
MicroRNA Regulation of Human Breast Cancer Stem Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Breast Cancer Stem Cells
2. Shared Properties between Breast CSCs and Normal Mammary Stem/Progenitor Cells
3. miRNAs Specific for Breast CSCs
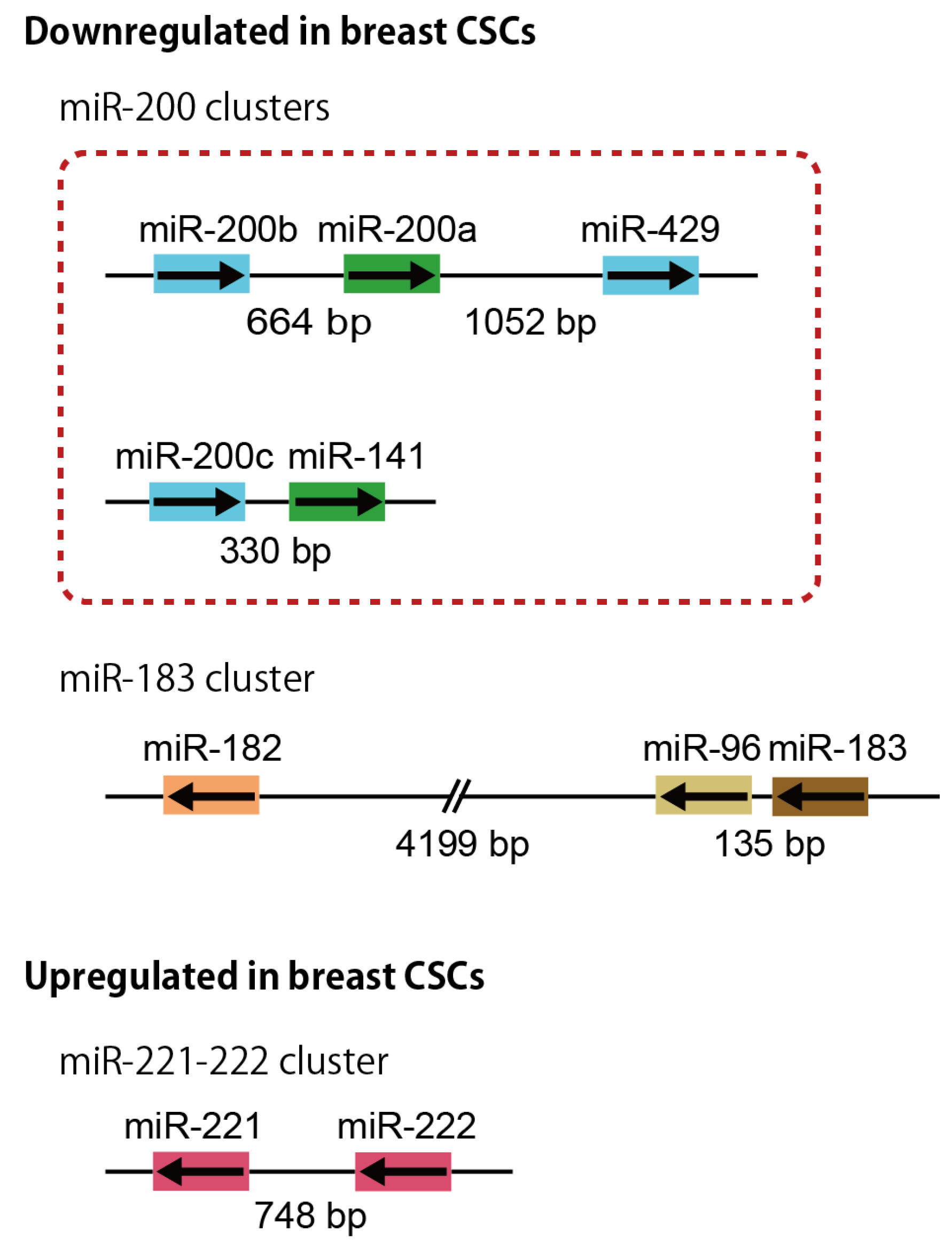
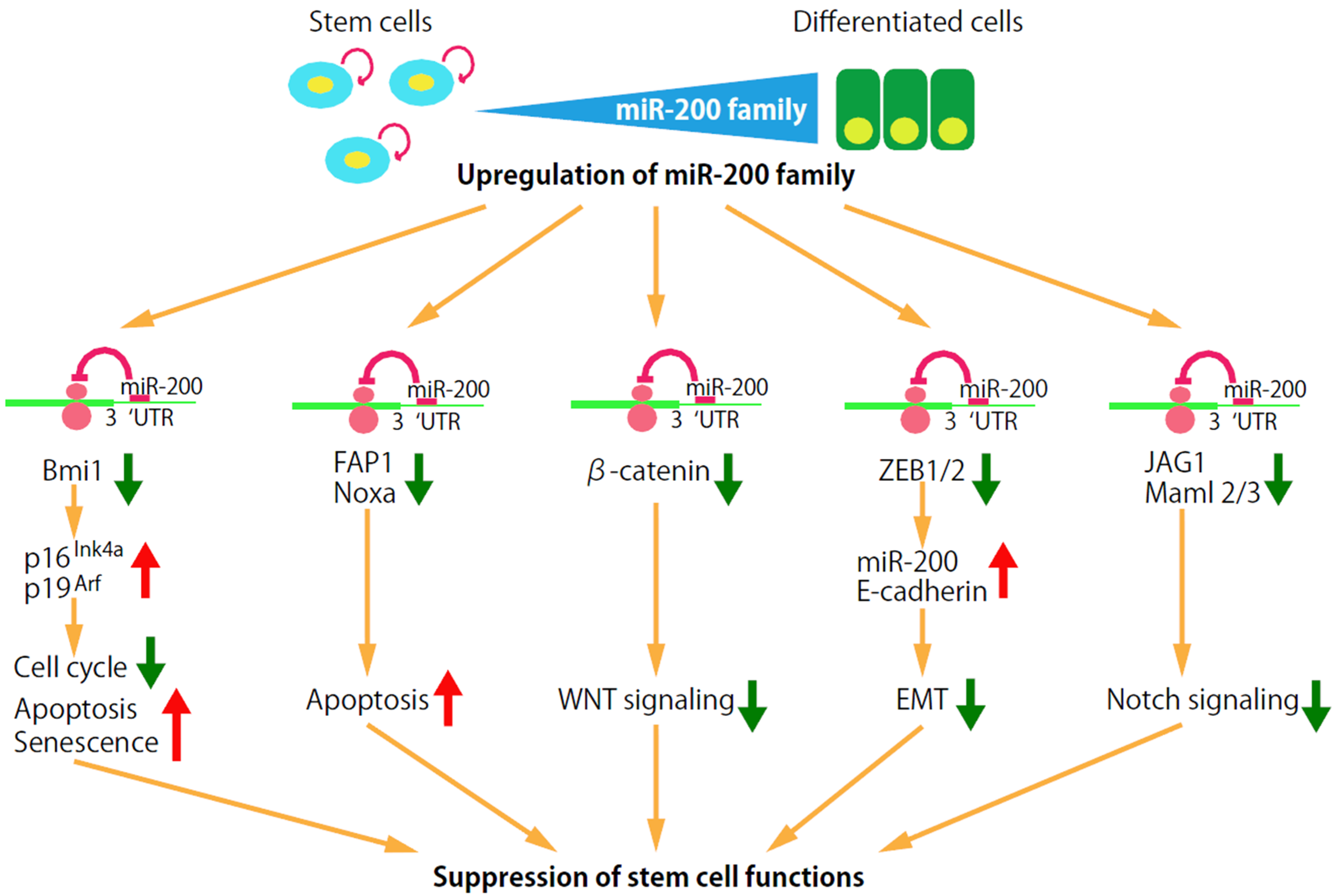
3.1. miR-200 Clusters

3.2. miR-183 Cluster
3.3. miR-221-222 Cluster
3.4. miR-142
3.5. miR-214
4. Signaling Pathways and Genes Targeted by miRNAs Specific to the Breast CSCs
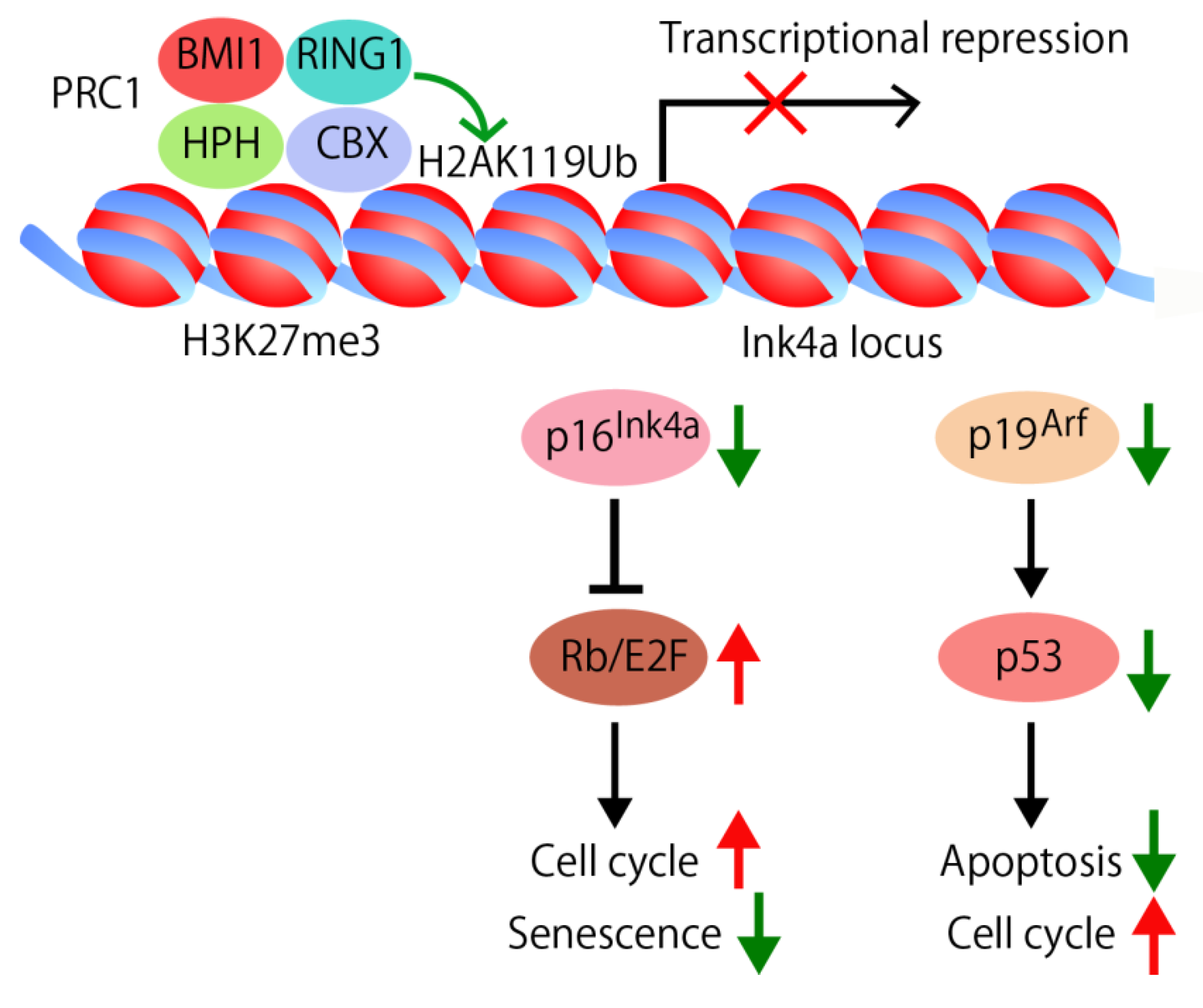
4.1. Self-Renewal Factor Bmi1


4.2. Apoptosis Signaling Pathway
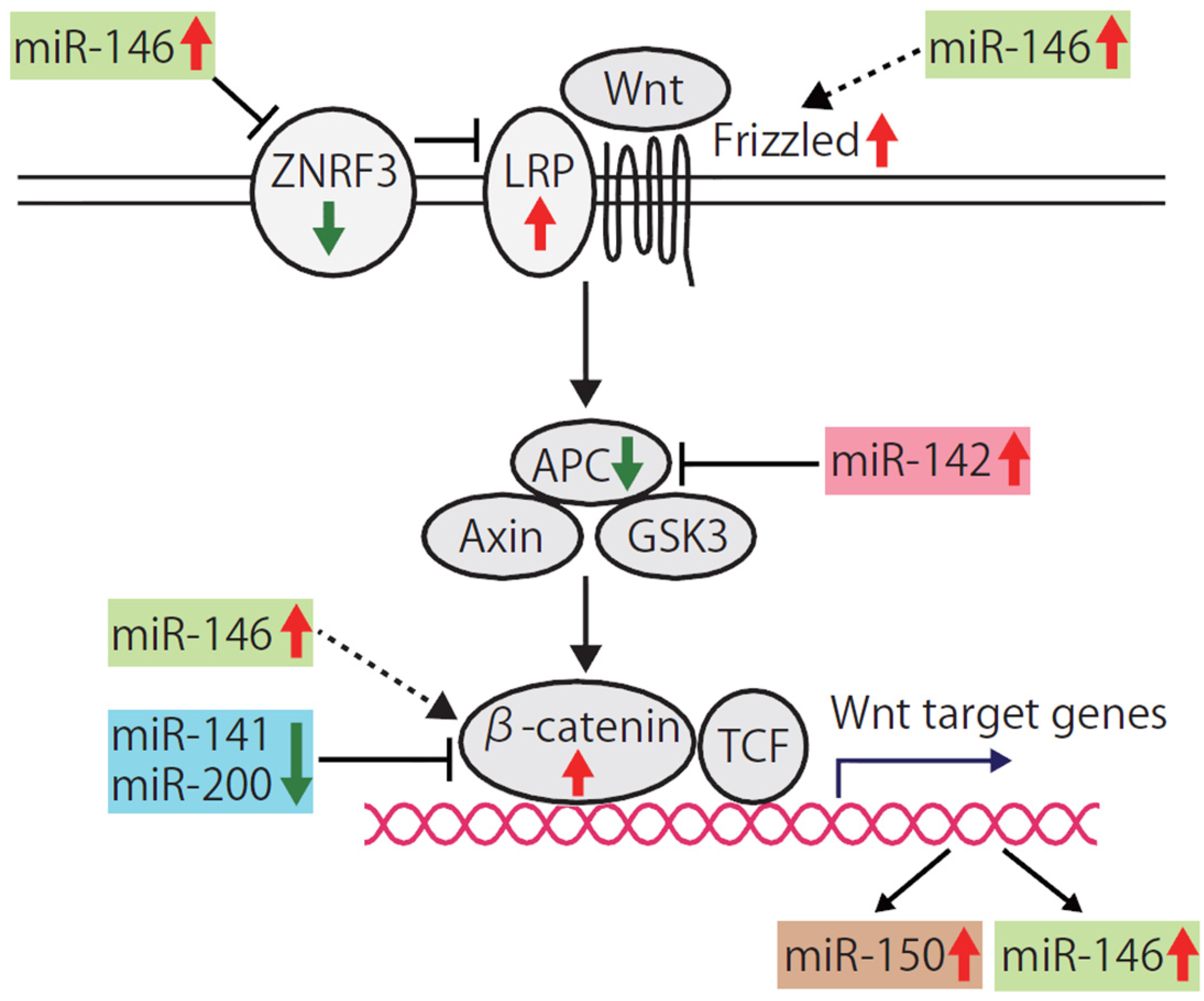
4.3. Wnt Signaling

4.4. EMT
4.5. Notch Signaling Pathway
5. Metastatic CSC Specific miRNAs
6. Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Sell, S. Stem cell origin of cancer and differentiation therapy. Crit. Rev. Oncol. Hematol. 2004, 51, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Cohnheim, J. Congenitales, quergestreiftes muskelsarkom der nieren. Arch. Pathol. Anatom. Physiol. Klin. Med. 1875, 65, 64–69. [Google Scholar] [CrossRef]
- Fialkow, P.J. Clonal origin of human tumors. Biochim. Biophys. Acta 1976, 458, 283–321. [Google Scholar] [CrossRef]
- Fialkow, P.J. Human tumors studied with genetic markers. Birth Defects Orig. Artic. Ser. 1976, 12, 123–132. [Google Scholar] [PubMed]
- Fialkow, P.J. Stem cell origin of human myeloid blood cell neoplasms. Verh. Dtsch. Ges. Pathol. 1990, 74, 43–47. [Google Scholar] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.U.; Miyazaki, H.; Ochiya, T. The role of microRNAs in the regulation of cancer stem cells. Front. Genet. 2014, 4, 295. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer stem cells—perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006, 66, 9339–9344. [Google Scholar] [CrossRef] [PubMed]
- Baccelli, I.; Trumpp, A. The evolving concept of cancer and metastasis stem cells. J. Cell Biol. 2012, 198, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, C.H.; Ailles, L.E.; Dylla, S.J.; Muijtjens, M.; Jones, C.; Zehnder, J.L.; Gotlib, J.; Li, K.; Manz, M.G.; Keating, A.; et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N. Engl. J. Med. 2004, 351, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Kikushige, Y.; Ishikawa, F.; Miyamoto, T.; Shima, T.; Urata, S.; Yoshimoto, G.; Mori, Y.; Iino, T.; Yamauchi, T.; Eto, T.; et al. Self-renewing hematopoietic stem cell is the primary target in pathogenesis of human chronic lymphocytic leukemia. Cancer Cell 2011, 20, 246–259. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D.; et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2014, 2, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Daniel, V.C.; Marchionni, L.; Hierman, J.S.; Rhodes, J.T.; Devereux, W.L.; Rudin, C.M.; Yung, R.; Parmigiani, G.; Dorsch, M.; Peacock, C.D.; et al. A primary xenograft model of small-cell lung cancer reveals irreversible changes in gene expression imposed by culture in vitro. Cancer Res. 2009, 69, 3364–3373. [Google Scholar] [CrossRef] [PubMed]
- Eirew, P.; Steif, A.; Khattra, J.; Ha, G.; Yap, D.; Farahani, H.; Gelmon, K.; Chia, S.; Mar, C.; Wan, A.; et al. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature 2015, 518, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Cowin, P.; Wysolmerski, J. Molecular mechanisms guiding embryonic mammary gland development. Cold Spring Harb. Perspect. Biol. 2010, 2, a003251. [Google Scholar] [CrossRef] [PubMed]
- Lechner, R.B.; Gurll, N.J.; Reynolds, D.G. Effects of naloxone on regional blood flow distribution in canine hemorrhagic shock. Proc. Soc. Exp. Biol. Med. 1985, 178, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.J.; Khaled, W.T. Mammary development in the embryo and adult: A journey of morphogenesis and commitment. Development 2008, 135, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Shackleton, M.; Vaillant, F.; Simpson, K.J.; Stingl, J.; Smyth, G.K.; Asselin-Labat, M.L.; Wu, L.; Lindeman, G.J.; Visvader, J.E. Generation of a functional mammary gland from a single stem cell. Nature 2006, 439, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Stingl, J.; Eirew, P.; Ricketson, I.; Shackleton, M.; Vaillant, F.; Choi, D.; Li, H.I.; Eaves, C.J. Purification and unique properties of mammary epithelial stem cells. Nature 2006, 439, 993–997. [Google Scholar] [CrossRef] [PubMed]
- Rios, A.C.; Fu, N.Y.; Lindeman, G.J.; Visvader, J.E. In situ identification of bipotent stem cells in the mammary gland. Nature 2014, 506, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Van Keymeulen, A.; Rocha, A.S.; Ousset, M.; Beck, B.; Bouvencourt, G.; Rock, J.; Sharma, N.; Dekoninck, S.; Blanpain, C. Distinct stem cells contribute to mammary gland development and maintenance. Nature 2011, 479, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Eirew, P.; Stingl, J.; Raouf, A.; Turashvili, G.; Aparicio, S.; Emerman, J.T.; Eaves, C.J. A method for quantifying normal human mammary epithelial stem cells with in vivo regenerative ability. Nat. Med. 2008, 14, 1384–1389. [Google Scholar] [CrossRef] [PubMed]
- Shimono, Y.; Zabala, M.; Cho, R.W.; Lobo, N.; Dalerba, P.; Qian, D.; Diehn, M.; Liu, H.; Panula, S.P.; Chiao, E.; et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell 2009, 138, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Keckesova, Z.; Donaher, J.L.; Shibue, T.; Tischler, V.; Reinhardt, F.; Itzkovitz, S.; Noske, A.; Zurrer-Hardi, U.; Bell, G.; et al. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell 2012, 148, 1015–1028. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K. Breast cancer: Origins and evolution. J. Clin. Invest. 2007, 117, 3155–3163. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Garcia, I.; Miska, E.A. MicroRNA functions in animal development and human disease. Development 2005, 132, 4653–4662. [Google Scholar] [CrossRef] [PubMed]
- Saini, H.K.; Griffiths-Jones, S.; Enright, A.J. Genomic analysis of human microRNA transcripts. Proc. Natl. Acad. Sci. USA 2007, 104, 17719–17724. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Griffiths-Jones, S.; Ashurst, J.L.; Bradley, A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004, 14, 1902–1910. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; Liao, Q.; Jiang, X.; Shao, Y.; Xiao, B.; Xi, Y.; Guo, J. Long noncoding RNA associated-competing endogenous RNAs in gastric cancer. Sci. Rep. 2014, 4, 6088. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.C.; Tavazoie, M.; Doetsch, F. Stem cells: From epigenetics to microRNAs. Neuron 2005, 46, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Chalfie, M.; Horvitz, H.R.; Sulston, J.E. Mutations that lead to reiterations in the cell lineages of C. elegans. Cell 1981, 24, 59–69. [Google Scholar] [CrossRef]
- Ambros, V.; Horvitz, H.R. Heterochronic mutants of the nematode Caenorhabditis elegans. Science 1984, 226, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Reinhart, B.J.; Slack, F.J.; Basson, M.; Pasquinelli, A.E.; Bettinger, J.C.; Rougvie, A.E.; Horvitz, H.R.; Ruvkun, G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 2000, 403, 901–906. [Google Scholar] [PubMed]
- Suh, M.R.; Lee, Y.; Kim, J.Y.; Kim, S.K.; Moon, S.H.; Lee, J.Y.; Cha, K.Y.; Chung, H.M.; Yoon, H.S.; Moon, S.Y.; et al. Human embryonic stem cells express a unique set of microRNAs. Dev. Biol. 2004, 270, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Houbaviy, H.B.; Murray, M.F.; Sharp, P.A. Embryonic stem cell-specific microRNAs. Dev. Cell 2003, 5, 351–358. [Google Scholar] [CrossRef]
- Sinkkonen, L.; Hugenschmidt, T.; Berninger, P.; Gaidatzis, D.; Mohn, F.; Artus-Revel, C.G.; Zavolan, M.; Svoboda, P.; Filipowicz, W. MicroRNAs control de novo DNA methylation through regulation of transcriptional repressors in mouse embryonic stem cells. Nat. Struct. Mol. Biol. 2008, 15, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Stadler, B.; Ivanovska, I.; Mehta, K.; Song, S.; Nelson, A.; Tan, Y.; Mathieu, J.; Darby, C.; Blau, C.A.; Ware, C.; et al. Characterization of microRNAs involved in embryonic stem cell states. Stem Cells Dev. 2010, 19, 935–950. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Yao, H.; Zhu, P.; Zhang, X.; Pan, Q.; Gong, C.; Huang, Y.; Hu, X.; Su, F.; Lieberman, J.; et al. Let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell 2007, 131, 1109–1123. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Hu, K.; Zhao, Z.; Chen, G.; Ou, X.; Zhang, H.; Zhang, X.; Wei, X.; Wang, D.; Cui, M.; et al. MicroRNA-1 down-regulates proliferation and migration of breast cancer stem cells by inhibiting the Wnt/β-catenin pathway. Oncotarget 2015, 6, 41638–41649. [Google Scholar] [PubMed]
- Takahashi, R.U.; Miyazaki, H.; Takeshita, F.; Yamamoto, Y.; Minoura, K.; Ono, M.; Kodaira, M.; Tamura, K.; Mori, M.; Ochiya, T. Loss of microRNA-27b contributes to breast cancer stem cell generation by activating ENPP1. Nat. Commun. 2015, 6, 7318. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Yu, F.; Yao, H.; Cui, X.; Jiao, Y.; Lin, L.; Chen, J.; Yin, D.; Song, E.; Liu, Q. miR-27a regulates endothelial differentiation of breast cancer stem like cells. Oncogene 2014, 33, 2629–2638. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Tang, D.G. MicroRNA regulation of cancer stem cells. Cancer Res. 2011, 71, 5950–5954. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbacher, D.; Balic, M.; Pichler, M. The role of microRNAs in breast cancer stem cells. Int. J. Mol. Sci. 2013, 14, 14712–14723. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.M.; Newman, M.; Parker, J.S.; Morin-Kensicki, E.M.; Wright, T.; Hammond, S.M. Extensive post-transcriptional regulation of microRNAs and its implications for cancer. Genes Dev. 2006, 20, 2202–2207. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, S.R.; Daley, G.Q.; Gregory, R.I. Selective blockade of microRNA processing by Lin28. Science 2008, 320, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Pode-Shakked, N.; Shukrun, R.; Mark-Danieli, M.; Tsvetkov, P.; Bahar, S.; Pri-Chen, S.; Goldstein, R.S.; Rom-Gross, E.; Mor, Y.; Fridman, E.; et al. The isolation and characterization of renal cancer initiating cells from human Wilms’ tumour xenografts unveils new therapeutic targets. EMBO Mol. Med. 2013, 5, 18–37. [Google Scholar] [CrossRef] [PubMed]
- Peter, M.E. Let-7 and miR-200 microRNAs: Guardians against pluripotency and cancer progression. Cell Cycle 2009, 8, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Altuvia, Y.; Landgraf, P.; Lithwick, G.; Elefant, N.; Pfeffer, S.; Aravin, A.; Brownstein, M.J.; Tuschl, T.; Margalit, H. Clustering and conservation patterns of human microRNAs. Nucleic Acids Res. 2005, 33, 2697–2706. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.C.; Ho, M.R.; Li, S.C.; Tsai, K.W.; Lai, C.H.; Hsu, C.N.; Lin, W.C. MetaMirClust: Discovery of miRNA cluster patterns using a data-mining approach. Genomics 2012, 100, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Mogilyansky, E.; Rigoutsos, I. The miR-17/92 cluster: A comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ. 2013, 20, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Small, E.M.; Sutherland, L.B.; Qi, X.; McAnally, J.; Plato, C.F.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes Dev. 2009, 23, 2166–2178. [Google Scholar] [CrossRef] [PubMed]
- Chivukula, R.R.; Shi, G.; Acharya, A.; Mills, E.W.; Zeitels, L.R.; Anandam, J.L.; Abdelnaby, A.A.; Balch, G.C.; Mansour, J.C.; Yopp, A.C.; et al. An essential mesenchymal function for miR-143/145 in intestinal epithelial regeneration. Cell 2014, 157, 1104–1116. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, B.M.; Heimberg, A.M.; Moy, V.N.; Sperling, E.A.; Holstein, T.W.; Heber, S.; Peterson, K.J. The deep evolution of metazoan microRNAs. Evol. Dev. 2009, 11, 50–68. [Google Scholar] [CrossRef] [PubMed]
- Trumbach, D.; Prakash, N. The conserved miR-8/miR-200 microRNA family and their role in invertebrate and vertebrate neurogenesis. Cell Tissue Res. 2015, 359, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Saini, H.K.; Enright, A.J.; Griffiths-Jones, S. Annotation of mammalian primary microRNAs. BMC Genomics 2008, 9, 564. [Google Scholar] [CrossRef] [PubMed]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Kolesnikoff, N.; Attema, J.L.; Roslan, S.; Bert, A.G.; Schwarz, Q.P.; Gregory, P.A.; Goodall, G.J. Specificity protein 1 (Sp1) maintains basal epithelial expression of the miR-200 family: Implications for epithelial-mesenchymal transition. J. Biol. Chem. 2014, 289, 11194–11205. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.M.; Cha, J.Y.; Kim, J.; Kim, D.; Trang, H.T.; Kim, Y.M.; Cho, Y.H.; Park, D.; Hong, S. Smad3 regulates E-cadherin via miRNA-200 pathway. Oncogene 2012, 31, 3051–3059. [Google Scholar] [CrossRef] [PubMed]
- Humphries, B.; Yang, C. The microRNA-200 family: Small molecules with novel roles in cancer development, progression and therapy. Oncotarget 2015, 6, 6472–6498. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, I.; Ganapathy, V.; Gillies, E.; Fonseca, I.; Sureban, S.M.; Houchen, C.W.; Reis, A.; Queimado, L. Wnt inhibitory factor 1 suppresses cancer stemness and induces cellular senescence. Cell Death Dis. 2014, 5, e1246. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Veronese, A.; Pichiorri, F.; Lee, T.J.; Jeon, Y.J.; Volinia, S.; Pineau, P.; Marchio, A.; Palatini, J.; Suh, S.S.; et al. p53 Regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J. Exp. Med. 2011, 208, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.Y.; Wright, J.A.; Attema, J.L.; Gregory, P.A.; Bert, A.G.; Smith, E.; Thomas, D.; Lopez, A.F.; Drew, P.A.; Khew-Goodall, Y.; et al. Epigenetic modulation of the miR-200 family is associated with transition to a breast cancer stem-cell-like state. J. Cell Sci. 2013, 126, 2256–2266. [Google Scholar] [CrossRef] [PubMed]
- Song, S.J.; Poliseno, L.; Song, M.S.; Ala, U.; Webster, K.; Ng, C.; Beringer, G.; Brikbak, N.J.; Yuan, X.; Cantley, L.C.; et al. MicroRNA-antagonism regulates breast cancer stemness and metastasis via TET-family-dependent chromatin remodeling. Cell 2013, 154, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Jackson, A.L.; Guo, J.; Linsley, P.S.; Eisenman, R.N. Myc-regulated microRNAs attenuate embryonic stem cell differentiation. EMBO J. 2009, 28, 3157–3170. [Google Scholar] [CrossRef] [PubMed]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; zur Hausen, A.; et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.G.; Langer, E.M.; Lindsley, R.C.; Cai, M.; Murphy, T.L.; Kyba, M.; Murphy, K.M. Snail and the microRNA-200 family act in opposition to regulate epithelial-to-mesenchymal transition and germ layer fate restriction in differentiating ESCs. Stem Cells 2011, 29, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, G.; Lu, C.; Guo, X.; Hong, W.; Kang, J.; Wang, J. Synergetic cooperation of microRNAs with transcription factors in iPS cell generation. PLoS ONE 2012, 7, e40849. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Guo, X.; Hong, W.; Liu, Q.; Wei, T.; Lu, C.; Gao, L.; Ye, D.; Zhou, Y.; Chen, J.; et al. Critical regulation of miR-200/ZEB2 pathway in Oct4/Sox2-induced mesenchymal-to-epithelial transition and induced pluripotent stem cell generation. Proc. Natl. Acad. Sci. USA 2013, 110, 2858–2863. [Google Scholar] [CrossRef] [PubMed]
- Uhlmann, S.; Zhang, J.D.; Schwager, A.; Mannsperger, H.; Riazalhosseini, Y.; Burmester, S.; Ward, A.; Korf, U.; Wiemann, S.; Sahin, O. miR-200bc/429 cluster targets PLCγ1 and differentially regulates proliferation and EGF-driven invasion than miR-200a/141 in breast cancer. Oncogene 2010, 29, 4297–4306. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhang, Z.; Xia, S.; Xing, C.; Ci, X.; Li, X.; Zhao, R.; Tian, S.; Ma, G.; Zhu, Z.; et al. KLF5 activates microRNA 200 transcription to maintain epithelial characteristics and prevent induced epithelial-mesenchymal transition in epithelial cells. Mol. Cell. Biol. 2013, 33, 4919–4935. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Pan, Y.; Zhou, Y. miR-96 promotes osteogenic differentiation by suppressing HBEGF-EGFR signaling in osteoblastic cells. FEBS Lett. 2014, 588, 4761–4768. [Google Scholar] [CrossRef] [PubMed]
- Dambal, S.; Shah, M.; Mihelich, B.; Nonn, L. The microRNA-183 cluster: The family that plays together stays together. Nucleic Acids Res. 2015, 43, 7173–7188. [Google Scholar] [CrossRef] [PubMed]
- Pierce, M.L.; Weston, M.D.; Fritzsch, B.; Gabel, H.W.; Ruvkun, G.; Soukup, G.A. MicroRNA-183 family conservation and ciliated neurosensory organ expression. Evol. Dev. 2008, 10, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Leung, W.K.; He, M.; Chan, A.W.; Law, P.T.; Wong, N. Wnt/β-Catenin activates miR-183/96/182 expression in hepatocellular carcinoma that promotes cell invasion. Cancer Lett. 2015, 362, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Xiang, H.; Peng, Y.L.; Peng, J.; Jiang, S.W. Mature miR-183, negatively regulated by transcription factor GATA3, promotes 3T3-L1 adipogenesis through inhibition of the canonical Wnt/β-catenin signaling pathway by targeting LRP6. Cell. Signal. 2014, 26, 1155–1165. [Google Scholar] [CrossRef] [PubMed]
- Donatelli, S.S.; Zhou, J.M.; Gilvary, D.L.; Eksioglu, E.A.; Chen, X.; Cress, W.D.; Haura, E.B.; Schabath, M.B.; Coppola, D.; Wei, S.; et al. TGF-β-inducible microRNA-183 silences tumor-associated natural killer cells. Proc. Natl. Acad Sci. USA 2014, 111, 4203–4208. [Google Scholar] [CrossRef] [PubMed]
- Li, X.L.; Hara, T.; Choi, Y.; Subramanian, M.; Francis, P.; Bilke, S.; Walker, R.L.; Pineda, M.; Zhu, Y.; Yang, Y.; et al. A p21-ZEB1 complex inhibits epithelial-mesenchymal transition through the microRNA 183-96-182 cluster. Mol. Cell Biol. 2014, 34, 533–550. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Sobenin, I.A.; Orekhov, A.N.; Bobryshev, Y.V. Human miR-221/222 in physiological and atherosclerotic vascular remodeling. Biomed. Res. Int. 2015, 2015, 354517. [Google Scholar] [CrossRef] [PubMed]
- Di Leva, G.; Gasparini, P.; Piovan, C.; Ngankeu, A.; Garofalo, M.; Taccioli, C.; Iorio, M.V.; Li, M.; Volinia, S.; Alder, H.; et al. MicroRNA cluster 221-222 and estrogen receptor α interactions in breast cancer. J. Natl. Cancer Inst. 2010, 102, 706–721. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.J.; Lin, J.; Yang, H.; Kong, W.; He, L.; Ma, X.; Coppola, D.; Cheng, J.Q. MicroRNA-221/222 negatively regulates estrogen receptor α and is associated with tamoxifen resistance in breast cancer. J. Biol. Chem. 2008, 283, 31079–31086. [Google Scholar] [CrossRef] [PubMed]
- Pallante, P.; Battista, S.; Pierantoni, G.M.; Fusco, A. Deregulation of microRNA expression in thyroid neoplasias. Nat. Rev. Endocrinol. 2014, 10, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, M.; Quintavalle, C.; Romano, G.; Croce, C.M.; Condorelli, G. miR221/222 in cancer: Their role in tumor progression and response to therapy. Curr. Mol. Med. 2012, 12, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Volinia, S.; Calin, G.A.; Liu, C.G.; Ambs, S.; Cimmino, A.; Petrocca, F.; Visone, R.; Iorio, M.; Roldo, C.; Ferracin, M.; et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. USA 2006, 103, 2257–2261. [Google Scholar] [CrossRef] [PubMed]
- Felli, N.; Fontana, L.; Pelosi, E.; Botta, R.; Bonci, D.; Facchiano, F.; Liuzzi, F.; Lulli, V.; Morsilli, O.; Santoro, S.; et al. MicroRNAs 221 and 222 inhibit normal erythropoiesis and erythroleukemic cell growth via kit receptor down-modulation. Proc. Natl. Acad. Sci. USA 2005, 102, 18081–18086. [Google Scholar] [CrossRef] [PubMed]
- Roscigno, G.; Quintavalle, C.; Donnarumma, E.; Puoti, I.; Diaz-Lagares, A.; Iaboni, M.; Fiore, D.; Russo, V.; Todaro, M.; Romano, G.; et al. miR-221 promotes stemness of breast cancer cells by targeting DNMT3b. Oncotarget 2015. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, L.; Ischenko, I.; Bao, Q.; Schwarz, B.; Niess, H.; Wang, Y.; Renner, A.; Mysliwietz, J.; Jauch, K.W.; et al. Antisense inhibition of microRNA-21 and microRNA-221 in tumor-initiating stem-like cells modulates tumorigenesis, metastasis, and chemotherapy resistance in pancreatic cancer. Target. Oncol. 2015, 10, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Aldaz, B.; Sagardoy, A.; Nogueira, L.; Guruceaga, E.; Grande, L.; Huse, J.T.; Aznar, M.A.; Diez-Valle, R.; Tejada-Solis, S.; Alonso, M.M.; et al. Involvement of miRNAs in the differentiation of human glioblastoma multiforme stem-like cells. PLoS ONE 2013, 8, e77098. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.Y.; Calin, G.A. MicroRNAs miR-221 and miR-222: A new level of regulation in aggressive breast cancer. Genome Med. 2011, 3, 56. [Google Scholar] [CrossRef] [PubMed]
- Stinson, S.; Lackner, M.R.; Adai, A.T.; Yu, N.; Kim, H.J.; O’Brien, C.; Spoerke, J.; Jhunjhunwala, S.; Boyd, Z.; Januario, T.; et al. TRPS1 targeting by miR-221/222 promotes the epithelial-to-mesenchymal transition in breast cancer. Sci. Signal. 2011, 4, ra41. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Z.; Lodish, H.F. MicroRNAs as regulators of mammalian hematopoiesis. Semin. Immunol. 2005, 17, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Ramkissoon, S.H.; Mainwaring, L.A.; Ogasawara, Y.; Keyvanfar, K.; McCoy, J.P., Jr.; Sloand, E.M.; Kajigaya, S.; Young, N.S. Hematopoietic-specific microRNA expression in human cells. Leuk. Res. 2006, 30, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Visone, R.; Rassenti, L.Z.; Veronese, A.; Taccioli, C.; Costinean, S.; Aguda, B.D.; Volinia, S.; Ferracin, M.; Palatini, J.; Balatti, V.; et al. Karyotype-specific microRNA signature in chronic lymphocytic leukemia. Blood 2009, 114, 3872–3879. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Neilson, J.R.; Kumar, P.; Manocha, M.; Shankar, P.; Sharp, P.A.; Manjunath, N. miRNA profiling of naive, effector and memory CD8 T cells. PLoS ONE 2007, 2, e1020. [Google Scholar] [CrossRef] [PubMed]
- Kwanhian, W.; Lenze, D.; Alles, J.; Motsch, N.; Barth, S.; Doll, C.; Imig, J.; Hummel, M.; Tinguely, M.; Trivedi, P.; et al. MicroRNA-142 is mutated in about 20% of diffuse large B-cell lymphoma. Cancer Med. 2012, 1, 141–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrestha, A.; Carraro, G.; El Agha, E.; Mukhametshina, R.; Chao, C.M.; Rizvanov, A.; Barreto, G.; Bellusci, S. Generation and validation of miR-142 knock out mice. PLoS ONE 2015, 10, e0136913. [Google Scholar] [CrossRef] [PubMed]
- Chapnik, E.; Rivkin, N.; Mildner, A.; Beck, G.; Pasvolsky, R.; Metzl-Raz, E.; Birger, Y.; Amir, G.; Tirosh, I.; Porat, Z.; et al. miR-142 orchestrates a network of actin cytoskeleton regulators during megakaryopoiesis. eLife 2014, 3, e01964. [Google Scholar] [CrossRef] [PubMed]
- Skarn, M.; Baroy, T.; Stratford, E.W.; Myklebost, O. Epigenetic regulation and functional characterization of microRNA-142 in mesenchymal cells. PLoS ONE 2013, 8, e79231. [Google Scholar] [CrossRef] [PubMed]
- Isobe, T.; Hisamori, S.; Hogan, D.J.; Zabala, M.; Hendrickson, D.G.; Dalerba, P.; Cai, S.; Scheeren, F.; Kuo, A.H.; Sikandar, S.S.; et al. miR-142 regulates the tumorigenicity of human breast cancer stem cells through the canonical WNT signaling pathway. eLife 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Penna, E.; Orso, F.; Taverna, D. miR-214 as a key hub that controls cancer networks: Small player, multiple functions. J. Invest. Dermatol. 2015, 135, 960–969. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Huang, J.; Yang, N.; Greshock, J.; Megraw, M.S.; Giannakakis, A.; Liang, S.; Naylor, T.L.; Barchetti, A.; Ward, M.R.; et al. MicroRNAs exhibit high frequency genomic alterations in human cancer. Proc. Natl. Acad. Sci. USA 2006, 103, 9136–9141. [Google Scholar] [CrossRef] [PubMed]
- Blenkiron, C.; Goldstein, L.D.; Thorne, N.P.; Spiteri, I.; Chin, S.F.; Dunning, M.J.; Barbosa-Morais, N.L.; Teschendorff, A.E.; Green, A.R.; Ellis, I.O.; et al. MicroRNA expression profiling of human breast cancer identifies new markers of tumor subtype. Genome Biol. 2007, 8, R214. [Google Scholar] [CrossRef] [PubMed]
- Sempere, L.F.; Christensen, M.; Silahtaroglu, A.; Bak, M.; Heath, C.V.; Schwartz, G.; Wells, W.; Kauppinen, S.; Cole, C.N. Altered MicroRNA expression confined to specific epithelial cell subpopulations in breast cancer. Cancer Res. 2007, 67, 11612–11620. [Google Scholar] [CrossRef] [PubMed]
- Aurora, A.B.; Mahmoud, A.I.; Luo, X.; Johnson, B.A.; van Rooij, E.; Matsuzaki, S.; Humphries, K.M.; Hill, J.A.; Bassel-Duby, R.; Sadek, H.A.; et al. MicroRNA-214 protects the mouse heart from ischemic injury by controlling Ca2+ overload and cell death. J. Clin. Invest. 2012, 122, 1222–1232. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Sato, T.; Amano, T.; Kawamura, Y.; Kawamura, N.; Kawaguchi, H.; Yamashita, N.; Kurihara, H.; Nakaoka, T. Dnm3os, a non-coding RNA, is required for normal growth and skeletal development in mice. Dev. Dyn. 2008, 237, 3738–3748. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.X.; Xu, M.; Tan, L.; Yang, H.; Permuth-Wey, J.; Kruk, P.A.; Wenham, R.M.; Nicosia, S.V.; Lancaster, J.M.; Sellers, T.A.; et al. MicroRNA miR-214 regulates ovarian cancer cell stemness by targeting p53/Nanog. J. Biol. Chem. 2012, 287, 34970–34978. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.C.; Li, Y.Y.; Wang, H.Y.; Fu, S.; Wang, X.P.; Zeng, M.S.; Zeng, Y.X.; Shao, J.Y. Knockdown of miR-214 promotes apoptosis and inhibits cell proliferation in nasopharyngeal carcinoma. PLoS ONE 2014, 9, e86149. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Lv, P.; Liu, X.; Zhu, M.; Qiu, X. MicroRNA-214 enhances the invasion ability of breast cancer cells by targeting p53. Int. J. Mol. Med. 2015, 35, 1395–1402. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Ooi, L.L.; Hui, K.M. miR-214 targets β-catenin pathway to suppress invasion, stem-like traits and recurrence of human hepatocellular carcinoma. PLoS ONE 2012, 7, e44206. [Google Scholar] [CrossRef] [PubMed]
- Van der Lugt, N.M.; Domen, J.; Linders, K.; van Roon, M.; Robanus-Maandag, E.; te Riele, H.; van der Valk, M.; Deschamps, J.; Sofroniew, M.; van Lohuizen, M.; et al. Posterior transformation, neurological abnormalities, and severe hematopoietic defects in mice with a targeted deletion of the bmi-1 proto-oncogene. Genes Dev. 1994, 8, 757–769. [Google Scholar] [CrossRef] [PubMed]
- Pietersen, A.M.; Evers, B.; Prasad, A.A.; Tanger, E.; Cornelissen-Steijger, P.; Jonkers, J.; van Lohuizen, M. Bmi1 regulates stem cells and proliferation and differentiation of committed cells in mammary epithelium. Curr. Biol. 2008, 18, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Adorno, M.; Sikandar, S.; Mitra, S.S.; Kuo, A.; Nicolis di Robilant, B.; Haro-Acosta, V.; Ouadah, Y.; Quarta, M.; Rodriguez, J.; Qian, D.; et al. Usp16 contributes to somatic stem-cell defects in Down’s syndrome. Nature 2013, 501, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Haupt, Y.; Bath, M.L.; Harris, A.W.; Adams, J.M. Bmi-1 transgene induces lymphomas and collaborates with myc in tumorigenesis. Oncogene 1993, 8, 3161–3164. [Google Scholar] [PubMed]
- Jacobs, J.J.; Kieboom, K.; Marino, S.; DePinho, R.A.; van Lohuizen, M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 1999, 397, 164–168. [Google Scholar] [PubMed]
- Park, I.K.; Morrison, S.J.; Clarke, M.F. Bmi1, stem cells, and senescence regulation. J. Clin. Invest. 2004, 113, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E.; DePinho, R.A. The INK4A/ARF locus and its two gene products. Curr. Opin. Genet. Dev. 1999, 9, 22–30. [Google Scholar] [CrossRef]
- Zindy, F.; Quelle, D.E.; Roussel, M.F.; Sherr, C.J. Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene 1997, 15, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Honda, R.; Yasuda, H. Association of p19ARF with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. EMBO J. 1999, 18, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.D.; Taylor, L.J.; Roussel, M.F.; Sherr, C.J.; Bar-Sagi, D. Nucleolar Arf sequesters Mdm2 and activates p53. Nat. Cell Biol. 1999, 1, 20–26. [Google Scholar] [PubMed]
- He, X.; Dong, Y.; Wu, C.W.; Zhao, Z.; Ng, S.S.; Chan, F.K.; Sung, J.J.; Yu, J. MicroRNA-218 inhibits cell cycle progression and promotes apoptosis in colon cancer by downregulating BMI1 polycomb ring finger oncogene. Mol. Med. 2012, 18, 1491–1498. [Google Scholar]
- Dimri, M.; Carroll, J.D.; Cho, J.H.; Dimri, G.P. MicroRNA-141 regulates BMI1 expression and induces senescence in human diploid fibroblasts. Cell Cycle 2013, 12, 3537–3546. [Google Scholar] [CrossRef] [PubMed]
- Dong, P.; Kaneuchi, M.; Watari, H.; Hamada, J.; Sudo, S.; Ju, J.; Sakuragi, N. MicroRNA-194 inhibits epithelial to mesenchymal transition of endometrial cancer cells by targeting oncogene BMI-1. Mol. Cancer 2011, 10, 99. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, R.; Nicoloso, M.; Arvizo, R.; Wang, E.; Cortez, A.; Rossi, S.; Calin, G.A.; Mukherjee, P. miR-15a and miR-16 control Bmi-1 expression in ovarian cancer. Cancer Res. 2009, 69, 9090–9095. [Google Scholar] [CrossRef] [PubMed]
- Godlewski, J.; Nowicki, M.O.; Bronisz, A.; Williams, S.; Otsuki, A.; Nuovo, G.; Raychaudhury, A.; Newton, H.B.; Chiocca, E.A.; Lawler, S. Targeting of the Bmi-1 oncogene/stem cell renewal factor by microRNA-128 inhibits glioma proliferation and self-renewal. Cancer Res. 2008, 68, 9125–9130. [Google Scholar] [CrossRef] [PubMed]
- Schickel, R.; Park, S.M.; Murmann, A.E.; Peter, M.E. miR-200c regulates induction of apoptosis through CD95 by targeting FAP-1. Mol. Cell 2010, 38, 908–915. [Google Scholar] [CrossRef] [PubMed]
- Lerner, M.; Haneklaus, M.; Harada, M.; Grander, D. miR-200c regulates Noxa expression and sensitivity to proteasomal inhibitors. PLoS ONE 2012, 7, e36490. [Google Scholar] [CrossRef] [PubMed]
- Medina, R.; Zaidi, S.K.; Liu, C.G.; Stein, J.L.; van Wijnen, A.J.; Croce, C.M.; Stein, G.S. MicroRNAs 221 and 222 bypass quiescence and compromise cell survival. Cancer Res. 2008, 68, 2773–2780. [Google Scholar] [CrossRef] [PubMed]
- Le Sage, C.; Nagel, R.; Egan, D.A.; Schrier, M.; Mesman, E.; Mangiola, A.; Anile, C.; Maira, G.; Mercatelli, N.; Ciafre, S.A.; et al. Regulation of the p27Kip1 tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. EMBO J. 2007, 26, 3699–3708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.Z.; Zhang, J.X.; Zhang, A.L.; Shi, Z.D.; Han, L.; Jia, Z.F.; Yang, W.D.; Wang, G.X.; Jiang, T.; You, Y.P.; et al. miR-221 and miR-222 target PUMA to induce cell survival in glioblastoma. Mol. Cancer 2010, 9, 229. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Clevers, H. Wnt signalling in stem cells and cancer. Nature 2005, 434, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Fuerer, C.; Ching, W.; Harnish, K.; Logan, C.; Zeng, A.; Ten Berge, D.; Kalani, Y. Wnt signaling and stem cell control. Cold Spring Harb. Symp. Quant. Biol. 2008, 73, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Anastas, J.N.; Moon, R.T. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 2013, 13, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, S.; Bicknell, D.; Kaklamanis, L.; Bodmer, W.F. Molecular analysis of APC mutations in familial adenomatous polyposis and sporadic colon carcinomas. Lancet 1992, 340, 626–630. [Google Scholar] [CrossRef]
- Kinzler, K.W.; Nilbert, M.C.; Su, L.K.; Vogelstein, B.; Bryan, T.M.; Levy, D.B.; Smith, K.J.; Preisinger, A.C.; Hedge, P.; McKechnie, D.; et al. Identification of FAP locus genes from chromosome 5q21. Science 1991, 253, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Nishisho, I.; Nakamura, Y.; Miyoshi, Y.; Miki, Y.; Ando, H.; Horii, A.; Koyama, K.; Utsunomiya, J.; Baba, S.; Hedge, P. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science 1991, 253, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Nagel, R.; le Sage, C.; Diosdado, B.; van der Waal, M.; Oude Vrielink, J.A.; Bolijn, A.; Meijer, G.A.; Agami, R. Regulation of the adenomatous polyposis coli gene by the miR-135 family in colorectal cancer. Cancer Res. 2008, 68, 5795–5802. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Xu, Z. miR-27 promotes osteoblast differentiation by modulating Wnt signaling. Biochem. Biophys. Res. Commun. 2010, 402, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wei, W.; Cheng, N.; Wang, K.; Li, B.; Jiang, X.; Sun, S. Hepatitis C Virus-induced upregulation of miR-155 promotes hepatocarcinogenesis by activating Wnt signaling. Hepatology 2012, 56, 1631–1640. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Ye, Y.; Zhang, W.; Wang, J.; Chen, A.; Guo, F. miR142-3p promotes osteoblast differentiation by modulating Wnt signaling. Mol. Med. Rep. 2013, 7, 689–693. [Google Scholar] [PubMed]
- Li, M.; Tian, L.; Wang, L.; Yao, H.; Zhang, J.; Lu, J.; Sun, Y.; Gao, X.; Xiao, H.; Liu, M. Down-regulation of miR-129-5p inhibits growth and induces apoptosis in laryngeal squamous cell carcinoma by targeting APC. PLoS ONE 2013, 8, e77829. [Google Scholar] [CrossRef] [PubMed]
- Shen, G.; Jia, H.; Tai, Q.; Li, Y.; Chen, D. miR-106b downregulates adenomatous polyposis coli and promotes cell proliferation in human hepatocellular carcinoma. Carcinogenesis 2013, 34, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Emmrich, S.; Rasche, M.; Schoning, J.; Reimer, C.; Keihani, S.; Maroz, A.; Xie, Y.; Li, Z.; Schambach, A.; Reinhardt, D.; et al. miR-99a/100~125b tricistrons regulate hematopoietic stem and progenitor cell homeostasis by shifting the balance between TGFβ and Wnt signaling. Genes Dev. 2014, 28, 858–874. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Park, M.G.; Lee, S.A.; Park, S.Y.; Kim, H.J.; Yu, S.K.; Kim, C.S.; Kim, S.G.; Oh, J.S.; You, J.S.; et al. Downregulation of adenomatous polyposis coli by microRNA-663 promotes odontogenic differentiation through activation of Wnt/β-catenin signaling. Biochem. Biophys. Res. Commun. 2014, 446, 894–900. [Google Scholar] [CrossRef] [PubMed]
- Carraro, G.; Shrestha, A.; Rostkovius, J.; Contreras, A.; Chao, C.M.; El Agha, E.; Mackenzie, B.; Dilai, S.; Guidolin, D.; Taketo, M.M.; et al. miR-142-3p balances proliferation and differentiation of mesenchymal cells during lung development. Development 2014, 141, 1272–1281. [Google Scholar] [CrossRef] [PubMed]
- Kapinas, K.; Kessler, C.; Ricks, T.; Gronowicz, G.; Delany, A.M. miR-29 modulates Wnt signaling in human osteoblasts through a positive feedback loop. J. Biol. Chem. 2010, 285, 25221–25231. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Huang, T.; Zhao, X.; Cheng, L. MicroRNAs modulate the Wnt signaling pathway through targeting its inhibitors. Biochem. Biophys. Res. Commun. 2011, 408, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Wang, S.; Mayr, C.; Bartel, D.P.; Lodish, H.F. miR-150, a microRNA expressed in mature B and T cells, blocks early B cell development when expressed prematurely. Proc. Natl. Acad. Sci. USA 2007, 104, 7080–7085. [Google Scholar] [CrossRef] [PubMed]
- Saydam, O.; Shen, Y.; Wurdinger, T.; Senol, O.; Boke, E.; James, M.F.; Tannous, B.A.; Stemmer-Rachamimov, A.O.; Yi, M.; Stephens, R.M.; et al. Downregulated microRNA-200a in meningiomas promotes tumor growth by reducing E-cadherin and activating the Wnt/β-catenin signaling pathway. Mol. Cell Biol. 2009, 29, 5923–5940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abedi, N.; Mohammadi-Yeganeh, S.; Koochaki, A.; Karami, F.; Paryan, M. miR-141 as potential suppressor of β-catenin in breast cancer. Tumour Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Kennell, J.A.; Gerin, I.; MacDougald, O.A.; Cadigan, K.M. The microRNA miR-8 is a conserved negative regulator of Wnt signaling. Proc. Natl. Acad. Sci. USA 2008, 105, 15417–15422. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Wu, B.; Xiao, K.; Kang, J.; Xie, J.; Zhang, X.; Fan, Y. miR-146b-5p promotes metastasis and induces epithelial-mesenchymal transition in thyroid cancer by targeting ZNRF3. Cell. Physiol. Biochem. 2015, 35, 71–82. [Google Scholar] [CrossRef] [PubMed]
- De Lau, W.; Peng, W.C.; Gros, P.; Clevers, H. The R-spondin/Lgr5/Rnf43 module: Regulator of Wnt signal strength. Genes Dev. 2014, 28, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.L.; Jiang, J.K.; Yang, S.H.; Huang, T.S.; Lan, H.Y.; Teng, H.W.; Yang, C.Y.; Tsai, Y.P.; Lin, C.H.; Wang, H.W.; et al. MicroRNA-146a directs the symmetric division of Snail-dominant colorectal cancer stem cells. Nat. Cell Biol. 2014, 16, 268–280. [Google Scholar] [CrossRef] [PubMed]
- Yook, J.I.; Li, X.Y.; Ota, I.; Hu, C.; Kim, H.S.; Kim, N.H.; Cha, S.Y.; Ryu, J.K.; Choi, Y.J.; Kim, J.; et al. A Wnt-Axin2-GSK3β cascade regulates Snail1 activity in breast cancer cells. Nat. Cell Biol. 2006, 8, 1398–1406. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Davalos, V.; Moutinho, C.; Villanueva, A.; Boque, R.; Silva, P.; Carneiro, F.; Esteller, M. Dynamic epigenetic regulation of the microRNA-200 family mediates epithelial and mesenchymal transitions in human tumorigenesis. Oncogene 2012, 31, 2062–2074. [Google Scholar] [CrossRef] [PubMed]
- Mongroo, P.S.; Rustgi, A.K. The role of the miR-200 family in epithelial-mesenchymal transition. Cancer Biol. Ther. 2010, 10, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, S.; Brabletz, T. The ZEB/miR-200 feedback loop—A motor of cellular plasticity in development and cancer? EMBO Rep. 2010, 11, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Song, S.J.; Ito, K.; Ala, U.; Kats, L.; Webster, K.; Sun, S.M.; Jongen-Lavrencic, M.; Manova-Todorova, K.; Teruya-Feldstein, J.; Avigan, D.E.; et al. The oncogenic microRNA miR-22 targets the TET2 tumor suppressor to promote hematopoietic stem cell self-renewal and transformation. Cell Stem Cell 2013, 13, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Wels, C.; Joshi, S.; Koefinger, P.; Bergler, H.; Schaider, H. Transcriptional activation of ZEB1 by Slug leads to cooperative regulation of the epithelial-mesenchymal transition-like phenotype in melanoma. J. Invest. Dermatol. 2011, 131, 1877–1885. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Tam, W.L.; Shibue, T.; Kaygusuz, Y.; Reinhardt, F.; Ng Eaton, E.; Weinberg, R.A. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature 2015, 525, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Mizutani, K.; Minami, A.; Nobutani, K.; Kurita, S.; Nagino, M.; Shimono, Y.; Takai, Y. Suppression of the TGF-β1-induced protein expression of SNAI1 and N-cadherin by miR-199a. Genes Cells 2014, 19, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Li, W.C.; Hellem, M.R.; Rostad, K.; Popa, M.; McCormack, E.; Oyan, A.M.; Kalland, K.H.; Ke, X.S. miR-182 and miR-203 induce mesenchymal to epithelial transition and self-sufficiency of growth signals via repressing SNAI2 in prostate cells. Int. J. Cancer 2013, 133, 544–555. [Google Scholar] [CrossRef] [PubMed]
- Dontu, G.; Jackson, K.W.; McNicholas, E.; Kawamura, M.J.; Abdallah, W.M.; Wicha, M.S. Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res. 2004, 6, R605–R615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, W.; Harris, A.L. Notch signaling in breast cancer and tumor angiogenesis: Cross-talk and therapeutic potentials. J. Mammary Gland Biol. Neoplasia 2006, 11, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Gangopadhyay, S.; Nandy, A.; Hor, P.; Mukhopadhyay, A. Breast cancer stem cells: A novel therapeutic target. Clin. Breast Cancer 2013, 13, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Borggrefe, T.; Oswald, F. The Notch signaling pathway: Transcriptional regulation at Notch target genes. Cell. Mol. Life Sci. 2009, 66, 1631–1646. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, S.; Bajdak, K.; Meidhof, S.; Burk, U.; Niedermann, G.; Firat, E.; Wellner, U.; Dimmler, A.; Faller, G.; Schubert, J.; et al. The ZEB1/miR-200 feedback loop controls Notch signalling in cancer cells. EMBO J. 2011, 30, 770–782. [Google Scholar] [CrossRef] [PubMed]
- Kuang, W.; Tan, J.; Duan, Y.; Duan, J.; Wang, W.; Jin, F.; Jin, Z.; Yuan, X.; Liu, Y. Cyclic stretch induced miR-146a upregulation delays C2C12 myogenic differentiation through inhibition of Numb. Biochem. Biophys. Res. Commun. 2009, 378, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Forloni, M.; Dogra, S.K.; Dong, Y.; Conte, D., Jr.; Ou, J.; Zhu, L.J.; Deng, A.; Mahalingam, M.; Green, M.R.; Wajapeyee, N. miR-146a promotes the initiation and progression of melanoma by activating Notch signaling. eLife 2014, 3, e01460. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Steeg, P.S.; Price, J.E.; Krishnamurthy, S.; Mani, S.A.; Reuben, J.; Cristofanilli, M.; Dontu, G.; Bidaut, L.; Valero, V.; et al. Breast cancer metastasis: Challenges and opportunities. Cancer Res. 2009, 69, 4951–4953. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Patel, M.R.; Prescher, J.A.; Patsialou, A.; Qian, D.; Lin, J.; Wen, S.; Chang, Y.F.; Bachmann, M.H.; Shimono, Y.; et al. Cancer stem cells from human breast tumors are involved in spontaneous metastases in orthotopic mouse models. Proc. Natl. Acad. Sci. USA 2010, 107, 18115–18120. [Google Scholar] [CrossRef] [PubMed]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Pang, R.; Law, W.L.; Chu, A.C.; Poon, J.T.; Lam, C.S.; Chow, A.K.; Ng, L.; Cheung, L.W.; Lan, X.R.; Lan, H.Y.; et al. A subpopulation of CD26+ cancer stem cells with metastatic capacity in human colorectal cancer. Cell Stem Cell 2010, 6, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Lawson, D.A.; Bhakta, N.R.; Kessenbrock, K.; Prummel, K.D.; Yu, Y.; Takai, K.; Zhou, A.; Eyob, H.; Balakrishnan, S.; Wang, C.Y.; et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature 2015, 526, 131–135. [Google Scholar] [CrossRef] [PubMed]
- DeRose, Y.S.; Wang, G.; Lin, Y.C.; Bernard, P.S.; Buys, S.S.; Ebbert, M.T.; Factor, R.; Matsen, C.; Milash, B.A.; Nelson, E.; et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat. Med. 2011, 17, 1514–1520. [Google Scholar] [CrossRef] [PubMed]
- Bockhorn, J.; Prat, A.; Chang, Y.F.; Liu, X.; Huang, S.; Shang, M.; Nwachukwu, C.; Gomez-Vega, M.J.; Harrell, J.C.; Olopade, O.I.; et al. Differentiation and loss of malignant character of spontaneous pulmonary metastases in patient-derived breast cancer models. Cancer Res. 2014, 74, 7406–7417. [Google Scholar] [CrossRef] [PubMed]
- Nobutani, K.; Shimono, Y.; Mizutani, K.; Ueda, Y.; Suzuki, T.; Kitayama, M.; Minami, A.; Momose, K.; Miyawaki, K.; Akashi, K.; et al. Downregulation of CXCR4 in metastasized breast cancer cells and implication in their dormancy. PLoS ONE 2015, 10, e0130032. [Google Scholar] [CrossRef] [PubMed]
- Nobutani, K.; Shimono, Y.; Yoshida, M.; Mizutani, K.; Minami, A.; Kono, S.; Mukohara, T.; Yamasaki, T.; Itoh, T.; Takao, S.; et al. Absence of primary cilia in cell cycle-arrested human breast cancer cells. Genes Cells 2014, 19, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Liu, A.Y.; Fan, C.; Zheng, H.; Li, Y.; Zhang, C.; Wu, S.; Yu, D.; Huang, Z.; Liu, F.; et al. MicroRNA-33b inhibits breast cancer metastasis by targeting HMGA2, SALL4 and Twist1. Sci. Rep. 2015, 5, 9995. [Google Scholar] [CrossRef] [PubMed]
- Cuiffo, B.G.; Campagne, A.; Bell, G.W.; Lembo, A.; Orso, F.; Lien, E.C.; Bhasin, M.K.; Raimo, M.; Hanson, S.E.; Marusyk, A.; et al. MSC-regulated microRNAs converge on the transcription factor FOXP2 and promote breast cancer metastasis. Cell Stem Cell 2014, 15, 762–774. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, Q.; Wang, Z.; Jiang, J.; Yu, S.C.; Ping, Y.F.; Yang, J.; Xu, S.L.; Ye, X.Z.; Xu, C.; et al. Metastatic consequences of immune escape from NK cell cytotoxicity by human breast cancer stem cells. Cancer Res. 2014, 74, 5746–5757. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Cai, K.; Wang, J.; Wang, X.; Cheng, K.; Shi, F.; Jiang, L.; Zhang, Y.; Dou, J. miR-7, inhibited indirectly by lincRNA HOTAIR, directly inhibits SETDB1 and reverses the EMT of breast cancer stem cells by downregulating the STAT3 pathway. Stem Cells 2014, 32, 2858–2868. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Zhao, Y.; Li, Z.; Yao, R.; Ma, M.; Gao, Y.; Zhao, L.; Zhang, Y.; Huang, B.; Lu, J. LincRNA-ROR induces epithelial-to-mesenchymal transition and contributes to breast cancer tumorigenesis and metastasis. Cell Death Dis. 2014, 5, e1287. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [PubMed]
- Falcone, G.; Felsani, A.; D’Agnano, I. Signaling by exosomal microRNAs in cancer. J. Exp. Clin. Cancer Res. 2015, 34, 32. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, N.; Kosaka, N.; Ono, M.; Katsuda, T.; Yoshioka, Y.; Tamura, K.; Lotvall, J.; Nakagama, H.; Ochiya, T. Brain metastatic cancer cells release microRNA-181c-containing extracellular vesicles capable of destructing blood-brain barrier. Nat. Commun. 2015, 6, 6716. [Google Scholar] [CrossRef] [PubMed]
- Le, M.T.; Hamar, P.; Guo, C.; Basar, E.; Perdigao-Henriques, R.; Balaj, L.; Lieberman, J. miR-200-containing extracellular vesicles promote breast cancer cell metastasis. J. Clin. Invest. 2014, 124, 5109–5128. [Google Scholar] [CrossRef] [PubMed]
- Rucci, N.; Teti, A. Osteomimicry: How tumor cells try to deceive the bone. Front. Biosci. (Schol. Ed.) 2010, 2, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Croset, M.; Kan, C.; Clezardin, P. Tumour-derived miRNAs and bone metastasis. Bonekey Rep. 2015, 4, 688. [Google Scholar] [CrossRef] [PubMed]
- Ell, B.; Kang, Y. MicroRNAs as regulators of bone homeostasis and bone metastasis. Bonekey Rep. 2014, 3, 549. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.Q.; Maeda, Y.; Taipaleenmaki, H.; Zhang, W.; Jafferji, M.; Gordon, J.A.; Li, Z.; Croce, C.M.; van Wijnen, A.J.; Stein, J.L.; et al. miR-218 directs a Wnt signaling circuit to promote differentiation of osteoblasts and osteomimicry of metastatic cancer cells. J. Biol. Chem. 2012, 287, 42084–42092. [Google Scholar] [CrossRef] [PubMed]
- Dykxhoorn, D.M.; Wu, Y.; Xie, H.; Yu, F.; Lal, A.; Petrocca, F.; Martinvalet, D.; Song, E.; Lim, B.; Lieberman, J. miR-200 enhances mouse breast cancer cell colonization to form distant metastases. PLoS ONE 2009, 4, e7181. [Google Scholar] [CrossRef] [PubMed]
- De Smet, M.D.; Meenken, C.J.; van den Horn, G.J. Fomivirsen—A phosphorothioate oligonucleotide for the treatment of CMV retinitis. Ocul. Immunol. Inflamm. 1999, 7, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Z.; Gemeinhart, R.A. Progress in microRNA delivery. J. Control Release 2013, 172, 962–974. [Google Scholar] [CrossRef] [PubMed]
- Bandiera, S.; Pfeffer, S.; Baumert, T.F.; Zeisel, M.B. miR-122—A key factor and therapeutic target in liver disease. J. Hepatol. 2015, 62, 448–457. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shimono, Y.; Mukohyama, J.; Nakamura, S.-i.; Minami, H. MicroRNA Regulation of Human Breast Cancer Stem Cells. J. Clin. Med. 2016, 5, 2. https://doi.org/10.3390/jcm5010002
Shimono Y, Mukohyama J, Nakamura S-i, Minami H. MicroRNA Regulation of Human Breast Cancer Stem Cells. Journal of Clinical Medicine. 2016; 5(1):2. https://doi.org/10.3390/jcm5010002
Chicago/Turabian StyleShimono, Yohei, Junko Mukohyama, Shun-ichi Nakamura, and Hironobu Minami. 2016. "MicroRNA Regulation of Human Breast Cancer Stem Cells" Journal of Clinical Medicine 5, no. 1: 2. https://doi.org/10.3390/jcm5010002