
NLRP3 Inflammasome and Pathobiology in AMD

Abstract
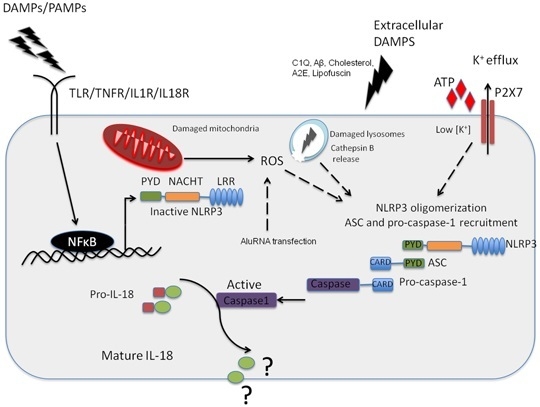
:
1. Introduction
2. The NLRP3 Inflammasome and Its Role in AMD
3. Effects of IL-1β and IL-18 on RPE in AMD
4. RPE Cell Death in AMD

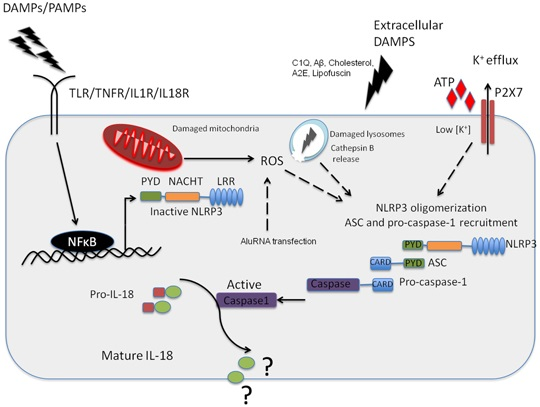{kind=link}
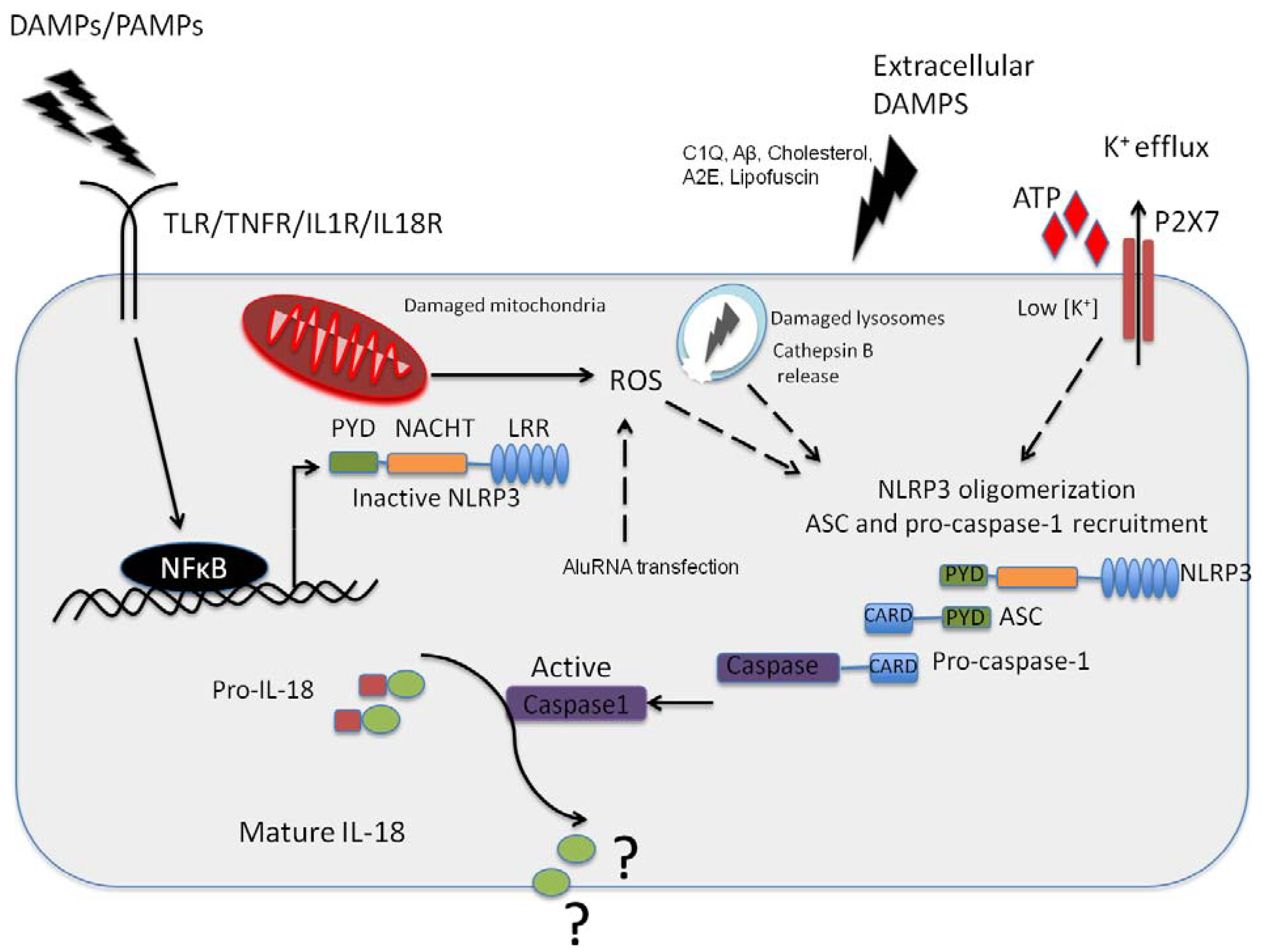{kind=link}
| Characteristics | Apoptosis | Pyroptosis | Necrosis | |
|---|---|---|---|---|
| Morphology | Cell lysis | NO | YES | YES |
| Cell swelling | NO | YES | YES | |
| Pore formation | NO | YES | YES | |
| Membrane blebbing | YES | NO | NO | |
| DNA fragmentation | YES | YES | YES | |
| Mechanism | Caspase-1 | NO | YES | NO |
| Caspase-3 | YES | NO | NO | |
| Cytochrome-c release | YES | NO | NO | |
| Outcome | Inflammation | NO | YES | YES |
| Programmed cell death | YES | YES | NO |
5. Role of Autophagy in Inflammasome Regulation in AMD

6. Discussion
7. Conclusions
- The NLRP3 inflammasome appears to play a central role in both atrophic and neovascular AMD.
- Mature IL-18 and IL-1β can be produced by both the RPE and systemic/resident myeloid derived cells, however whether mature cytokines can induce RPE specific apoptosis is still unclear.
- A robust elucidation of the role played by the NLRP3 inflammasome in atrophic and neovascular AMD may lead to potential strategies to treat both clinical forms of the condition.
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fritsche, L.G.; Chen, W.; Schu, M.; Yaspan, B.L.; Yu, Y.; Thorleifsson, G.; Zack, D.J.; Arakawa, S.; Cipriani, V.; Ripke, S.; et al. Seven new loci associated with age-related macular degeneration. Nat. Genet. 2013, 45, 433–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Age-Related Eye Disease Study Research Group. Risk factors associated with age-related macular degeneration. A case-control study in the age-related eye disease study: Age-related eye disease study report number 3. Ophthalmology 2000, 107, 2224–2232. [Google Scholar]
- Clemons, T.E.; Milton, R.C.; Klein, R.; Seddon, J.M.; Ferris, F.L., III. Risk factors for the incidence of advanced age-related macular degeneration in the age-related eye disease study (AREDS) AREDS report no. 19. Ophthalmology 2005, 112, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Dasari, B.; Prasanthi, J.R.; Marwarha, G.; Singh, B.B.; Ghribi, O. Cholesterol-enriched diet causes age-related macular degeneration-like pathology in rabbit retina. BMC Ophthalmol. 2011, 11. [Google Scholar] [CrossRef]
- Parekh, N.; Voland, R.P.; Moeller, S.M.; Blodi, B.A.; Ritenbaugh, C.; Chappell, R.J.; Wallace, R.B.; Mares, J.A. Association between dietary fat intake and age-related macular degeneration in the carotenoids in age-related eye disease study (CAREDS): An ancillary study of the women’s health initiative. Arch. Ophthalmol. 2009, 127, 1483–1493. [Google Scholar] [CrossRef] [PubMed]
- Jager, R.D.; Mieler, W.F.; Miller, J.W. Age-related macular degeneration. N. Engl. J. Med. 2008, 358, 2606–2617. [Google Scholar] [CrossRef] [PubMed]
- Sui, G.Y.; Liu, G.C.; Liu, G.Y.; Gao, Y.Y.; Deng, Y.; Wang, W.Y.; Tong, S.H.; Wang, L. Is sunlight exposure a risk factor for age-related macular degeneration? A systematic review and meta-analysis. Br. J. Ophthalmol. 2013, 97, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Millen, A.E.; Voland, R.; Sondel, S.A.; Parekh, N.; Horst, R.L.; Wallace, R.B.; Hageman, G.S.; Chappell, R.; Blodi, B.A.; Klein, M.L.; et al. Vitamin D status and early age-related macular degeneration in postmenopausal women. Arch. Ophthalmol. 2011, 129, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Nita, M.; Grzybowski, A.; Ascaso, F.J.; Huerva, V. Age-related macular degeneration in the aspect of chronic low-grade inflammation (pathophysiological parainflammation). Med. Inflamm. 2014, 2014. [Google Scholar] [CrossRef]
- Whitcup, S.M.; Sodhi, A.; Atkinson, J.P.; Holers, V.M.; Sinha, D.; Rohrer, B.; Dick, A.D. The role of the immune response in age-related macular degeneration. Int. J. Inflamm. 2013, 2013. [Google Scholar] [CrossRef]
- Mullins, R.F.; Russell, S.R.; Anderson, D.H.; Hageman, G.S. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J. 2000, 14, 835–846. [Google Scholar] [PubMed]
- Johnson, L.V.; Leitner, W.P.; Rivest, A.J.; Staples, M.K.; Radeke, M.J.; Anderson, D.H. The Alzheimer’s a beta -peptide is deposited at sites of complement activation in pathologic deposits associated with aging and age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2002, 99, 11830–11835. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Clark, M.E.; Crossman, D.K.; Kojima, K.; Messinger, J.D.; Mobley, J.A.; Curcio, C.A. Abundant lipid and protein components of drusen. PLoS One 2010, 5, e10329. [Google Scholar] [CrossRef] [PubMed]
- Crabb, J.W.; Miyagi, M.; Gu, X.; Shadrach, K.; West, K.A.; Sakaguchi, H.; Kamei, M.; Hasan, A.; Yan, L.; Rayborn, M.E.; et al. Drusen proteome analysis: An approach to the etiology of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2002, 99, 14682–14687. [Google Scholar] [CrossRef] [PubMed]
- Sparrow, J.R.; Boulton, M. RPE lipofuscin and its role in retinal pathobiology. Exp.Eye Res. 2005, 80, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Pascolini, D.; Mariotti, S.P. Global estimates of visual impairment: 2010. Br. J. Ophthalmol. 2012, 96, 614–618. [Google Scholar] [CrossRef] [PubMed]
- Access Economics. The Global Economic Cost of Visual Impairment. Available online: http://www.icoph.org/resources/146/The-Global-Economic-Cost-of-Visual-Impairment.html (accessed on 17 September 2014).
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2. [Google Scholar] [CrossRef]
- Bright Focus Foundation. Available online: http://www.brightfocus.org/macular/about/under-standing/facts.html (accessed on 17 September 2014).
- National Council for the Blind of Ireland. The Cost of Blindness in the Republic of Ireland 2010–2020. Available online: https://www.ncbi.ie/sites/default/files/cost_of_blindness_pdf_nov_2012.pdf (accessed on 17 September 2014).
- National Council for the Blind of Ireland. The Economic Impact of Vision Impairment and Blindness in the Republic of Ireland. Available online: http://www.ncbi.ie/files/Cost_of_Sight_Loss_Full_Report_July_2011.pdf (accessed on 18 September 2014).
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Dowling, J.K.; O’Neill, L.A. Biochemical regulation of the inflammasome. Crit. Rev. Biochem. Mol. Biol. 2012, 47, 424–443. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, K.; Muto, J.; Taylor, K.R.; Cogen, A.L.; Audish, D.; Bertin, J.; Grant, E.P.; Coyle, A.J.; Misaghi, A.; Hoffman, H.M.; et al. NLRP3/cryopyrin is necessary for interleukin-1beta (IL-1beta) release in response to hyaluronan, an endogenous trigger of inflammation in response to injury. J. Biol. Chem. 2009, 284, 12762–12771. [Google Scholar] [CrossRef] [PubMed]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Petrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nunez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [PubMed]
- Compan, V.; Baroja-Mazo, A.; Lopez-Castejon, G.; Gomez, A.I.; Martinez, C.M.; Angosto, D.; Montero, M.T.; Herranz, A.S.; Bazan, E.; Reimers, D.; et al. Cell volume regulation modulates NLRP3 inflammasome activation. Immunity 2012, 37, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Rajamaki, K.; Nordstrom, T.; Nurmi, K.; Akerman, K.E.; Kovanen, P.T.; Oorni, K.; Eklund, K.K. Extracellular acidosis is a novel danger signal alerting innate immunity via the NLRP3 inflammasome. J. Biol. Chem. 2013, 288, 13410–13419. [Google Scholar] [CrossRef] [PubMed]
- Babelova, A.; Moreth, K.; Tsalastra-Greul, W.; Zeng-Brouwers, J.; Eickelberg, O.; Young, M.F.; Bruckner, P.; Pfeilschifter, J.; Schaefer, R.M.; Grone, H.J.; et al. Biglycan, a danger signal that activates the NLRP3 inflammasome via toll-like and P2X receptors. J. Biol. Chem. 2009, 284, 24035–24048. [Google Scholar] [CrossRef] [PubMed]
- Eisenbarth, S.C.; Colegio, O.R.; O’Connor, W.; Sutterwala, F.S.; Flavell, R.A. Crucial role for the NALP3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature 2008, 453, 1122–1126. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Dostert, C.; Petrilli, V.; van Bruggen, R.; Steele, C.; Mossman, B.T.; Tschopp, J. Innate immune activation through NALP3 inflammasome sensing of asbestos and silica. Science 2008, 320, 674–677. [Google Scholar] [CrossRef] [PubMed]
- Yazdi, A.S.; Guarda, G.; Riteau, N.; Drexler, S.K.; Tardivel, A.; Couillin, I.; Tschopp, J. Nanoparticles activate the nlr pyrin domain containing 3 (NLRP3) inflammasome and cause pulmonary inflammation through release of IL-1alpha and IL-1beta. Proc. Natl. Acad. Sci. USA 2010, 107, 19449–19454. [Google Scholar] [CrossRef] [PubMed]
- Kahlenberg, J.M.; Dubyak, G.R. Mechanisms of caspase-1 activation by P2X7 receptor-mediated K+ release. Am. J. Physiol. Cell Physiol. 2004, 286, C1100–C1108. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Planillo, R.; Kuffa, P.; Martinez-Colon, G.; Smith, B.L.; Rajendiran, T.M.; Nunez, G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef] [PubMed]
- Petrilli, V.; Papin, S.; Dostert, C.; Mayor, A.; Martinon, F.; Tschopp, J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007, 14, 1583–1589. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef] [PubMed]
- Cruz, C.M.; Rinna, A.; Forman, H.J.; Ventura, A.L.; Persechini, P.M.; Ojcius, D.M. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J. Biol. Chem. 2007, 282, 2871–2879. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Castejon, G.; Luheshi, N.M.; Compan, V.; High, S.; Whitehead, R.C.; Flitsch, S.; Kirov, A.; Prudovsky, I.; Swanton, E.; Brough, D. Deubiquitinases regulate the activity of caspase-1 and interleukin-1beta secretion via assembly of the inflammasome. J. Biol. Chem. 2013, 288, 2721–2733. [Google Scholar] [CrossRef] [PubMed]
- Hoegen, T.; Tremel, N.; Klein, M.; Angele, B.; Wagner, H.; Kirschning, C.; Pfister, H.W.; Fontana, A.; Hammerschmidt, S.; Koedel, U. The NLRP3 inflammasome contributes to brain injury in pneumococcal meningitis and is activated through ATP-dependent lysosomal cathepsin B release. J. Immunol. 2011, 187, 5440–5451. [Google Scholar] [CrossRef] [PubMed]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: NF-κB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef] [PubMed]
- Doyle, S.L.; Campbell, M.; Ozaki, E.; Salomon, R.G.; Mori, A.; Kenna, P.F.; Farrar, G.J.; Kiang, A.S.; Humphries, M.M.; Lavelle, E.C.; et al. NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nat. Med. 2012, 18, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Tseng, W.A.; Thein, T.; Kinnunen, K.; Lashkari, K.; Gregory, M.S.; D’Amore, P.A.; Ksander, B.R. NLRP3 inflammasome activation in retinal pigment epithelial cells by lysosomal destabilization: Implications for age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2013, 54, 110–120. [Google Scholar] [CrossRef]
- Kauppinen, A.; Niskanen, H.; Suuronen, T.; Kinnunen, K.; Salminen, A.; Kaarniranta, K. Oxidative stress activates NLRP3 inflammasomes in ARPE-19 cells—Implications for age-related macular degeneration (AMD). Immunol. Lett. 2012, 147, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Tarallo, V.; Hirano, Y.; Gelfand, B.D.; Dridi, S.; Kerur, N.; Kim, Y.; Cho, W.G.; Kaneko, H.; Fowler, B.J.; Bogdanovich, S.; et al. Dicer1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MYD88. Cell 2012, 149, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.T.; Gao, J.; Cao, S.; Sandhu, N.; Cui, J.Z.; Chou, C.L.; Fang, E.; Matsubara, J.A. Inflammatory mediators induced by amyloid-beta in the retina and RPE in vivo: Implications for inflammasome activation in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2013, 54, 2225–2237. [Google Scholar] [CrossRef]
- Anderson, O.A.; Finkelstein, A.; Shima, D.T. A2E induces IL-1ss production in retinal pigment epithelial cells via the NLRP3 inflammasome. PLoS One 2013, 8, e67263. [Google Scholar] [CrossRef] [PubMed]
- Marneros, A.G. NLRP3 inflammasome blockade inhibits VEGF-a-induced age-related macular degeneration. Cell Rep. 2013, 4, 945–958. [Google Scholar] [CrossRef] [PubMed]
- Puren, A.J.; Fantuzzi, G.; Dinarello, C.A. Gene expression, synthesis, and secretion of interleukin 18 and interleukin 1beta are differentially regulated in human blood mononuclear cells and mouse spleen cells. Proc. Natl. Acad. Sci. USA 1999, 96, 2256–2261. [Google Scholar] [CrossRef] [PubMed]
- Hiscott, J.; Marois, J.; Garoufalis, J.; D’Addario, M.; Roulston, A.; Kwan, I.; Pepin, N.; Lacoste, J.; Nguyen, H.; Bensi, G.; et al. Characterization of a functional NF-κB site in the human interleukin 1 beta promoter: Evidence for a positive autoregulatory loop. Mol. Cell. Biol. 1993, 13, 6231–6240. [Google Scholar] [PubMed]
- Dinarello, C.A. Interleukin 1 and interleukin 18 as mediators of inflammation and the aging process. Am. J. Clin. Nutr. 2006, 83 (Suppl. S2), 447–455. [Google Scholar]
- Dinarello, C.A. Immunological and inflammatory functions of the interleukin-1 family. Ann. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef]
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The interleukin-1 family: Back to the future. Immunity 2013, 39, 1003–1018. [Google Scholar] [CrossRef] [PubMed]
- Ben-Av, P.; Crofford, L.J.; Wilder, R.L.; Hla, T. Induction of vascular endothelial growth factor expression in synovial fibroblasts by prostaglandin e and interleukin-1: A potential mechanism for inflammatory angiogenesis. FEBS Lett. 1995, 372, 83–87. [Google Scholar] [CrossRef] [PubMed]
- El Awad, B.; Kreft, B.; Wolber, E.M.; Hellwig-Burgel, T.; Metzen, E.; Fandrey, J.; Jelkmann, W. Hypoxia and interleukin-1beta stimulate vascular endothelial growth factor production in human proximal tubular cells. Kidney Int. 2000, 58, 43–50. [Google Scholar]
- Jung, Y.D.; Liu, W.; Reinmuth, N.; Ahmad, S.A.; Fan, F.; Gallick, G.E.; Ellis, L.M. Vascular endothelial growth factor is upregulated by interleukin-1 beta in human vascular smooth muscle cells via the p38 mitogen-activated protein kinase pathway. Angiogenesis 2001, 4, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Voronov, E.; Shouval, D.S.; Krelin, Y.; Cagnano, E.; Benharroch, D.; Iwakura, Y.; Dinarello, C.A.; Apte, R.N. IL-1 is required for tumor invasiveness and angiogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 2645–2650. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Nokihara, H.; Yamamoto, A.; Goto, H.; Ogawa, H.; Kanematsu, T.; Miki, T.; Uehara, H.; Saijo, Y.; Nukiwa, T.; et al. Multifunctional interleukin-1beta promotes metastasis of human lung cancer cells in scid mice via enhanced expression of adhesion-, invasion- and angiogenesis-related molecules. Cancer Sci. 2003, 94, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Sola-Villa, D.; Camacho, M.; Sola, R.; Soler, M.; Diaz, J.M.; Vila, L. IL-1beta induces VEGF, independently of PGE2 induction, mainly through the pi3-k/mtor pathway in renal mesangial cells. Kidney Int. 2006, 70, 1935–1941. [Google Scholar] [PubMed]
- Nakanishi, K.; Yoshimoto, T.; Tsutsui, H.; Okamura, H. Interleukin-L8 regulates both TH1 and TH2 responses. Ann. Rev. Immunol. 2001, 19, 423–474. [Google Scholar] [CrossRef]
- Nakanishi, K.; Yoshimoto, T.; Tsutsui, H.; Okamura, H. Interleukin-18 is a unique cytokine that stimulates both TH1 and TH2 responses depending on its cytokine milieu. Cytokine Growth Factor Rev. 2001, 12, 53–72. [Google Scholar] [CrossRef] [PubMed]
- Boraschi, D.; Dinarello, C.A. IL-18 in autoimmunity: Review. Eur. Cytokine Netw. 2006, 17, 224–252. [Google Scholar] [PubMed]
- Dinarello, C.A.; Novick, D.; Kim, S.; Kaplanski, G. Interleukin-L8 and IL-18 binding protein. Front. Immunol. 2013, 4, 1–10. [Google Scholar] [PubMed]
- Bamias, G.; Corridoni, D.; Pizarro, T.T.; Cominelli, F. New insights into the dichotomous role of innate cytokines in gut homeostasis and inflammation. Cytokine 2012, 59, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Bian, Z.M.; Elner, S.G.; Yoshida, A.; Kunkel, S.L.; Su, J.; Elner, V.M. Activation of p38, ERK1/2 and NIK pathways is required for IL-1beta and TNF-alpha-induced chemokine expression in human retinal pigment epithelial cells. Exp. Eye Res. 2001, 73, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Sugano, E.; Saigo, Y.; Tamai, M. Interleukin-1beta and barrier function of retinal pigment epithelial cells (ARPE-19): Aberrant expression of junctional complex molecules. Investig. Ophthalmol. Vis. Sci. 2003, 44, 4097–4104. [Google Scholar] [CrossRef]
- Peng, S.; Gan, G.; Rao, V.S.; Adelman, R.A.; Rizzolo, L.J. Effects of proinflammatory cytokines on the claudin-19 rich tight junctions of human retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 2012, 53, 5016–5028. [Google Scholar] [CrossRef]
- Olson, J.L.; Courtney, R.J.; Rouhani, B.; Mandava, N.; Dinarello, C.A. Intravitreal anakinra inhibits choroidal neovascular membrane growth in a rat model. Ocul. Immunol. Inflamm. 2009, 17, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Doyle, S.L.; Ozaki, E.; Brennan, K.; Humphries, M.M.; Mulfaul, K.; Keaney, J.; Kenna, P.F.; Maminishkis, A.; Kiang, A.S.; Saunders, S.P.; et al. IL-18 attenuates experimental choroidal neovascularization as a potential therapy for wet age-related macular degeneration. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef]
- Doyle, S.L.; Adamson, P.; Lopez, F.J.; Humphries, P.; Campbell, M. Reply to IL-18 is not therapeutic for neovascular age-related macular degeneration. Nat. Med. 2014, 20, 1376–1377. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Choy, D.F.; Yoshida, T.; Iwase, T.; Hafiz, G.; Xie, B.; Hackett, S.F.; Arron, J.R.; Campochiaro, P.A. Interleukin-18 has antipermeablity and antiangiogenic activities in the eye: Reciprocal suppression with vegf. J. Cell. Physiol. 2014, 229, 974–983. [Google Scholar] [CrossRef] [PubMed]
- Miao, E.A.; Leaf, I.A.; Treuting, P.M.; Mao, D.P.; Dors, M.; Sarkar, A.; Warren, S.E.; Wewers, M.D.; Aderem, A. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol. 2010, 11, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Fink, S.L.; Cookson, B.T. Apoptosis, pyroptosis, and necrosis: Mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Holler, N.; Zaru, R.; Micheau, O.; Thome, M.; Attinger, A.; Valitutti, S.; Bodmer, J.L.; Schneider, P.; Seed, B.; Tschopp, J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase rip as effector molecule. Nat. Immunol. 2000, 1, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Matsumoto, H.; Roh, M.; Giani, A.; Kataoka, K.; Morizane, Y.; Kayama, M.; Thanos, A.; Nakatake, S.; Notomi, S.; et al. Programmed necrosis, not apoptosis, is a key mediator of cell loss and damp-mediated inflammation in dsrna-induced retinal degeneration. Cell Death Differ. 2014, 21, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Shelley, E.J.; Madigan, M.C.; Natoli, R.; Penfold, P.L.; Provis, J.M. Cone degeneration in aging and age-related macular degeneration. Arch. Ophthalmol. 2009, 127, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, H.; Dridi, S.; Tarallo, V.; Gelfand, B.D.; Fowler, B.J.; Cho, W.G.; Kleinman, M.E.; Ponicsan, S.L.; Hauswirth, W.W.; Chiodo, V.A.; et al. Dicer1 deficit induces alu rna toxicity in age-related macular degeneration. Nature 2011, 471, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Mills, K.H.; Harris, J. Autophagy and inflammatory diseases. Immunol. Cell Biol. 2013, 91, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Harris, J. Autophagy and IL-1 family cytokines. Front. Immunol. 2013, 4. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.; Hartman, M.; Roche, C.; Zeng, S.G.; O’Shea, A.; Sharp, F.A.; Lambe, E.M.; Creagh, E.M.; Golenbock, D.T.; Tschopp, J.; et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J. Biol. Chem. 2011, 286, 9587–9597. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.S.; Shenderov, K.; Huang, N.N.; Kabat, J.; Abu-Asab, M.; Fitzgerald, K.A.; Sher, A.; Kehrl, J.H. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012, 13, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Byrne, B.G.; Dubuisson, J.F.; Joshi, A.D.; Persson, J.J.; Swanson, M.S. Inflammasome components coordinate autophagy and pyroptosis as macrophage responses to infection. MBIO 2013, 4. [Google Scholar] [CrossRef]
- Wang, A.L.; Lukas, T.J.; Yuan, M.; Du, N.; Tso, M.O.; Neufeld, A.H. Autophagy and exosomes in the aged retinal pigment epithelium: Possible relevance to drusen formation and age-related macular degeneration. PLoS One 2009, 4, e4160. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Zhao, H.; Martinez, J.; Doggett, T.A.; Kolesnikov, A.V.; Tang, P.H.; Ablonczy, Z.; Chan, C.C.; Zhou, Z.; Green, D.R.; et al. Noncanonical autophagy promotes the visual cycle. Cell 2013, 154, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.F.; Maguire, M.G.; Ying, G.S.; Grunwald, J.E.; Fine, S.L.; Jaffe, G.J. Ranibizumab and bevacizumab for neovascular age-related macular degeneration. N. Engl. J. Med. 2011, 364, 1897–1908. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Marneros, A.G. Macrophages are essential for the early wound healing response and the formation of a fibrovascular scar. Am. J. Pathol. 2013, 182, 2407–2417. [Google Scholar] [CrossRef] [PubMed]
- Dupaul-Chicoine, J.; Yeretssian, G.; Doiron, K.; Bergstrom, K.S.; McIntire, C.R.; LeBlanc, P.M.; Meunier, C.; Turbide, C.; Gros, P.; Beauchemin, N.; et al. Control of intestinal homeostasis, colitis, and colitis-associated colorectal cancer by the inflammatory caspases. Immunity 2010, 32, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Allen, I.C.; TeKippe, E.M.; Woodford, R.M.; Uronis, J.M.; Holl, E.K.; Rogers, A.B.; Herfarth, H.H.; Jobin, C.; Ting, J.P. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J. Exp. Med. 2010, 207, 1045–1056. [Google Scholar] [CrossRef] [PubMed]
- Zaki, M.H.; Boyd, K.L.; Vogel, P.; Kastan, M.B.; Lamkanfi, M.; Kanneganti, T.D. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 2010, 32, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Zaki, M.H.; Vogel, P.; Body-Malapel, M.; Lamkanfi, M.; Kanneganti, T.D. IL-18 production downstream of the NLRP3 inflammasome confers protection against colorectal tumor formation. J. Immunol. 2010, 185, 4912–4920. [Google Scholar] [CrossRef] [PubMed]
- Qiao, H.; Sonoda, K.H.; Sassa, Y.; Hisatomi, T.; Yoshikawa, H.; Ikeda, Y.; Murata, T.; Akira, S.; Ishibashi, T. Abnormal retinal vascular development in IL-18 knockout mice. Lab. Investig. J. Tech. Methods Pathol. 2004, 84, 973–980. [Google Scholar] [CrossRef]
- Qiao, H.; Sonoda, K.H.; Ikeda, Y.; Yoshimura, T.; Hijioka, K.; Jo, Y.J.; Sassa, Y.; Tsutsumi-Miyahara, C.; Hata, Y.; Akira, S.; et al. Interleukin-L8 regulates pathological intraocular neovascularization. J. Leukoc. Biol. 2007, 81, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Mallat, Z.; Silvestre, J.S.; Le Ricousse-Roussanne, S.; Lecomte-Raclet, L.; Corbaz, A.; Clergue, M.; Duriez, M.; Barateau, V.; Akira, S.; Tedgui, A.; et al. Interleukin-L8/interleukin-L8 binding protein signaling modulates ischemia-induced neovascularization in mice hindlimb. Circ. Res. 2002, 91, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, C.M.; Salhany, K.E.; Wysocka, M.; Aruga, E.; Kurzawa, H.; Chang, A.E.; Hunter, C.A.; Fox, J.C.; Trinchieri, G.; Lee, W.M. Interleukin-12 and interleukin-18 synergistically induce murine tumor regression which involves inhibition of angiogenesis. J. Clin. Investig. 1998, 101, 1441–1452. [Google Scholar] [CrossRef] [PubMed]
- Kerur, N.; Hirano, Y.; Tarallo, V.; Fowler, B.J.; Bastos-Carvalho, A.; Yasuma, T.; Yasuma, R.; Kim, Y.; Hinton, D.R.; Kirschning, C.J.; et al. TLR-independent and P2X7-dependent signaling mediate alu rna-induced NLRP3 inflammasome activation in geographic atrophy. Investig. Ophthalmol. Vis. Sci. 2013, 54, 7395–7401. [Google Scholar] [CrossRef]
- Kindzelskii, A.L.; Elner, V.M.; Elner, S.G.; Yang, D.; Hughes, B.A.; Petty, H.R. Toll-like receptor 4 (TLR4) of retinal pigment epithelial cells participates in transmembrane signaling in response to photoreceptor outer segments. J. Gen. Physiol. 2004, 124, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Fernandes-Alnemri, T.; Wu, J.; Yu, J.W.; Datta, P.; Miller, B.; Jankowski, W.; Rosenberg, S.; Zhang, J.; Alnemri, E.S. The pyroptosome: A supramolecular assembly of Asc dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 2007, 14, 1590–1604. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.; Magupalli, V.G.; Ruan, J.; Yin, Q.; Atianand, M.K.; Vos, M.R.; Schroder, G.F.; Fitzgerald, K.A.; Wu, H.; Egelman, E.H. Unified polymerization mechanism for the assembly of asc-dependent inflammasomes. Cell 2014, 156, 1193–1206. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Chen, J.; Xu, H.; Liu, S.; Jiang, Q.X.; Halfmann, R.; Chen, Z.J. Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell 2014, 156, 1207–1222. [Google Scholar] [CrossRef] [PubMed]
- Grunwald, G.B. Structure and function of the retinal pigment epithelium. In Duane’s Ophthalmology, 6th ed.; Tasman, W., Jaeger, E.A., Eds.; Lippincott Williams & Wilkins: Philadelphia, Pennsylvania, USA, 2006. [Google Scholar]
- O’Neill, L.A.; Bowie, A.G. The family of five: Tir-domain-containing adaptors in toll-like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Fowler, B.J.; Gelfand, B.D.; Kim, Y.; Kerur, N.; Tarallo, V.; Hirano, Y.; Amarnath, S.; Fowler, D.H.; Radwan, M.; Young, M.T.; et al. Nucleoside reverse transcriptase inhibitors possess intrinsic anti-inflammatory activity. Science 2014, 346, 1000–1003. [Google Scholar] [CrossRef] [PubMed]
- Ablonczy, Z.; Dahrouj, M.; Marneros, A.G. Progressive dysfunction of the retinal pigment epithelium and retina due to increased VEGF-A levels. FASEB J. 2014, 28, 2369–2379. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.T.; Wang, A.; To, E.; Gao, J.; Cao, S.; Cui, J.Z.; Matsubara, J.A. Vinpocetine inhibits amyloid-beta induced activation of NF-kappaB, NLRP3 inflammasome and cytokine production in retinal pigment epithelial cells. Exp. Eye Res. 2014, 127C, 49–58. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Celkova, L.; Doyle, S.L.; Campbell, M. NLRP3 Inflammasome and Pathobiology in AMD. J. Clin. Med. 2015, 4, 172-192. https://doi.org/10.3390/jcm4010172
Celkova L, Doyle SL, Campbell M. NLRP3 Inflammasome and Pathobiology in AMD. Journal of Clinical Medicine. 2015; 4(1):172-192. https://doi.org/10.3390/jcm4010172
Chicago/Turabian StyleCelkova, Lucia, Sarah L. Doyle, and Matthew Campbell. 2015. "NLRP3 Inflammasome and Pathobiology in AMD" Journal of Clinical Medicine 4, no. 1: 172-192. https://doi.org/10.3390/jcm4010172