Signaling Pathway in Pancreatic Carcinogenesis and Its Clinical Significance. J. Clin. Med. 2017, 6, 5. https://doi.org/10.3390/jcm6010005
Ahmed S, Bradshaw A-D, Gera S, Dewan MZ, Xu R. The TGF-β/Smad4 Signaling Pathway in Pancreatic Carcinogenesis and Its Clinical Significance. Journal of Clinical Medicine. 2017; 6(1):5. https://doi.org/10.3390/jcm6010005
Chicago/Turabian StyleAhmed, Sunjida, Azore-Dee Bradshaw, Shweta Gera, M. Zahidunnabi Dewan, and Ruliang Xu. 2017. "The TGF-β/Smad4 Signaling Pathway in Pancreatic Carcinogenesis and Its Clinical Significance" Journal of Clinical Medicine 6, no. 1: 5. https://doi.org/10.3390/jcm6010005