
Primary Coenzyme Q10 Deficiency-Related Ataxias

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Material and Methods
3. Case Report
4. Primary Coenzyme Q10 Deficiency-Related Ataxia: The Example of CoQ8A

5. Other Causes of Primary Coenzyme Q10 Deficiency-Related Ataxia
6. Treatment Options
7. Non-Pharmacological Management
8. Discussion and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Orsucci, D.; Caldarazzo Ienco, E.; Rossi, A.; Siciliano, G.; Mancuso, M. Mitochondrial Syndromes Revisited. J. Clin. Med. 2021, 10, 1249. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hidalgo-Gutiérrez, A.; González-García, P.; Díaz-Casado, M.E.; Barriocanal-Casado, E.; López-Herrador, S.; Quinzii, C.M.; López, L.C. Metabolic Targets of Coenzyme Q10 in Mitochondria. Antioxidants 2021, 10, 520. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Baschiera, E.; Sorrentino, U.; Calderan, C.; Desbats, M.A.; Salviati, L. The multiple roles of coenzyme Q in cellular homeostasis and their relevance for the pathogenesis of coenzyme Q deficiency. Free Radic. Biol. Med. 2021, 166, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Emmanuele, V.; López, L.C.; Berardo, A.; Naini, A.; Tadesse, S.; Wen, B.; D’Agostino, E.; Solomon, M.; DiMauro, S.; Quinzii, C.; et al. Heterogeneity of coenzyme Q10 deficiency: Patient study and literature review. Arch. Neurol. 2012, 69, 978–983, Erratum in Arch. Neurol. 2012, 69, 886. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Radmard, S.; Zesiewicz, T.A.; Kuo, S.H. Evaluation of Cerebellar Ataxic Patients. Neurol. Clin. 2023, 41, 21–44. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Salviati, L.; Trevisson, E.; Agosto, C.; Doimo, M.; Navas, P. Primary Coenzyme Q10 Deficiency Overview. In GeneReviews®[Internet]; updated 2023 June 8; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2017. [Google Scholar] [PubMed]
- Stefely, J.A.; Licitra, F.; Laredj, L.; Reidenbach, A.G.; Kemmerer, Z.A.; Grangeray, A.; Jaeg-Ehret, T.; Minogue, C.E.; Ulbrich, A.; Hutchins, P.D.; et al. Cerebellar Ataxia and Coenzyme Q Deficiency through Loss of Unorthodox Kinase Activity. Mol. Cell 2016, 63, 608–620. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Stefely, J.A.; Reidenbach, A.G.; Ulbrich, A.; Oruganty, K.; Floyd, B.J.; Jochem, A.; Saunders, J.M.; Johnson, I.E.; Minogue, C.E.; Wrobel, R.L.; et al. Mitochondrial ADCK3 employs an atypical protein kinase-like fold to enable coenzyme Q biosynthesis. Mol. Cell 2015, 57, 83–94. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Manolaras, I.; Del Bondio, A.; Griso, O.; Reutenauer, L.; Eisenmann, A.; Habermann, B.H.; Puccio, H. Mitochondrial dysfunction and calcium dysregulation in COQ8A-ataxia Purkinje neurons are rescued by CoQ10 treatment. Brain 2023, 146, 3836–3850. [Google Scholar] [CrossRef] [PubMed]
- Mollet, J.; Delahodde, A.; Serre, V.; Chretien, D.; Schlemmer, D.; Lombes, A.; Boddaert, N.; Desguerre, I.; de Lonlay, P.; de Baulny, H.O.; et al. CABC1 gene mutations cause ubiquinone deficiency with cerebellar ataxia and seizures. Am. J. Hum. Genet. 2008, 82, 623–630. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Traschütz, A.; Schirinzi, T.; Laugwitz, L.; Murray, N.H.; Bingman, C.A.; Reich, S.; Kern, J.; Heinzmann, A.; Vasco, G.; Bertini, E.; et al. Clinico-Genetic, Imaging and Molecular Delineation of COQ8A-Ataxia: A Multicenter Study of 59 Patients. Ann. Neurol. 2020, 88, 251–263. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Blumkin, L.; Leshinsky-Silver, E.; Zerem, A.; Yosovich, K.; Lerman-Sagie, T.; Lev, D. Heterozygous Mutations in the ADCK3 Gene in Siblings with Cerebellar Atrophy and Extreme Phenotypic Variability. JIMD Rep. 2014, 12, 103–107. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Horvath, R.; Czermin, B.; Gulati, S.; Demuth, S.; Houge, G.; Pyle, A.; Dineiger, C.; Blakely, E.L.; Hassani, A.; Foley, C.; et al. Adult-onset cerebellar ataxia due to mutations in CABC1/ADCK3. J. Neurol. Neurosurg. Psychiatry 2012, 83, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Mignot, C.; Apartis, E.; Durr, A.; Marques Lourenço, C.; Charles, P.; Devos, D.; Moreau, C.; de Lonlay, P.; Drouot, N.; Burglen, L.; et al. Phenotypic variability in ARCA2 and identification of a core ataxic phenotype with slow progression. Orphanet J. Rare Dis. 2013, 8, 173. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Galosi, S.; Barca, E.; Carrozzo, R.; Schirinzi, T.; Quinzii, C.M.; Lieto, M.; Vasco, G.; Zanni, G.; Di Nottia, M.; Galatolo, D.; et al. Dystonia-Ataxia with early handwriting deterioration in COQ8A mutation carriers: A case series and literature review. Park. Relat. Disord. 2019, 68, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, L.; Morbiato, L.; Burgardt, A.; Tonello, F.; Bartlett, A.K.; Guerra, R.M.; Ferizhendi, K.K.; Desbats, M.A.; Rascalou, B.; Marchi, M.; et al. COQ4 is required for the oxidative decarboxylation of the C1 carbon of coenzyme Q in eukaryotic cells. Mol. Cell 2024, 84, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Salviati, L.; Trevisson, E.; Rodriguez Hernandez, M.A.; Casarin, A.; Pertegato, V.; Doimo, M.; Cassina, M.; Agosto, C.; Desbats, M.A.; Sartori, G.; et al. Haploinsufficiency of COQ4 causes coenzyme Q10 deficiency. J. Med. Genet. 2012, 49, 187–191. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Laugwitz, L.; Seibt, A.; Herebian, D.; Peralta, S.; Kienzle, I.; Buchert, R.; Falb, R.; Gauck, D.; Müller, A.; Grimmel, M.; et al. Human COQ4 deficiency: Delineating the clinical, metabolic and neuroimaging phenotypes. J. Med. Genet. 2022, 59, 878–887. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Brea-Calvo, G.; Haack, T.B.; Karall, D.; Ohtake, A.; Invernizzi, F.; Carrozzo, R.; Kremer, L.; Dusi, S.; Fauth, C.; Scholl-Bürgi, S.; et al. COQ4 mutations cause a broad spectrum of mitochondrial disorders associated with CoQ10 deficiency. Am. J. Hum. Genet. 2015, 96, 309–317. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bosch, A.M.; Kamsteeg, E.J.; Rodenburg, R.J.; van Deutekom, A.W.; Buis, D.R.; Engelen, M.; Cobben, J.M. Coenzyme Q10 deficiency due to a COQ4 gene defect causes childhood-onset spinocerebellar ataxia and stroke-like episodes. Mol. Genet. Metab. Rep. 2018, 17, 19–21. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Caglayan, A.O.; Gumus, H.; Sandford, E.; Kubisiak, T.L.; Ma, Q.; Ozel, A.B.; Per, H.; Li, J.Z.; Shakkottai, V.G.; Burmeister, M. COQ4 Mutation Leads to Childhood-Onset Ataxia Improved by CoQ10 Administration. Cerebellum 2019, 18, 665–669. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mero, S.; Salviati, L.; Leuzzi, V.; Rubegni, A.; Calderan, C.; Nardecchia, F.; Galatolo, D.; Desbats, M.A.; Naef, V.; Gemignani, F.; et al. New pathogenic variants in COQ4 cause ataxia and neurodevelopmental disorder without detectable CoQ10 deficiency in muscle or skin fibroblasts. J. Neurol. 2021, 268, 3381–3389. [Google Scholar] [CrossRef] [PubMed]
- Cordts, I.; Semmler, L.; Prasuhn, J.; Seibt, A.; Herebian, D.; Navaratnarajah, T.; Park, J.; Deininger, N.; Laugwitz, L.; Göricke, S.L.; et al. Bi-Allelic COQ4 Variants Cause Adult-Onset Ataxia-Spasticity Spectrum Disease. Mov. Disord. 2022, 37, 2147–2153. [Google Scholar] [CrossRef] [PubMed]
- Ashby, M.N.; Kutsunai, S.Y.; Ackerman, S.; Tzagoloff, A.; Edwards, P.A. COQ2 is a candidate for the structural gene encoding para-hydroxybenzoate:polyprenyltransferase. J. Biol. Chem. 1992, 267, 4128–4136. [Google Scholar] [CrossRef] [PubMed]
- Quinzii, C.; Naini, A.; Salviati, L.; Trevisson, E.; Navas, P.; Dimauro, S.; Hirano, M. A mutation in para-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency. Am. J. Hum. Genet. 2006, 78, 345–459. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Desbats, M.A.; Morbidoni, V.; Silic-Benussi, M.; Doimo, M.; Ciminale, V.; Cassina, M.; Sacconi, S.; Hirano, M.; Basso, G.; Pierrel, F.; et al. The COQ2 genotype predicts the severity of coenzyme Q10 deficiency. Hum. Mol. Genet. 2016, 25, 4256–4265. [Google Scholar] [CrossRef] [PubMed]
- Multiple-System Atrophy Research Collaboration. Mutations in COQ2 in familial and sporadic multiple-system atrophy. N. Engl. J. Med. 2013, 369, 233–244, Erratum in N. Engl. J. Med. 2014, 371, 94. [Google Scholar] [CrossRef] [PubMed]
- Porto, K.J.; Hirano, M.; Mitsui, J.; Chikada, A.; Matsukawa, T.; Ishiura, H.; Japan Multiple System Atrophy Registry Consortium; Toda, T.; Kusunoki, S.; Tsuji, S. COQ2 V393A confers high risk susceptibility for multiple system atrophy in East Asian population. J. Neurol. Sci. 2021, 429, 117623. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Ohta, Y.; Yamashita, T.; Sato, K.; Takemoto, M.; Hishikawa, N.; Abe, K. New susceptible variant of COQ2 gene in Japanese patients with sporadic multiple system atrophy. Neurol. Genet. 2016, 2, e54. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jiménez-Jiménez, F.J.; Alonso-Navarro, H.; García-Martín, E.; Agúndez, J.A.G. Coenzyme Q10 and Parkinsonian Syndromes: A Systematic Review. J. Pers. Med. 2022, 12, 975. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Malicdan, M.C.V.; Vilboux, T.; Ben-Zeev, B.; Guo, J.; Eliyahu, A.; Pode-Shakked, B.; Dori, A.; Kakani, S.; Chandrasekharappa, S.C.; Ferreira, C.R.; et al. A novel inborn error of the coenzyme Q10 biosynthesis pathway: Cerebellar ataxia and static encephalomyopathy due to COQ5 C-methyltransferase deficiency. Hum. Mutat. 2018, 39, 69–79. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Montini, G.; Malaventura, C.; Salviati, L. Early coenzyme Q10 supplementation in primary coenzyme Q10 deficiency. N. Engl. J. Med. 2008, 358, 2849–2850. [Google Scholar] [CrossRef] [PubMed]
- Mantle, D.; Millichap, L.; Castro-Marrero, J.; Hargreaves, I.P. Primary Coenzyme Q10 Deficiency: An Update. Antioxidants 2023, 12, 1652. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Alcázar-Fabra, M.; Rodríguez-Sánchez, F.; Trevisson, E.; Brea-Calvo, G. Primary Coenzyme Q deficiencies: A literature review and online platform of clinical features to uncover genotype-phenotype correlations. Free Radic. Biol. Med. 2021, 167, 141–180. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hekimi, S. The efficacy of coenzyme Q10treatment in alleviating the symptoms of primary coenzyme Q10 deficiency: A systematic review. J. Cell Mol. Med. 2022, 26, 4635–4644. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, L.; Ashizawa, T.; Peng, D. Primary coenzyme Q10 deficiency due to COQ8A gene mutations. Mol. Genet. Genomic Med. 2020, 8, e1420. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chang, A.; Ruiz-Lopez, M.; Slow, E.; Tarnopolsky, M.; Lang, A.E.; Munhoz, R.P. ADCK3-related Coenzyme Q10 Deficiency: A Potentially Treatable Genetic Disease. Mov. Disord. Clin. Pract. 2018, 5, 635–639. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Artuch, R.; Brea-Calvo, G.; Briones, P.; Aracil, A.; Galván, M.; Espinós, C.; Corral, J.; Volpini, V.; Ribes, A.; Andreu, A.L.; et al. Cerebellar ataxia with coenzyme Q10 deficiency: Diagnosis and follow-up after coenzyme Q10 supplementation. J. Neurol. Sci. 2006, 246, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Prasuhn, J.; Göttlich, M.; Ebeling, B.; Bodemann, C.; Großer, S.; Wellach, I.; Reuther, K.; Hanssen, H.; Brüggemann, N. The cerebellar bioenergetic state predicts treatment response in COQ8A-related ataxia. Park. Relat. Disord. 2022, 99, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Pineda, M.; Montero, R.; Aracil, A.; O’Callaghan, M.M.; Mas, A.; Espinos, C.; Martinez-Rubio, D.; Palau, F.; Navas, P.; Briones, P.; et al. Coenzyme Q(10)-responsive ataxia: 2-year-treatment follow-up. Mov. Disord. 2010, 25, 1262–1268. [Google Scholar] [CrossRef] [PubMed]
- Schirinzi, T.; Favetta, M.; Romano, A.; Sancesario, A.; Summa, S.; Minosse, S.; Zanni, G.; Castelli, E.; Bertini, E.; Petrarca, M.; et al. One-year outcome of coenzyme Q10 supplementation in ADCK3 ataxia (ARCA2). Cerebellum Ataxias 2019, 6, 15. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mitsui, J.; Koguchi, K.; Momose, T.; Takahashi, M.; Matsukawa, T.; Yasuda, T.; Tokushige, S.I.; Ishiura, H.; Goto, J.; Nakazaki, S.; et al. Three-Year Follow-Up of High-Dose Ubiquinol Supplementation in a Case of Familial Multiple System Atrophy with Compound Heterozygous COQ2 Mutations. Cerebellum 2017, 16, 664–672. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mitsui, J.; Matsukawa, T.; Uemura, Y.; Kawahara, T.; Chikada, A.; Porto, K.J.L.; Naruse, H.; Tanaka, M.; Ishiura, H.; Toda, T.; et al. High-dose ubiquinol supplementation in multiple-system atrophy: A multicentre, randomised, double-blinded, placebo-controlled phase 2 trial. EClinicalMedicine 2023, 59, 101920. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lo, R.Y.; Figueroa, K.P.; Pulst, S.M.; Lin, C.Y.; Perlman, S.; Wilmot, G.; Gomez, C.; Schmahmann, J.; Paulson, H.; Shakkottai, V.G.; et al. Coenzyme Q10 and spinocerebellar ataxias. Mov. Disord. 2015, 30, 214–220. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Parikh, S.; Goldstein, A.; Karaa, A.; Koenig, M.K.; Anselm, I.; Brunel-Guitton, C.; Christodoulou, J.; Cohen, B.H.; Dimmock, D.; Enns, G.M.; et al. Patient care standards for primary mitochondrial disease: A consensus statement from the Mitochondrial Medicine Society. Genet. Med. 2017, 19, 1380–1397. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Orsucci, D.; Ienco, E.C.; Siciliano, G.; Mancuso, M. Mitochondrial disorders and drugs: What every physician should know. Drugs Context 2019, 8, 212588. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ilg, W.; Bastian, A.J.; Boesch, S.; Burciu, R.G.; Celnik, P.; Claaßen, J.; Feil, K.; Kalla, R.; Miyai, I.; Nachbauer, W.; et al. Consensus paper: Management of degenerative cerebellar disorders. Cerebellum 2014, 13, 248–268. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Montero, R.; Sánchez-Alcázar, J.A.; Briones, P.; Hernández, A.R.; Cordero, M.D.; Trevisson, E.; Salviati, L.; Pineda, M.; García-Cazorla, A.; Navas, P.; et al. Analysis of coenzyme Q10 in muscle and fibroblasts for the diagnosis of CoQ10 deficiency syndromes. Clin. Biochem. 2008, 41, 697–700. [Google Scholar] [CrossRef] [PubMed]
- Yubero, D.; Montero, R.; Armstrong, J.; Espinós, C.; Palau, F.; Santos-Ocaña, C.; Salviati, L.; Navas, P.; Artuch, R. Molecular diagnosis of coenzyme Q10 deficiency. Expert. Rev. Mol. Diagn. 2015, 15, 1049–1059. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Clarke, C.F.; Hirano, M. 176th ENMC International Workshop: Diagnosis and treatment of coenzyme Q10 deficiency. Neuromuscul. Disord. 2012, 22, 76–86. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]



{kind=link}
{kind=link}
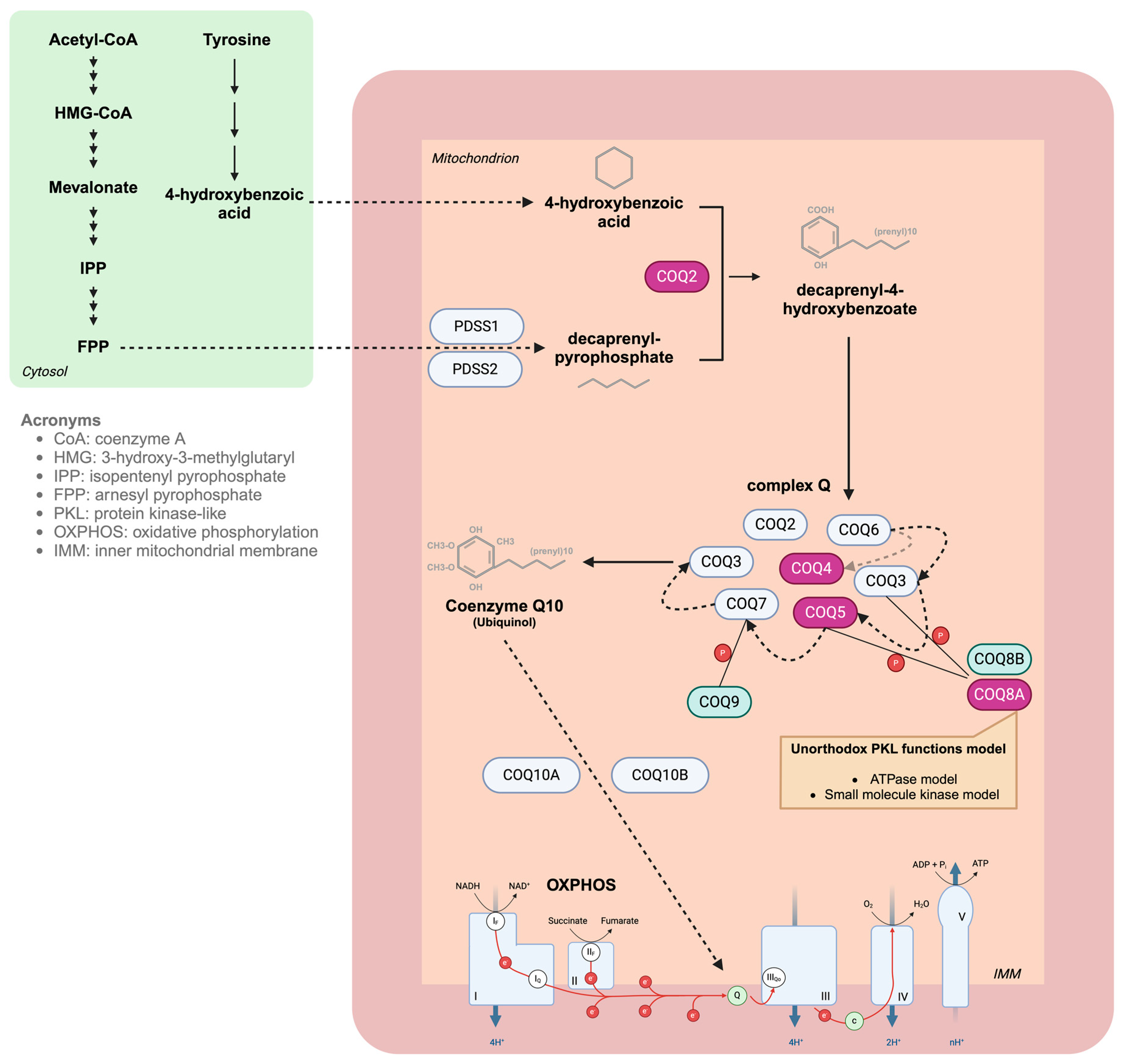{kind=link}
| Gene | DNA Variant | Protein Variant | Zygosity | Mutation Type | Phenotype | References |
|---|---|---|---|---|---|---|
| COQ4 | c.190C>T | p.Pro64Ser | Homozygous | Missense | Infantile-onset spastic ataxia, epilepsy | [19] |
| COQ4 | c.230C>T | p.Thr77Ile | Homozygous | Missense | Childhood-onset spinocerebellar ataxia with stroke-like episodes | [20] |
| COQ4 | c.164G>T | p.Gly55Val | Homozygous | Missense | Childhood-onset ataxia with partial epilepsy and cognitive impairment | [21] |
| COQ4 | c.305G>A | p.Arg102His | Compound het in association with c.284G>A | Missense | Childhood-onset ataxia, neurodevelopmental disorder | [22] |
| COQ4 | c.284G>A | p.Gly95Asp | Compound het in association with c.305G>A | Missense | Childhood-onset ataxia, neurodevelopmental disorder | [22] |
| COQ4 | c.577C>T | p.Pro193Ser | Compound het in association with c.718C>T | Missense | Childhood onset ataxia, progressive spasticity | [22] |
| COQ4 | c.718C>T | p.Arg240Cys | Compound het in association with c.577C>T | Missense | Childhood-onset ataxia, progressive spasticity | [22] |
| COQ4 | c.305G>A | p.Arg102His | Compound het in association with c.473G>A | Missense | Childhood-onset spastic ataxia, mild cognitive impairment | [23] |
| COQ4 | c.473G>A | p.Arg158Gln | Compound het in association with c.305G>A | Missense | Childhood-onset spastic ataxia, mild cognitive impairment | [23] |
| COQ4 | c.434G>A | p.Arg145His | Compound het in association with c.437T>G | Missense | Childhood-onset spastic ataxia, postural tremor | [23] |
| COQ4 | c.437T>G | p.Phe146Cys | Compound het in association with c.434G>A | Missense | Childhood-onset spastic ataxia, postural tremor | [23] |
| COQ4 | c.202+4A>C | - | Homozygous | Intronic | Childhood-onset ataxia, mild cognitive impairment | [23] |
| COQ2 | c.382A>G | p.Met128Val | Homozygous in association with hom c.1178T>C | Missense | MSA-p (definite), cerebellar signs, retinitis pigmentosa | [27] |
| COQ2 | c.1178T>C | p.Val393Ala | Homozygous in association with hom c.382A>G | Missense | MSA-p (definite), cerebellar signs, retinitis pigmentosa | [27] |
| COQ2 | c.1159C>T | p.Arg337Ter | Compound het in association with c.1178T>C | Nonsense | MSA-c (probable) | [27] |
| COQ2 | c.1178T>C | p.Val393Ala | Compound het in association with c.1159C>T | Missense | MSA-c (probable) | [27] |
| COQ5 | [Chr12(GRCh37):120940098-120949687] | - | Homozygous | Tandem duplication (9590 bp) | Childhood-onset cerebellar ataxia, encephalopathy, generalized tonic-clonic seizure, developmental delay | [31] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopriore, P.; Vista, M.; Tessa, A.; Giuntini, M.; Caldarazzo Ienco, E.; Mancuso, M.; Siciliano, G.; Santorelli, F.M.; Orsucci, D. Primary Coenzyme Q10 Deficiency-Related Ataxias. J. Clin. Med. 2024, 13, 2391. https://doi.org/10.3390/jcm13082391
Lopriore P, Vista M, Tessa A, Giuntini M, Caldarazzo Ienco E, Mancuso M, Siciliano G, Santorelli FM, Orsucci D. Primary Coenzyme Q10 Deficiency-Related Ataxias. Journal of Clinical Medicine. 2024; 13(8):2391. https://doi.org/10.3390/jcm13082391
Chicago/Turabian StyleLopriore, Piervito, Marco Vista, Alessandra Tessa, Martina Giuntini, Elena Caldarazzo Ienco, Michelangelo Mancuso, Gabriele Siciliano, Filippo Maria Santorelli, and Daniele Orsucci. 2024. "Primary Coenzyme Q10 Deficiency-Related Ataxias" Journal of Clinical Medicine 13, no. 8: 2391. https://doi.org/10.3390/jcm13082391