
The Application of Genetic Risk Scores in Age-Related Macular Degeneration: A Review

 ,
,
Abstract
:1. Introduction
2. Experimental Section
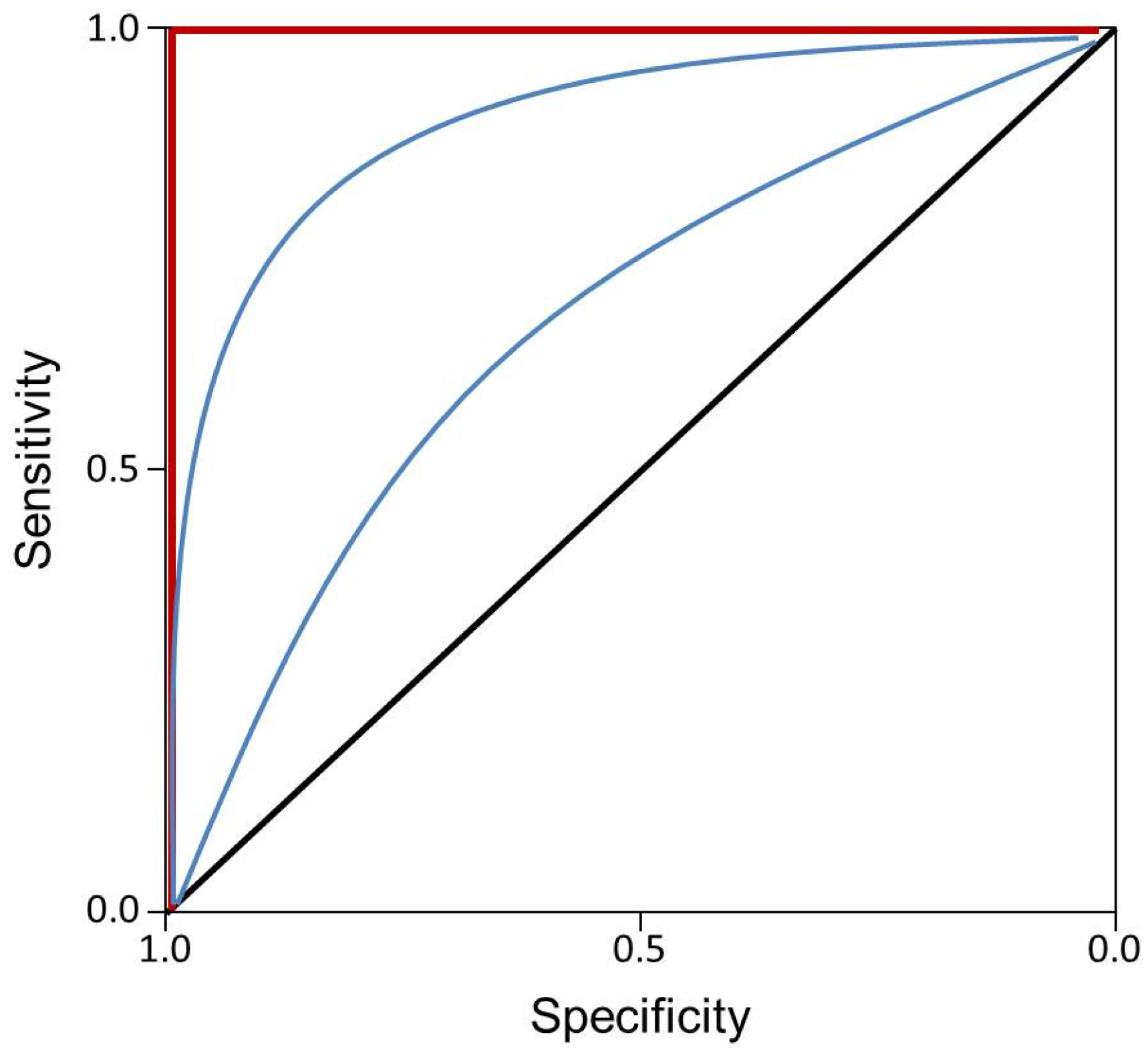
3. Discussion
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Smith, W.; Assink, J.; Klein, R.; Mitchell, P.; Klaver, C.C.; Klein, B.E.; Hofman, A.; Jensen, S.; Wang, J.J.; de Jong, P.T. Risk factors for age-related macular degeneration: Pooled findings from three continents. Ophthalmology 2001, 108, 697–704. [Google Scholar] [CrossRef]
- Global Data on Visual Impairments 2010. Available online: http://www.who.int/blindness/GLOBALDATAFINALforweb.pdf (accessed on 7 March 2014).
- Age-related Macular Degeneration (AMD). Available online: http://www.nei.nih.gov/eyedata/amd.asp#1 (accessed on 7 March 2014).
- Friedman, D.S.; O’Colmain, B.J.; Munoz, B.; Tomany, S.C.; McCarty, C.; de Jong, P.T.; Nemesure, B.; Mitchell, P.; Kempen, J. Prevalence of age-related macular degeneration in the United States. Arch. Ophthalmol. 2004, 122, 564–572. [Google Scholar] [PubMed]
- Brown, D.M.; Regillo, C.D. Anti-VEGF agents in the treatment of neovascular age-related macular degeneration: Applying clinical trial results to the treatment of everyday patients. Am. J. Ophthalmol. 2007, 144, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Cukras, C.; Agron, E.; Klein, M.L.; Ferris, F.L., III; Chew, E.Y.; Gensler, G.; Wong, W.T. Natural history of drusenoid pigment epithelial detachment in age-related macular degeneration: Age-Related Eye Disease Study Report No. 28. Ophthalmology 2010, 117, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Janssens, A.C.; Moonesinghe, R.; Yang, Q.; Steyerberg, E.W.; van Duijn, C.M.; Khoury, M.J. The impact of genotype frequencies on the clinical validity of genomic profiling for predicting common chronic diseases. Genet. Med. 2007, 9, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Ratnapriya, R.; Chew, E.Y. Age-related macular degeneration-clinical review and genetics update. Clin. Genet. 2013, 84, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Klein, B.E.; Klein, R. Lifestyle exposures and eye diseases in adults. Am. J. Ophthalmol. 2007, 144, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Tikellis, G.; Robman, L.D.; Dimitrov, P.; Nicolas, C.; McCarty, C.A.; Guymer, R.H. Characteristics of progression of early age-related macular degeneration: The cardiovascular health and age-related maculopathy study. Eye 2007, 21, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Ingelman-Sundberg, M. Personalized medicine into the next generation. J. Intern. Med. 2014, 277, 152–154. [Google Scholar] [CrossRef] [PubMed]
- Seddon, J.M.; Cote, J.; Page, W.F.; Aggen, S.H.; Neale, M.C. The US twin study of age-related macular degeneration: Relative roles of genetic and environmental influences. Arch. Ophthalmol. 2005, 123, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Hammond, C.J.; Webster, A.R.; Snieder, H.; Bird, A.C.; Gilbert, C.E.; Spector, T.D. Genetic influence on early age-related maculopathy: A twin study. Ophthalmology 2002, 109, 730–736. [Google Scholar] [CrossRef]
- Klaver, C.C.; Wolfs, R.C.; Assink, J.J.; van Duijn, C.M.; Hofman, A.; de Jong, P.T. Genetic risk of age-related maculopathy. Population-based familial aggregation study. Arch. Ophthalmol. 1998, 116, 1646–1651. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.L.; Mauldin, W.M.; Stoumbos, V.D. Heredity and age-related macular degeneration. Observations in monozygotic twins. Arch. Ophthalmol. 1994, 112, 932–937. [Google Scholar] [CrossRef] [PubMed]
- Meyers, S.M. A twin study on age-related macular degeneration. Trans. Am. Ophthalmol. Soc. 1994, 92, 775–843. [Google Scholar] [CrossRef]
- Heiba, I.M.; Elston, R.C.; Klein, B.E.; Klein, R. Sibling correlations and segregation analysis of age-related maculopathy: The Beaver Dam Eye Study. Genet. Epidemiol. 1994, 11, 51–67. [Google Scholar] [CrossRef] [PubMed]
- Seddon, J.M.; Cote, J.; Davis, N.; Rosner, B. Progression of age-related macular degeneration: Association with body mass index, waist circumference, and waist-hip ratio. Arch. Ophthalmol. 2003, 121, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Cooke Bailey, J.N.; Pericak-Vance, M.A.; Haines, J.L. Genome-Wide Association Studies: Getting to Pathogenesis, the Role of Inflammation/Complement in AMD. Cold Spring Harb. Perspect. Med. 2014. [Google Scholar] [CrossRef] [PubMed]
- Fisher, S.A.; Abecasis, G.R.; Yashar, B.M.; Zareparsi, S.; Swaroop, A.; Iyengar, S.K.; Klein, B.E.; Klein, R.; Lee, K.E.; Majewski, J.; et al. Meta-analysis of genome scans of age-related macular degeneration. Hum. Mol. Genet. 2005, 14, 2257–2264. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, S.K.; Song, D.; Klein, B.E.; Klein, R.; Schick, J.H.; Humphrey, J.; Millard, C.; Liptak, R.; Russo, K.; Jun, G.; et al. Dissection of genomewide-scan data in extended families reveals a major locus and oligogenic susceptibility for age-related macular degeneration. Am. J. Hum. Genet. 2004, 74, 20–39. [Google Scholar] [CrossRef] [PubMed]
- Majewski, J.; Schultz, D.W.; Weleber, R.G.; Schain, M.B.; Edwards, A.O.; Matise, T.C.; Acott, T.S.; Ott, J.; Klein, M.L. Age-related macular degeneration—A genome scan in extended families. Am. J. Hum. Genet. 2003, 73, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Schick, J.H.; Iyengar, S.K.; Klein, B.E.; Klein, R.; Reading, K.; Liptak, R.; Millard, C.; Lee, K.E.; Tomany, S.C.; Moore, E.L.; et al. A whole-genome screen of a quantitative trait of age-related maculopathy in sibships from the Beaver Dam Eye Study. Am. J. Hum. Genet. 2003, 72, 1412–1424. [Google Scholar] [CrossRef] [PubMed]
- Seddon, J.M.; Santangelo, S.L.; Book, K.; Chong, S.; Cote, J. A genomewide scan for age-related macular degeneration provides evidence for linkage to several chromosomal regions. Am. J. Hum. Genet. 2003, 73, 780–790. [Google Scholar] [CrossRef] [PubMed]
- Weeks, D.E.; Conley, Y.P.; Mah, T.S.; Paul, T.O.; Morse, L.; Ngo-Chang, J.; Dailey, J.P.; Ferrell, R.E.; Gorin, M.B. A full genome scan for age-related maculopathy. Hum. Mol. Genet. 2000, 9, 1329–1349. [Google Scholar] [CrossRef] [PubMed]
- Weeks, D.E.; Conley, Y.P.; Tsai, H.J.; Mah, T.S.; Rosenfeld, P.J.; Paul, T.O.; Eller, A.W.; Morse, L.S.; Dailey, J.P.; Ferrell, R.E.; et al. Age-related maculopathy: An expanded genome-wide scan with evidence of susceptibility loci within the 1q31 and 17q25 regions. Am. J. Ophthalmol. 2001, 132, 682–692. [Google Scholar] [CrossRef]
- Weeks, D.E.; Conley, Y.P.; Tsai, H.J.; Mah, T.S.; Schmidt, S.; Postel, E.A.; Agarwal, A.; Haines, J.L.; Pericak-Vance, M.A.; Rosenfeld, P.J.; et al. Age-related maculopathy: A genomewide scan with continued evidence of susceptibility loci within the 1q31, 10q26, and 17q25 regions. Am. J. Hum. Genet. 2004, 75, 174–189. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, L.G.; Igl, W.; Bailey, J.N.; Grassmann, F.; Sengupta, S.; Bragg-Gresham, J.L.; Burdon, K.P.; Hebbring, S.J.; Wen, C.; Gorski, M.; et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat.Genet. 2016, 48, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.M. Genetic association studies: Design, analysis and interpretation. Brief. Bioinform. 2002, 3, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Sahebi, L.; Dastgiri, S.; Ansarin, K.; Sahebi, R.; Mohammadi, S.A. Study Designs in Genetic Epidemiology. ISRN Genet. 2013, 2013. [Google Scholar] [CrossRef]
- Jakobsdottir, J.; Gorin, M.B.; Conley, Y.P.; Ferrell, R.E.; Weeks, D.E. Interpretation of genetic association studies: Markers with replicated highly significant odds ratios may be poor classifiers. PLoS. Genet. 2009, 5, e1000337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, K.L.; Olson, L.M.; Schnetz-Boutaud, N.; Gallins, P.; Agarwal, A.; Iannaccone, A.; Kritchevsky, S.B.; Garcia, M.; Nalls, M.A.; Newman, A.B.; et al. Using genetic variation and environmental risk factor data to identify individuals at high risk for age-related macular degeneration. PLoS ONE 2011, 6, e17784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, G.S.; Maguire, M.G. Development of a risk score for geographic atrophy in complications of the age-related macular degeneration prevention trial. Ophthalmology 2011, 118, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.J.; Mitchell, P.; Klein, R.; Klein, B.E.; Chang, M.L.; Gensler, G.; Taylor, A. A risk score for the prediction of advanced age-related macular degeneration: Development and validation in 2 prospective cohorts. Ophthalmology 2014, 121, 1421–1427. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, L.G.; Chen, W.; Schu, M.; Yaspan, B.L.; Yu, Y.; Thorleifsson, G.; Zack, D.J.; Arakawa, S.; Cipriani, V.; Ripke, S.; et al. Seven new loci associated with age-related macular degeneration. Nat. Genet. 2013, 45, 433–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, M.L.; Francis, P.J.; Ferris, F.L., III; Hamon, S.C.; Clemons, T.E. Risk assessment model for development of advanced age-related macular degeneration. Arch. Ophthalmol. 2011, 129, 1543–1550. [Google Scholar] [CrossRef] [PubMed]
- Seddon, J.M.; Reynolds, R.; Yu, Y.; Daly, M.J.; Rosner, B. Risk models for progression to advanced age-related macular degeneration using demographic, environmental, genetic, and ocular factors. Ophthalmology 2011, 118, 2203–2211. [Google Scholar] [CrossRef] [PubMed]
- Seddon, J.M.; Silver, R.E.; Kwong, M.; Rosner, B. Risk Prediction for Progression of Macular Degeneration: 10 Common and Rare Genetic Variants, Demographic, Environmental, and Macular Covariates. Investig. Ophthalmol. Vis. Sci. 2015, 56, 2192–2202. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zeng, J.; Zhao, C.; Wang, K.; Trood, E.; Buehler, J.; Weed, M.; Kasuga, D.; Bernstein, P.S.; Hughes, G.; et al. Assessing susceptibility to age-related macular degeneration with genetic markers and environmental factors. Arch. Ophthalmol. 2011, 129, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Perlee, L.T.; Bansal, A.T.; Gehrs, K.; Heier, J.S.; Csaky, K.; Allikmets, R.; Oeth, P.; Paladino, T.; Farkas, D.H.; Rawlings, P.L.; et al. Inclusion of genotype with fundus phenotype improves accuracy of predicting choroidal neovascularization and geographic atrophy. Ophthalmology 2013, 120, 1880–1892. [Google Scholar] [CrossRef] [PubMed]
- Hageman, G.S.; Gehrs, K.; Lejnine, S.; Bansal, A.T.; DeAngelis, M.M.; Guymer, R.H.; Baird, P.N.; Allikmets, R.; Deciu, C.; Oeth, P.; et al. Clinical validation of a genetic model to estimate the risk of developing choroidal neovascular age-related macular degeneration. Hum. Genom. 2011, 5, 420–440. [Google Scholar] [CrossRef]
- Grassmann, F.; Fritsche, L.G.; Keilhauer, C.N.; Heid, I.M.; Weber, B.H. Modelling the genetic risk in age-related macular degeneration. PLoS ONE 2012, 7, e37979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buitendijk, G.H.; Rochtchina, E.; Myers, C.; van Duijn, C.M.; Lee, K.E.; Klein, B.E.; Meuer, S.M.; de Jong, P.T.; Holliday, E.G.; Tan, A.G.; et al. Prediction of age-related macular degeneration in the general population: The three sontinent AMD consortium. Ophthalmology 2013, 120, 2644–2655. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.W. Age-related macular degeneration revisited—Piecing the puzzle: The LXIX Edward Jackson memorial lecture. Am. J. Ophthalmol. 2013, 155, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Seddon, J.M.; Reynolds, R.; Yu, Y.; Rosner, B. Validation of a prediction algorithm for progression to advanced macular degeneration subtypes. JAMA Ophthalmol. 2013, 131, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, J.D.; Cooke Bailey, J.N.; D’Aoust, L.; Cade, W.; Ayala-Haedo, J.; Fuzzell, D.; Laux, R.; Adams, L.D.; Reinhart-Mercer, L.; Caywood, L.; et al. Rare complement factor H variant associated with age-related macular degeneration in the Amish. Investig. Ophthalmol. Vis. Sci. 2014, 55, 4455–4460. [Google Scholar] [CrossRef] [PubMed]
- Restrepo, N.A.; Spencer, K.L.; Goodloe, R.; Garrett, T.A.; Heiss, G.; Buzkova, P.; Jorgensen, N.; Jensen, R.A.; Matise, T.C.; Hindorff, L.A.; et al. Genetic Determinants of Age-Related Macular Degeneration in Diverse Populations From the PAGE Study. Investig. Ophthalmol. Vis. Sci. 2014, 55, 6839–6850. [Google Scholar] [CrossRef] [PubMed]
- Spencer, K.L.; Glenn, K.; Brown-Gentry, K.; Haines, J.L.; Crawford, D.C. Population differences in genetic risk for age-related macular degeneration and implications for genetic testing. Arch. Ophthalmol. 2012, 130, 116–117. [Google Scholar] [CrossRef] [PubMed]
- Awh, C.C.; Lane, A.M.; Hawken, S.; Zanke, B.; Kim, I.K. CFH and ARMS2 genetic polymorphisms predict response to antioxidants and zinc in patients with age-related macular degeneration. Ophthalmology 2013, 120, 2317–2323. [Google Scholar] [CrossRef] [PubMed]
- Awh, C.C.; Hawken, S.; Zanke, B.W. Treatment Response to Antioxidants and Zinc Based on CFH and ARMS2 Genetic Risk Allele Number in the Age-Related Eye Disease Study. Ophthalmology 2015, 122, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Awh, C.C.; Zanke, B.W. No clinically significant association between CFH and ARMS2 genotypes and response to nutritional supplements: AREDS report number 38. Ophthalmology 2014, 121, 2173–2180. [Google Scholar]
- Chew, E.Y.; Klein, M.L.; Clemons, T.E.; Agron, E.; Ratnapriya, R.; Edwards, A.O.; Fritsche, L.G.; Swaroop, A.; Abecasis, G.R. No Clinically Significant Association between CFH and ARMS2 Genotypes and Response to Nutritional Supplements: AREDS Report Number 38. Ophthalmology 2014, 121, 2173–2180. [Google Scholar] [CrossRef] [PubMed]
- Smailhodzic, D.; Muether, P.S.; Chen, J.; Kwestro, A.; Zhang, A.Y.; Omar, A.; van de Ven, J.P.; Keunen, J.E.; Kirchhof, B.; Hoyng, C.B.; et al. Cumulative effect of risk alleles in CFH, ARMS2, and VEGFA on the response to ranibizumab treatment in age-related macular degeneration. Ophthalmology 2012, 119, 2304–2311. [Google Scholar] [CrossRef] [PubMed]
- Chew, E.Y.; Klein, M.L.; Clemons, T.E.; Agron, E.; Abecasis, G.R. Genetic testing in persons with age-related macular degeneration and the use of the AREDS supplements: To test or not to test? Ophthalmology 2015, 122, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.; McKeigue, P.M.; Skerka, C.; Hayward, C.; Rudan, I.; Vitart, V.; Polasek, O.; Armbrecht, A.M.; Yates, J.R.; Vatavuk, Z.; et al. Genetic influences on plasma CFH and CFHR1 concentrations and their role in susceptibility to age-related macular degeneration. Hum. Mol. Genet. 2013, 22, 4857–4869. [Google Scholar] [CrossRef] [PubMed]
- Hageman, G.S.; Luthert, P.J.; Victor Chong, N.H.; Johnson, L.V.; Anderson, D.H.; Mullins, R.F. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degeneration. Prog. Retin. Eye Res. 2001, 20, 705–732. [Google Scholar] [CrossRef]
- Spencer, K.L.; Hauser, M.A.; Olson, L.M.; Schmidt, S.; Scott, W.K.; Gallins, P.; Agarwal, A.; Postel, E.A.; Pericak-Vance, M.A.; Haines, J.L. Deletion of CFHR3 and CFHR1 genes in age-related macular degeneration. Hum. Mol. Genet. 2008, 17, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Williamson, J.F.; McLure, C.A.; Guymer, R.H.; Baird, P.N.; Millman, J.; Cantsilieris, S.; Dawkins, R.L. Almost total protection from age-related macular degeneration by haplotypes of the Regulators of Complement Activation. Genomics 2011, 98, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Helgason, H.; Sulem, P.; Duvvari, M.R.; Luo, H.; Thorleifsson, G.; Stefansson, H.; Jonsdottir, I.; Masson, G.; Gudbjartsson, D.F.; Walters, G.B.; et al. A rare nonsynonymous sequence variant in C3 is associated with high risk of age-related macular degeneration. Nat. Genet. 2013, 45, 1371–1374. [Google Scholar] [CrossRef] [PubMed]
- Seddon, J.M.; Yu, Y.; Miller, E.C.; Reynolds, R.; Tan, P.L.; Gowrisankar, S.; Goldstein, J.I.; Triebwasser, M.; Anderson, H.E.; Zerbib, J.; et al. Rare variants in CFI, C3 and C9 are associated with high risk of advanced age-related macular degeneration. Nat. Genet. 2013, 45, 1366–1370. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, S.; Iartchouk, O.; Chin, K.; Tan, P.L.; Tai, A.K.; Ripke, S.; Gowrisankar, S.; Vemuri, S.; Montgomery, K.; Yu, Y.; et al. A rare penetrant mutation in CFH confers high risk of age-related macular degeneration. Nat. Genet. 2011, 43, 1232–1236. [Google Scholar] [CrossRef] [PubMed]
- Van de Ven, J.P.; Nilsson, S.C.; Tan, P.L.; Buitendijk, G.H.; Ristau, T.; Mohlin, F.C.; Nabuurs, S.B.; Schoenmaker-Koller, F.E.; Smailhodzic, D.; Campochiaro, P.A.; et al. A functional variant in the CFI gene confers a high risk of age-related macular degeneration. Nat. Genet. 2013, 45, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Larson, D.E.; Wang, C.; Koboldt, D.C.; Sergeev, Y.V.; Fulton, R.S.; Fulton, L.L.; Fronick, C.C.; Branham, K.E.; Bragg-Gresham, J.; et al. Identification of a rare coding variant in complement 3 associated with age-related macular degeneration. Nat. Genet. 2013, 45, 1375–1379. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, L.G.; Lauer, N.; Hartmann, A.; Stippa, S.; Keilhauer, C.N.; Oppermann, M.; Pandey, M.K.; Kohl, J.; Zipfel, P.F.; Weber, B.H.; et al. An imbalance of human complement regulatory proteins CFHR1, CFHR3 and factor H influences risk for age-related macular degeneration (AMD). Hum. Mol. Genet. 2010, 19, 4694–4704. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.E.; Orr, N.; Esfandiary, H.; Diaz-Torres, M.; Goodship, T.; Chakravarthy, U. A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degeneration. Nat. Genet. 2006, 38, 1173–1177. [Google Scholar] [CrossRef] [PubMed]
- Sawitzke, J.; Im, K.M.; Kostiha, B.; Dean, M.; Gold, B. Association assessment of copy number polymorphism and risk of age-related macular degeneration. Ophthalmology 2011, 118, 2442–2446. [Google Scholar] [CrossRef] [PubMed]
- Canter, J.A.; Olson, L.M.; Spencer, K.; Schnetz-Boutaud, N.; Anderson, B.; Hauser, M.A.; Schmidt, S.; Postel, E.A.; Agarwal, A.; Pericak-Vance, M.A.; et al. Mitochondrial DNA polymorphism A4917G is independently associated with age-related macular degeneration. PLoS ONE 2008, 3, e2091. [Google Scholar] [CrossRef] [PubMed]
- Scott, W.K.; Schmidt, S.; Hauser, M.A.; Gallins, P.; Schnetz-Boutaud, N.; Spencer, K.L.; Gilbert, J.R.; Agarwal, A.; Postel, E.A.; Haines, J.L.; et al. Independent effects of complement factor H Y402H polymorphism and cigarette smoking on risk of age-related macular degeneration. Ophthalmology 2007, 114, 1151–1156. [Google Scholar] [CrossRef] [PubMed]
- Spencer, K.L.; Hauser, M.A.; Olson, L.M.; Schnetz-Boutaud, N.; Scott, W.K.; Schmidt, S.; Gallins, P.; Agarwal, A.; Postel, E.A.; Pericak-Vance, M.A.; et al. Haplotypes spanning the complement factor H gene are protective against age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4277–4283. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Tian, L.; Huang, Y. Correlation between the interactions of ABCA4 polymorphisms and smoking with the susceptibility to age-related macular degeneration. Int. J. Clin. Exp. Pathol. 2015, 8, 7403–7408. [Google Scholar] [PubMed]
- Scholl, H.P.; Peto, T.; Dandekar, S.; Bunce, C.; Xing, W.; Jenkins, S.; Bird, A.C. Inter- and intra-observer variability in grading lesions of age-related maculopathy and macular degeneration. Graefes Arch. Clin. Exp. Ophthalmol. 2003, 241, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Seddon, J.M.; Reynolds, R.; Maller, J.; Fagerness, J.A.; Daly, M.J.; Rosner, B. Prediction model for prevalence and incidence of advanced age-related macular degeneration based on genetic, demographic, and environmental variables. Investig. Ophthalmol. Vis. Sci. 2009, 50, 2044–2053. [Google Scholar] [CrossRef] [PubMed]
- For Insureres. Available online: http://www.macularisk.com/for-insurers.html (accessed on 7 March 2014).
- Age-Related Macular Degeneration Test. Available online: https://www.genetests.org/tests/details.php?id=138048 (accessed on 7 March 2014).
- Anthem Blue Cross Blue Shield Medical Policy. Available online: https://www.anthem.com/medicalpolicies/policies/mp_pw_c172203.htm (accessed on 7 March 2014).


{kind=link}
| Genetic Loci | Environmental/Demographic/Lifestyle Data | Outcome | Predictive Ability in Initial Sample | Predictive Ability in Validation Sample | Study |
|---|---|---|---|---|---|
| None | Age, smoking status, hypertension, AREDS simple severity score, night vision score | Geographic atrophy | 0.76 | x | [33] |
| None | Age, sex, education level, race, smoking status, pigment abnormality presence, soft drusen, maximum drusen size | Advanced AMD | 0.88 | 0.91 | [34] |
| CFH, ARMS2-HTRA1, C2-CFB, C3 | None | AMD | 0.734 | [35] | |
| CFH, ARMS2/HTRA1 | Age, smoking hx, first degree family hx of AMD, modified AREDS phenotype | Advanced AMD | 0.872 | Hosmer-Lemeshow statistic = 15.00, p = 0.09 | [36] |
| CFH, ARMS2/HTRA1, CFB, C3 | Age, smoking history | AMD high risk | 0.84 | x | [32] |
| CFH, ARMS2/HTRA1, C2, CFB, C3 | Age, smoking, BMI, antioxidant treatment, presence of advanced AMD in one eye, drusen size in both eyes | 10-year progression to advanced AMD | 0.915 | 0.908 | [37] |
| CFH, ARMS2/HTRA1, C2, CFB, C3, COL8A, RAD51B | Smoking status, BMI (adjusted for age, sex, education, 4 AREDS treatment groups) | 10-year progression to advanced AMD | 0.911 | 0.907 | [38] |
| CFH, C2, CFB, HTRA1/LOC387715, C3 | Age, smoking status | AMD | 0.82 | x | [39] |
| CFH, CFHR4, CFHR5, F13B, C2, CFB, ARMS2, C3, HTRA1 | Baseline grade (AREDS simplified severity scale), smoking, BMI, age, sex, treatment (AREDS vitamin-mineral treatment), education | CNV/GA | CNV 0.96; GA 0.94 | [40] | |
| CFH, CFHR4, CFHR5, F13B, C2, CFB, LOC387155/ARMS2, C3 | None | CNV | 0.82 | 0.80 | [41] |
| CFH, ARMS2, CFB, C3, APOE, PLA2G12A, LIPC, SYN3/TIMP3 | None | AMD | 0.820 | 0.813 | [42] |
| CFH, ARMS2, C2/CFB, C3, CFI, LPL, LIPC, MYRIP, SKIV2L, ABAC1, CETP, TIMP3, VEGFA, COL8A1, TNFRSF10A, FRK/COL10A1, SLC16A8, ADAMTS9, TGFBR1, RAD51B, IER3/DDR1, B3GALTL | Age, sex, smoking, BMI, baseline AMD phenotype | AMD | 0.88 | 0.85 | [43] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cooke Bailey, J.N.; Hoffman, J.D.; Sardell, R.J.; Scott, W.K.; Pericak-Vance, M.A.; Haines, J.L. The Application of Genetic Risk Scores in Age-Related Macular Degeneration: A Review. J. Clin. Med. 2016, 5, 31. https://doi.org/10.3390/jcm5030031
Cooke Bailey JN, Hoffman JD, Sardell RJ, Scott WK, Pericak-Vance MA, Haines JL. The Application of Genetic Risk Scores in Age-Related Macular Degeneration: A Review. Journal of Clinical Medicine. 2016; 5(3):31. https://doi.org/10.3390/jcm5030031
Chicago/Turabian StyleCooke Bailey, Jessica N., Joshua D. Hoffman, Rebecca J. Sardell, William K. Scott, Margaret A. Pericak-Vance, and Jonathan L. Haines. 2016. "The Application of Genetic Risk Scores in Age-Related Macular Degeneration: A Review" Journal of Clinical Medicine 5, no. 3: 31. https://doi.org/10.3390/jcm5030031