
Arrhythmogenic Left Ventricular Cardiomyopathy: From Diagnosis to Risk Management
 , , and
, , and
Abstract
:1. Introduction
2. Diagnosis
2.1. Echocardiography
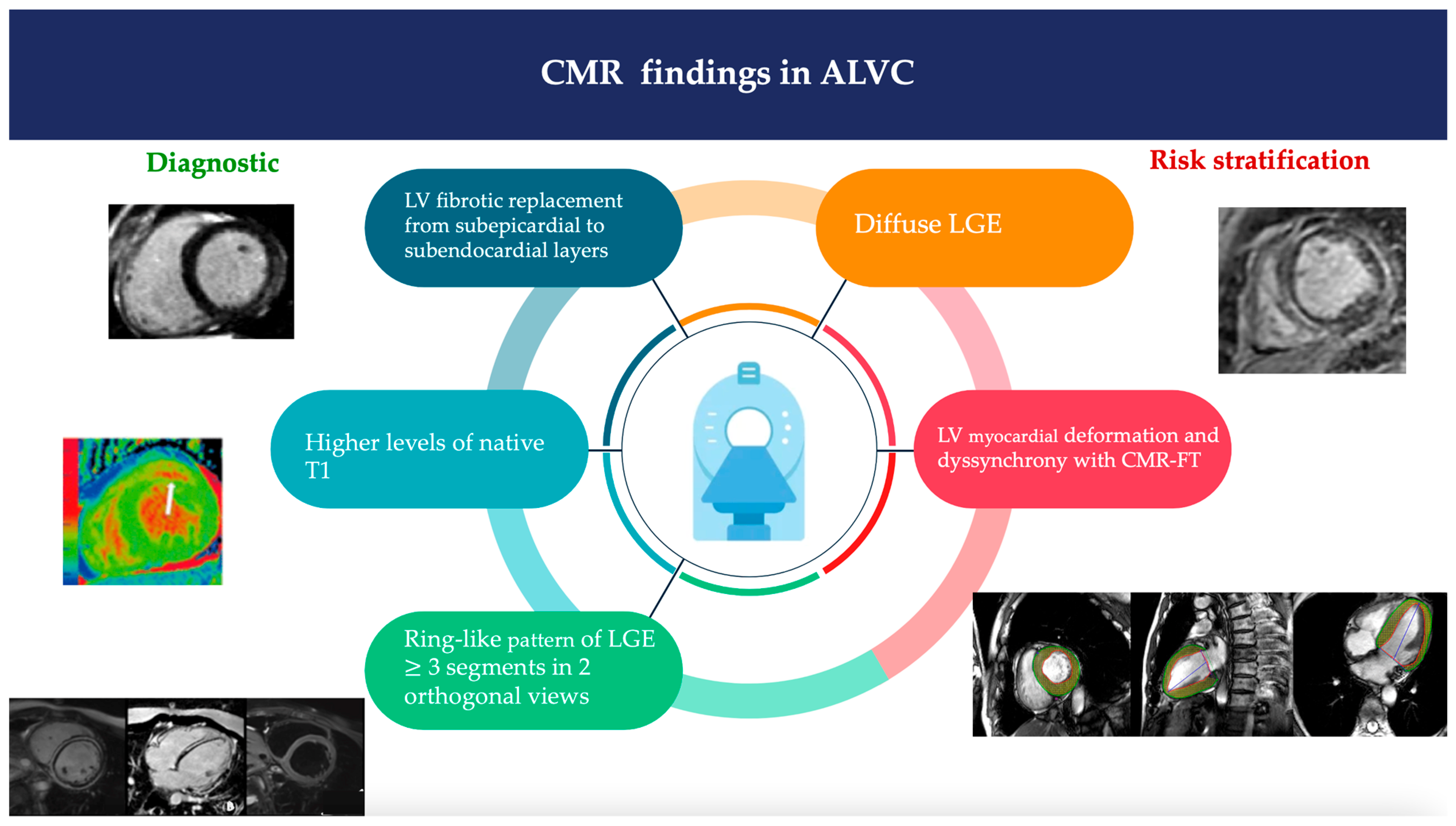
2.2. Cardiac Magnetic Resonance
2.3. Other Imaging
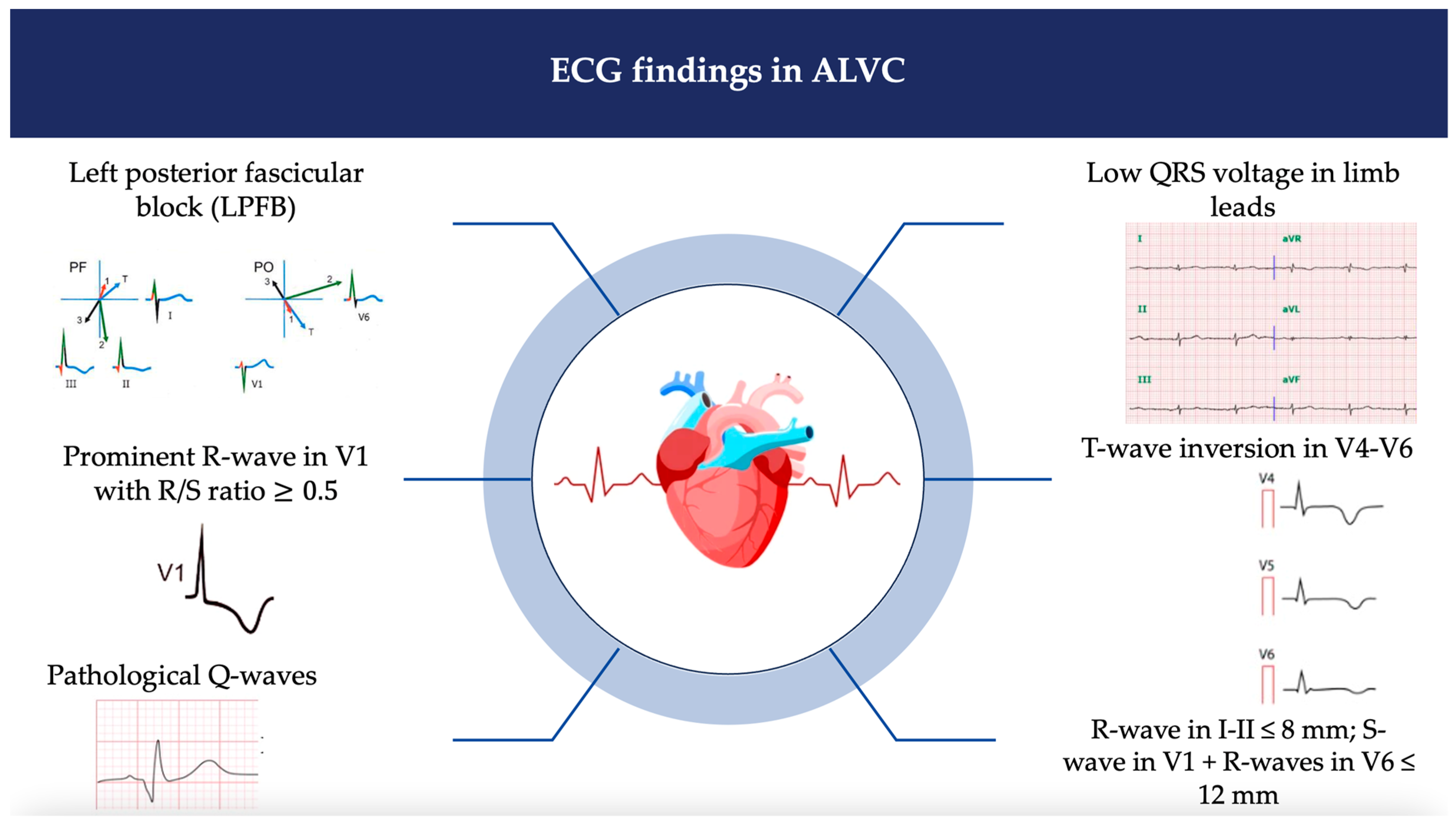
2.4. Twelve-Lead Electrocardiography
2.5. Holter ECG and Signal-Avereraged ECG
2.6. Genetic Findings and Family History
3. Differential Diagnosis
3.1. Dilated Cardiomyopathy (Early Stage)
3.2. Cardiac Sarcoidosis
3.3. Chronic Myocarditis
3.4. Neuromuscular Disorders
3.4.1. Dystrophinopathies
3.4.2. Emery–Dreifuss Muscular Dystrophy (EDMD)
3.4.3. Myotonic Dystrophy Type 1 (DM1)
3.5. Chagas Disease
4. Genotype–Phenotype Correlation for SCD Prevention Strategy
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Elliott, P.M.; Anastasakis, A.; Asimaki, A.; Basso, C.; Bauce, B.; Brooke, M.A.; Calkins, H.; Corrado, D.; Duru, F.; Green, K.J.; et al. Definition and treatment of arrhythmogenic cardiomyopathy: An updated expert panel report. Eur. J. Heart Fail. 2019, 21, 955–964. [Google Scholar] [CrossRef]
- Corrado, D.; Basso, C.; Thiene, G.; McKenna, W.J.; Davies, M.J.; Fontaliran, F.; Nava, A.; Silvestri, F.; Blomstrom-Lundqvist, C.; Wlodarska, E.K.; et al. Spectrum of Clinicopathologic Manifestations of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: A Multicenter Study. J. Am. Coll. Cardiol. 1997, 30, 1512–1520. [Google Scholar] [CrossRef]
- Marcus, F.I.; Fontaine, G.H.; Guiraudon, G.; Frank, R.; Laurenceau, J.L.; Malergue, C.; Grosgogeat, Y. Right ventricular dysplasia: A report of 24 adult cases. Circulation 1982, 65, 384–398. [Google Scholar] [CrossRef]
- Sen-Chowdhry, S.; Syrris, P.; Ward, D.; Asimaki, A.; Sevdalis, E.; McKenna, W.J. Clinical and Genetic Characterization of Families With Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Provides Novel Insights Into Patterns of Disease Expression. Circulation 2007, 115, 1710–1720. [Google Scholar] [CrossRef]
- Corrado, D.; Basso, C.; Judge, D.P. Arrhythmogenic Cardiomyopathy. Circ. Res. 2017, 121, 784–802. [Google Scholar] [CrossRef]
- Towbin, J.A.; McKenna, W.J.; Abrams, D.J.; Ackerman, M.J.; Calkins, H.; Darrieux, F.C.; Daubert, J.P.; de Chillou, C.; DePasquale, E.C.; Desai, M.Y.; et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm 2019, 16, e301–e372. [Google Scholar] [CrossRef]
- Arbelo, E.; Protonotarios, A.; Gimeno, J.R.; Arbustini, E.; Barriales-Villa, R.; Basso, C.; Bezzina, C.R.; Biagini, E.; Blom, N.A.; de Boer, R.A.; et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur. Heart J. 2023, 44, 3503–3626. [Google Scholar] [CrossRef] [PubMed]
- Quarta, G.; Syrris, P.; Ashworth, M.; Jenkins, S.; Alapi, K.Z.; Morgan, J.; Muir, A.; Pantazis, A.; McKenna, W.J.; Elliott, P.M. Mutations in the Lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2012, 33, 1128–1136. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Genga, M.F.; Cuenca, S.; Ferro, M.D.; Zorio, E.; Salgado-Aranda, R.; Climent, V.; Padrón-Barthe, L.; Duro-Aguado, I.; Jiménez-Jáimez, J.; Hidalgo-Olivares, V.M.; et al. Truncating FLNC Mutations Are Associated With High-Risk Dilated and Arrhythmogenic Cardiomyopathies. J. Am. Coll. Cardiol. 2016, 68, 2440–2451. [Google Scholar] [CrossRef] [PubMed]
- Te Riele, A.S.J.; Agullo-Pascual, E.; James, C.A.; Leo-Macias, A.; Cerrone, M.; Zhang, M.; Lin, X.; Lin, B.; Rothenberg, E.; Sobreira, N.L.; et al. Multilevel analyses of SCN5A mutations in arrhythmogenic right ventricular dysplasia/cardiomyopathy suggest non-canonical mechanisms for disease pathogenesis. Cardiovasc. Res. 2017, 113, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Zeppenfeld, K.; Tfelt-Hansen, J.; de Riva, M.; Winkel, B.G.; Behr, E.R.; A Blom, N.; Charron, P.; Corrado, D.; Dagres, N.; de Chillou, C.; et al. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur. Heart J. 2022, 43, 3997–4126. [Google Scholar] [CrossRef]
- Marcus, F.I.; McKenna, W.J.; Sherrill, D.; Basso, C.; Bauce, B.; Bluemke, D.A.; Calkins, H.; Corrado, D.; Cox, M.G.; Daubert, J.P.; et al. Diagnosis of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia. Circulation 2010, 121, 1533–1541. [Google Scholar] [CrossRef]
- Tsui, H.; van Kampen, S.J.; Han, S.J.; Meraviglia, V.; van Ham, W.B.; Casini, S.; van der Kraak, P.; Vink, A.; Yin, X.; Mayr, M.; et al. Desmosomal protein degradation as an underlying cause of arrhythmogenic cardiomyopathy. Sci. Transl. Med. 2023, 15, eadd4248. [Google Scholar] [CrossRef]
- Monda, E.; Lioncino, M.; Rubino, M.; Caiazza, M.; Cirillo, A.; Fusco, A.; Pacileo, R.; Fimiani, F.; Amodio, F.; Borrelli, N.; et al. The Risk of Sudden Unexpected Cardiac Death in Children. Heart Fail. Clin. 2022, 18, 115–123. [Google Scholar] [CrossRef]
- Corrado, D.; Perazzolo Marra, M.; Zorzi, A.; Beffagna, G.; Cipriani, A.; Lazzari, M.; Migliore, F.; Pilichou, K.; Rampazzo, A.; Rigato, I.; et al. Diagnosis of arrhythmogenic cardiomyopathy: The Padua criteria. Int. J. Cardiol. 2020, 319, 106–114. [Google Scholar] [CrossRef]
- Corrado, D.; Van Tintelen, P.J.; McKenna, W.J.; Hauer, R.N.W.; Anastastakis, A.; Asimaki, A.; Basso, C.; Bauce, B.; Brunckhorst, C.; Bucciarelli-Ducci, C.; et al. International Experts. Arrhythmogenic right ventricular cardiomyopathy: Evalu-ation of the current diagnostic criteria and differential diagnosis. Eur. Heart J. 2020, 41, 1414–1429. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Anastasakis, A.; Basso, C.; Bauce, B.; Blomström-Lundqvist, C.; Bucciarelli-Ducci, C.; Cipriani, A.; De Asmundis, C.; Gandjbakhch, E.; Jiménez-Jáimez, J.; et al. Proposed diagnostic criteria for arrhythmogenic cardiomyopathy: European Task Force consensus report. Int. J. Cardiol. 2024, 395, 131447. [Google Scholar] [CrossRef] [PubMed]
- Haugaa, K.H.; Haland, T.F.; Leren, I.S.; Saberniak, J.; Edvardsen, T. Arrhythmogenic right ventricular cardiomyopathy, clinical manifestations, and diagnosis. EP Eur. 2016, 18, 965–972. [Google Scholar] [CrossRef]
- Segura-Rodríguez, D.; Bermúdez-Jiménez, F.J.; González-Camacho, L.; Escobar, E.M.; García-Orta, R.; Alcalá-López, J.E.; Pavés, A.B.; Oyonarte-Ramírez, J.M.; López-Fernández, S.; Álvarez, M.; et al. Layer-Specific Global Longitudinal Strain Predicts Arrhythmic Risk in Arrhythmogenic Cardiomyopathy. Front. Cardiovasc. Med. 2021, 8, 748003. [Google Scholar] [CrossRef]
- Te Riele, A.S.; Tandri, H.; Bluemke, D.A. Arrhythmogenic right ventricular cardiomyopathy (ARVC): Cardiovascular magnetic resonance update. J. Cardiovasc. Magn. Reson. 2014, 16, 50. [Google Scholar] [CrossRef] [PubMed]
- Borgquist, R.; Haugaa, K.H.; Gilljam, T.; Bundgaard, H.; Hansen, J.; Eschen, O.; Jensen, H.K.; Holst, A.G.; Edvardsen, T.; Svendsen, J.H.; et al. The diagnostic performance of imaging methods in ARVC using the 2010 Task Force criteria. Eur. Heart J.-Cardiovasc. Imaging 2014, 15, 1219–1225. [Google Scholar] [CrossRef]
- Haugaa, K.H.; Basso, C.; Badano, L.P.; Bucciarelli-Ducci, C.; Cardim, N.; Gaemperli, O.; Galderisi, M.; Habib, G.; Knuuti, J.; Lancellotti, P.; et al. Comprehensive multi-modality imaging approach in arrhythmogenic cardiomyopathy—An expert consensus document of the European Association of Cardiovascular Imaging. Eur. Heart J.-Cardiovasc. Imaging 2017, 18, 237–253. [Google Scholar] [CrossRef]
- De Lazzari, M.; Zorzi, A.; Cipriani, A.; Susana, A.; Mastella, G.; Rizzo, A.; Rigato, I.; Bauce, B.; Giorgi, B.; Lacognata, C.; et al. Relationship Between Electrocardiographic Findings and Cardiac Magnetic Resonance Phenotypes in Arrhythmogenic Cardiomyopathy. J. Am. Heart Assoc. 2018, 7, e009855. [Google Scholar] [CrossRef]
- Bariani, R.; Cipriani, A.; Rizzo, S.; Celeghin, R.; Bueno Marinas, M.; Giorgi, B.; De Gaspari, M.; Rigato, I.; Leoni, L.; Zorzi, A.; et al. “Hot phase” clinical presentation in arrhythmogenic cardiomyopathy. EP Eur. 2021, 23, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.D.; Compagnucci, P.; Zorzi, A.; Cavarretta, E.; Castelletti, S.; Contursi, M.; D’Aleo, A.; D’Ascenzi, F.; Mos, L.; Palmieri, V.; et al. Electroanatomic mapping in athletes: Why and when. An expert opinion paper from the Italian Society of Sports Cardiology. Int. J. Cardiol. 2023, 383, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, A.; Bauce, B.; De Lazzari, M.; Rigato, I.; Bariani, R.; Meneghin, S.; Pilichou, K.; Motta, R.; Aliberti, C.; Thiene, G.; et al. Arrhythmogenic Right Ventricular Cardiomyopathy: Characterization of Left Ventricular Phenotype and Differential Diagnosis with Dilated Cardiomyopathy. J. Am. Heart Assoc. 2020, 9, e014628. [Google Scholar] [CrossRef] [PubMed]
- Augusto, J.B.; Eiros, R.; Nakou, E.; Moura-Ferreira, S.; A Treibel, T.; Captur, G.; Akhtar, M.M.; Protonotarios, A.; Gossios, T.D.; Savvatis, K.; et al. Dilated cardiomyopathy and arrhythmogenic left ventricular cardiomyopathy: A comprehensive genotype-imaging phenotype study. Eur. Heart J.-Cardiovasc. Imaging 2020, 21, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.L.; Akhtar, M.M.; Sabater-Molina, M.; Futema, M.; Asimaki, A.; Protonotarios, A.; Dalageorgou, C.; Pittman, A.M.; Suarez, M.P.; Aguilera, B.; et al. Filamin C variants are associated with a distinctive clinical and immunohistochemical arrhythmogenic cardiomyopathy phenotype. Int. J. Cardiol. 2020, 307, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Segura-Rodríguez, D.; Bermúdez-Jiménez, F.J.; Carriel, V.; López-Fernández, S.; González-Molina, M.; Ramírez, J.M.O.; Fernández-Navarro, L.; García-Roa, M.D.; Cabrerizo, E.M.; Durand-Herrera, D.; et al. Myocardial fibrosis in arrhythmogenic cardiomyopathy: A genotype–phenotype correlation study. Eur. Heart J.-Cardiovasc. Imaging 2020, 21, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Sen-Chowdhry, S.; Syrris, P.; Prasad, S.K.; Hughes, S.E.; Merrifield, R.; Ward, D.; Pennell, D.J.; McKenna, W.J. Left-Dominant Arrhythmogenic Cardiomyopathy: An Under-Recognized Clinical Entity. J. Am. Coll. Cardiol. 2008, 52, 2175–2187. [Google Scholar] [CrossRef]
- Aquaro, G.D.; Barison, A.; Todiere, G.; Grigoratos, C.; Ali, L.A.; Di Bella, G.; Emdin, M.; Festa, P. Usefulness of Combined Functional Assessment by Cardiac Magnetic Resonance and Tissue Characterization Versus Task Force Criteria for Diagnosis of Arrhythmogenic Right Ventricular Cardiomyopathy. Am. J. Cardiol. 2016, 118, 1730–1736. [Google Scholar] [CrossRef]
- Rubino, M.; Scatteia, A.; Frisso, G.; Pacileo, G.; Caiazza, M.; Pascale, C.E.; Guarini, P.; Limongelli, G.; Dellegrottaglie, S. Imaging the “Hot Phase” of a Familiar Left-Dominant Arrhythmogenic Cardiomyopathy. Genes 2021, 12, 1933. [Google Scholar] [CrossRef]
- Bourfiss, M.; Prakken, N.H.; van der Heijden, J.F.; Kamel, I.; Zimmerman, S.L.; Asselbergs, F.W.; Leiner, T.; Velthuis, B.K.; Riele, A.S.T. Diagnostic Value of Native T1 Mapping in Arrhythmogenic Right Ventricular Cardiomyopathy. JACC Cardiovasc. Imaging 2019, 12, 1580–1582. [Google Scholar] [CrossRef]
- He, J.; Xu, J.; Li, G.; Zhou, D.; Li, S.; Zhuang, B.; Chen, X.; Duan, X.; Li, L.; Fan, X.; et al. Arrhythmogenic Left Ventricular Cardiomyopathy: A Clinical and CMR Study. Sci. Rep. 2020, 10, 533. [Google Scholar] [CrossRef] [PubMed]
- Monda, E.; Frisso, G.; Rubino, M.; Caiazza, M.; Esposito, A.; Cirillo, A.; Fusco, A.; Palmiero, G.; Mazzaccara, C.; Pacileo, R.; et al. Potential role of imaging markers in predicting future disease expression of arrhythmogenic cardiomyopathy. Futur. Cardiol. 2021, 17, 647–654. [Google Scholar] [CrossRef]
- Tat, E.; Ball, C.; Camren, G.P.; Wroblewski, I.; Dajani, K.A.; Goldberg, A.; Kinno, M.; Sanagala, T.; Syed, M.A.; Wilber, D.J.; et al. Impact of late gadolinium enhancement extent, location, and pattern on ventricular tachycardia and major adverse cardiac events in patients with ischemic vs. non-ischemic cardiomyopathy. Front. Cardiovasc. Med. 2022, 9, 1026215. [Google Scholar] [CrossRef]
- Aquaro, G.D.; De Luca, A.; Cappelletto, C.; Raimondi, F.; Bianco, F.; Botto, N.; Lesizza, P.; Grigoratos, C.; Minati, M.; Dell’omodarme, M.; et al. Prognostic Value of Magnetic Resonance Phenotype in Patients with Arrhythmogenic Right Ventricular Cardiomyopathy. J. Am. Coll. Cardiol. 2020, 75, 2753–2765. [Google Scholar] [CrossRef]
- Corrado, D.; Wichter, T.; Link, M.S.; Hauer, R.N.; Marchlinski, F.E.; Anastasakis, A.; Bauce, B.; Basso, C.; Brunckhorst, C.; Tsatsopoulou, A.; et al. Treatment of arrhythmogenic right ventricular cardiomyopathy/dysplasia: An international task force consensus statement. Eur. Heart J. 2015, 132, 441–453. [Google Scholar] [CrossRef]
- Song, Y.; Li, L.; Chen, X.; Ji, K.; Lu, M.; Hauer, R.; Chen, L.; Zhao, S. Left Ventricular Longitudinal Dyssynchrony by CMR Feature Tracking Is Related to Adverse Prognosis in Advanced Arrhythmogenic Cardiomyopathy. Front. Cardiovasc. Med. 2021, 8, 712832. [Google Scholar] [CrossRef]
- Protonotarios, A.; Wicks, E. The role of FDG-PET imaging in arrhythmogenic cardiomyopathy. Int. J. Cardiol. 2023, 391, 131275. [Google Scholar] [CrossRef]
- Protonotarios, A.; Wicks, E.; Ashworth, M.; Stephenson, E.; Guttmann, O.; Savvatis, K.; Sekhri, N.; Mohiddin, S.A.; Syrris, P.; Menezes, L.; et al. Prevalence of 18F-fluorodeoxyglucose positron emission tomography abnormalities in patients with arrhythmogenic right ventricular cardiomyopathy. Int. J. Cardiol. 2018, 284, 99–104. [Google Scholar] [CrossRef]
- Neves, R.; Tseng, A.S.; Garmany, R.; Fink, A.L.; McLeod, C.J.; Cooper, L.T.; MacIntyre, C.J.; Homb, A.C.; Rosenbaum, A.N.; Bois, J.P.; et al. Cardiac fludeoxyglucose-18 positron emission tomography in genotype-positive arrhythmogenic cardiomyopathy. Int. J. Cardiol. 2023, 389, 131173. [Google Scholar] [CrossRef]
- Monda, E.; Rubino, M.; Palmiero, G.; Verrillo, F.; Lioncino, M.; Diana, G.; Cirillo, A.; Fusco, A.; Dongiglio, F.; Caiazza, M.; et al. Multimodality Imaging in Arrhythmogenic Left Ventricular Cardiomyopathy. J. Clin. Med. 2023, 12, 1568. [Google Scholar] [CrossRef]
- Calore, C.; Zorzi, A.; Sheikh, N.; Nese, A.; Facci, M.; Malhotra, A.; Zaidi, A.; Schiavon, M.; Pelliccia, A.; Sharma, S.; et al. Electrocardiographic anterior T-wave inversion in athletes of different ethnicities: Differential diagnosis between athlete’s heart and cardiomyopathy. Eur. Heart J. 2016, 37, 2515–2527. [Google Scholar] [CrossRef]
- D’Ascenzi, F.; Anselmi, F.; Adami, P.E.; Pelliccia, A. Interpretation of T-wave inversion in physiological and pathological conditions: Current state and future perspectives. Clin. Cardiol. 2020, 43, 827–833. [Google Scholar] [CrossRef]
- Wilson, M.G.; Sharma, S.; Carré, F.; Charron, P.; Richard, P.; O’Hanlon, R.; Prasad, S.K.; Heidbuchel, H.; Brugada, J.; Salah, O.; et al. Significance of deep T-wave inversions in asymptomatic athletes with normal cardiovascular examinations: Practical solutions for managing the diagnostic conundrum. Br. J. Sports Med. 2012, 46 (Suppl. S1), i51–i58. [Google Scholar] [CrossRef] [PubMed]
- Calò, L.; Oliviero, G.; Crescenzi, C.; Romeo, F.; Martino, A.; Bressi, E.; Stefanini, M.; Silvetti, E.; Danza, L.; Rebecchi, M.; et al. Electrocardiogram in arrhytmogenic cardiomyopathy. Eur. Heart J. Suppl. 2023, 25 (Suppl. C), C169–C172. [Google Scholar] [CrossRef]
- Cadrin-Tourigny, J.; Bosman, L.P.; Nozza, A.; Wang, W.; Tadros, R.; Bhonsale, A.; Bourfiss, M.; Fortier, A.; Lie, Ø.H.; Saguner, A.M.; et al. A new prediction model for ventricular arrhythmias in arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2022, 43, E1–E9. [Google Scholar] [CrossRef] [PubMed]
- Jordà, P.; Bosman, L.P.; Gasperetti, A.; Mazzanti, A.; Gourraud, J.B.; Davies, B.; Frederiksen, T.C.; Weidmann, Z.M.; Di Marco, A.; Roberts, J.D.; et al. Arrhythmic risk prediction in arrhythmogenic right ventricular cardiomyopathy: External validation of the arrhythmogenic right ventricular cardiomyopathy risk calculator. Eur. Heart J. 2022, 43, 3041–3052. [Google Scholar] [CrossRef] [PubMed]
- Platonov, P.G.; Calkins, H.; Hauer, R.N.; Corrado, D.; Svendsen, J.H.; Wichter, T.; Biernacka, E.K.; Saguner, A.M.; Riele, A.S.T.; Zareba, W. High interobserver variability in the assessment of epsilon waves: Implications for diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart Rhythm. 2016, 13, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Calò, L.; Della Bona, R.; Martino, A.; Crescenzi, C.; Panattoni, G.; D’amati, G.; Gaita, F.; Mango, R.; Sciarra, L.; Laredo, M. Left Posterior Fascicular Block and Increased Risk of Sudden Cardiac Death in Young People. J. Am. Coll. Cardiol. 2021, 77, 1143–1145. [Google Scholar] [CrossRef]
- Laredo, M.; Tovia-Brodie, O.; Milman, A.; Michowitz, Y.; Roudijk, R.W.; Peretto, G.; Badenco, N.; Riele, A.S.J.M.T.; Sala, S.; Duthoit, G.; et al. Electrocardiographic findings in patients with arrhythmogenic cardiomyopathy and right bundle branch block ventricular tachycardia. Europace 2023, 25, 1025–1034. [Google Scholar] [CrossRef]
- Corrado, D.; Basso, C. Arrhythmogenic left ventricular cardiomyopathy. Heart 2022, 108, 733–743. [Google Scholar] [CrossRef]
- Zorzi, A.; Marra, M.P.; Rigato, I.; De Lazzari, M.; Susana, A.; Niero, A.; Pilichou, K.; Migliore, F.; Rizzo, S.; Giorgi, B.; et al. Nonischemic Left Ventricular Scar as a Substrate of Life-Threatening Ventricular Arrhythmias and Sudden Cardiac Death in Competitive Athletes. Circ. Arrhythm. Electrophysiol. 2016, 9, e004229. [Google Scholar] [CrossRef] [PubMed]
- Pearman, C.M.; Lee, D.; Davies, B.; Khan, H.; Tadros, R.; Cadrin-Tourigny, J.; Roberts, J.D.; Sanatani, S.; Simpson, C.; Angaran, P.; et al. Incremental value of the signal-averaged ECG for diagnosing arrhythmogenic cardiomyopathy. Heart Rhythm. 2023, 20, 224–230. [Google Scholar] [CrossRef]
- Kumar, S.; Baldinger, S.H.; Kapur, S.; Romero, J.; Mehta, N.K.; Mahida, S.; Fujii, A.; Tedrow, U.B.; Stevenson, W.G. Right ventricular scar-related ventricular tachycardia in nonischemic cardiomyopathy: Electrophysiological characteristics, mapping, and ablation of underlying heart disease. J. Cardiovasc. Electrophysiol. 2018, 29, 79–89. [Google Scholar] [CrossRef]
- Bosman, L.P.; Cadrin-Tourigny, J.; Bourfiss, M.; Ghasabeh, M.A.; Sharma, A.; Tichnell, C.; Roudijk, R.W.; Murray, B.; Tandri, H.; Khairy, P.; et al. Diagnosing arrhythmogenic right ventricular cardiomyopathy by 2010 Task Force Criteria: Clinical performance and simplified practical implementation. EP Eur. 2020, 22, 787–796. [Google Scholar] [CrossRef]
- Casella, M.; Gasperetti, A.; Sicuso, R.; Conte, E.; Catto, V.; Sommariva, E.; Bergonti, M.; Vettor, G.; Rizzo, S.; Pompilio, G.; et al. Characteristics of Patients with Arrhythmogenic Left Ventricular Cardiomyopathy. Circ. Arrhythm. Electrophysiol. 2020, 13, e009005. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Ackerman, M.J.; Priori, S.G.; Willems, S.; Berul, C.; Brugada, R.; Calkins, H.; Camm, A.J.; Ellinor, P.T.; Gollob, M.; Hamilton, R.; et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Europace 2011, 8, 1308–1339. [Google Scholar] [CrossRef]
- Corrado, D.; Link, M.S.; Calkins, H. Arrhythmogenic Right Ventricular Cardiomyopathy. N. Engl. J. Med. 2017, 376, 61–72. [Google Scholar] [CrossRef]
- Smith, E.D.; Lakdawala, N.K.; Papoutsidakis, N.; Aubert, G.; Mazzanti, A.; McCanta, A.C.; Agarwal, P.P.; Arscott, P.; Dellefave-Castillo, L.M.; Vorovich, E.E.; et al. Desmoplakin Cardiomyopathy, a Fibrotic and Inflammatory Form of Cardiomyopathy Distinct from Typical Dilated or Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation 2020, 141, 1872–1884. [Google Scholar] [CrossRef] [PubMed]
- Verstraelen, T.E.; Van Lint, F.H.; Bosman, L.P.; De Brouwer, R.; Proost, V.M.; Abeln, B.G.; Taha, K.; Zwinderman, A.H.; Dickhoff, C.; Oomen, T.; et al. Prediction of ventricular arrhythmia in phospholamban p.Arg14del mutation carriers–reaching the frontiers of individual risk prediction. Eur. Heart J. 2021, 42, 2842–2850. [Google Scholar] [CrossRef] [PubMed]
- Bermúdez-Jiménez, F.J.; Carriel, V.; Brodehl, A.; Alaminos, M.; Campos, A.; Schirmer, I.; Milting, H.; Abril, B.; Álvarez, M.; López-Fernández, S.; et al. Novel Desmin Mutation p.Glu401Asp Impairs Filament Formation, Disrupts Cell Membrane Integrity, and Causes Severe Arrhythmogenic Left Ventricular Cardiomyopathy/Dysplasia. Circulation 2018, 137, 1595–1610. [Google Scholar] [CrossRef]
- Thiene, G.; Corrado, D.; Nava, A.; Rossi, L.; Poletti, A.; Boffa, G.M.; Daliento, L.; Pennelli, N. Right ventricular cardiomyopathy: Is there evidence of an inflammatory aetiology? Eur. Heart J. 1991, 12 (Suppl. D), 22–25. [Google Scholar] [CrossRef]
- Groeneweg, J.A.; van der Zwaag, P.A.; Olde Nordkamp, L.R.; Bikker, H.; Jongbloed, J.D.; Jongbloed, R.; Wiesfeld, A.C.; Cox, M.G.; van der Heijden, J.F.; Atsma, D.E.; et al. Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy According to Revised 2010 Task Force Criteria with Inclusion of Non-Desmosomal Phospholamban Mutation Carriers. Am. J. Cardiol. 2013, 112, 1197–1206. [Google Scholar] [CrossRef]
- Te Rijdt, W.P.; Ten Sande, J.N.; Gorter, T.M.; van der Zwaag, P.A.; van Rijsingen, I.A.; Boekholdt, S.M.; van Tintelen, J.P.; van Haelst, P.L.; Planken, R.N.; de Boer, R.A.; et al. Myocardial fibrosis as an early feature in phospholamban p.Arg14del mutation carriers: Phenotypic insights from cardiovascular magnetic resonance imaging. Eur. Heart J. Cardiovasc. Imaging 2019, 20, 92–100. [Google Scholar] [CrossRef]
- Protonotarios, A.; Elliott, P.M. Arrhythmogenic cardiomyopathies (ACs): Diagnosis, risk stratification and management. Heart 2019, 105, 1117–1128. [Google Scholar] [CrossRef]
- Taha, K.; E Verstraelen, T.; de Brouwer, R.; Bruin-Bon, R.H.A.C.M.d.; Cramer, M.J.; Rijdt, W.P.T.; Bouma, B.J.; A de Boer, R.; A Doevendans, P.; Asselbergs, F.W.; et al. Optimal echocardiographic assessment of myocardial dysfunction for arrhythmic risk stratification in phospholamban mutation carriers. Eur. Heart J.-Cardiovasc. Imaging 2023, 23, 1492–1501. [Google Scholar] [CrossRef]
- Pinto, Y.M.; Elliott, P.M.; Arbustini, E.; Adler, Y.; Anastasakis, A.; Böhm, M.; Duboc, D.; Gimeno, J.; De Groote, P.; Imazio, M.; et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: A position statement of the ESC working group on myocardial and pericardial diseases. Eur. Heart J. 2016, 37, 1850–1858. [Google Scholar] [CrossRef]
- Merlo, M.; Cannatà, A.; Gobbo, M.; Stolfo, D.; Elliott, P.M.; Sinagra, G. Evolving concepts in dilated cardiomyopathy. Eur. J. Heart Fail. 2018, 20, 228–239. [Google Scholar] [CrossRef]
- Hulten, E.; Aslam, S.; Osborne, M.; Abbasi, S.; Bittencourt, M.S.; Blankstein, R. Cardiac sarcoidosis—State of the art review. Cardiovasc. Diagn. Ther. 2016, 6, 50–63. [Google Scholar] [CrossRef]
- Limongelli, G.; Adorisio, R.; Baggio, C.; Bauce, B.; Biagini, E.; Castelletti, S.; Favilli, S.; Imazio, M.; Lioncino, M.; Merlo, M.; et al. Diagnosis and Management of Rare Cardiomyopathies in Adult and Paediatric Patients. A Position Paper of the Italian Society of Cardiology (SIC) and Italian Society of Paediatric Cardiology (SICP). Int. J. Cardiol. 2022, 357, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Vita, T.; Okada, D.R.; Veillet-Chowdhury, M.; Bravo, P.E.; Mullins, E.; Hulten, E.; Agrawal, M.; Madan, R.; Taqueti, V.R.; Steigner, M.; et al. Complementary Value of Cardiac Magnetic Resonance Imaging and Positron Emission Tomography/Computed Tomography in the Assessment of Cardiac Sarcoidosis. Circ. Cardiovasc. Imaging 2018, 11, e007030. [Google Scholar] [CrossRef] [PubMed]
- Ammirati, E.; Frigerio, M.; Adler, E.D.; Basso, C.; Birnie, D.H.; Brambatti, M.; Friedrich, M.G.; Klingel, K.; Lehtonen, J.; Moslehi, J.J.; et al. Management of Acute Myocarditis and Chronic Inflammatory Cardiomyopathy. Circ. Heart Fail. 2020, 13, e007405. [Google Scholar] [CrossRef]
- Monda, E.; Limongelli, G. Is There a Role for Genetic Testing in Patients With Myocarditis? Circ. Genom. Precis. Med. 2022, 15, e003824. [Google Scholar] [CrossRef]
- van der Bijl, P.; Delgado, V.; Bootsma, M.; Bax, J.J. Risk Stratification of Genetic, Dilated Cardiomyopathies Associated With Neuromuscular Disorders. Circulation 2018, 137, 2514–2527. [Google Scholar] [CrossRef]
- Kamdar, F.; Garry, D.J. Dystrophin-Deficient Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 2533–2546. [Google Scholar] [CrossRef]
- Yilmaz, A.; Gdynia, H.-J.; Baccouche, H.; Mahrholdt, H.; Meinhardt, G.; Basso, C.; Thiene, G.; Sperfeld, A.-D.; Ludolph, A.C.; Sechtem, U. Cardiac involvement in patients with Becker muscular dystrophy: New diagnostic and pathophysiological insights by a CMR approach. J. Cardiovasc. Magn. Reson. 2008, 10, 50. [Google Scholar] [CrossRef]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 2021, 7, 13. [Google Scholar] [CrossRef]
- Palladino, A.; Papa, A.A.; Morra, S.; Russo, V.; Ergoli, M.; Rago, A.; Orsini, C.; Nigro, G.; Politano, L. Are there real benefits to implanting cardiac devices in patients with end-stage dilated dystrophinopathic cardiomyopathy? Review of literature and personal results. Acta Myol. 2019, 38, 1–7. [Google Scholar]
- Blaszczyk, E.; Gröschel, J.; Schulz-Menger, J. Role of CMR Imaging in Diagnostics and Evaluation of Cardiac Involvement in Muscle Dystrophies. Curr. Heart Fail. Rep. 2021, 18, 211–224. [Google Scholar] [CrossRef]
- Nigro, G.; Comi, L.; Politano, L.; Bain, R. The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int. J. Cardiol. 1990, 26, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Luetkens, J.A.; von Landenberg, C.; Isaak, A.; Faron, A.; Kuetting, D.; Gliem, C.; Dabir, D.; Kornblum, C.; Thomas, D. Comprehensive Cardiac Magnetic Resonance for Assessment of Cardiac Involvement in Myotonic Muscular Dystrophy Type 1 and 2 without Known Cardiovascular Disease. Circ. Cardiovasc. Imaging 2019, 12, e009100. [Google Scholar] [CrossRef] [PubMed]
- Angulski, A.B.B.; Hosny, N.; Cohen, H.; Martin, A.A.; Hahn, D.; Bauer, J.; Metzger, J.M. Duchenne muscular dystrophy: Disease mechanism and therapeutic strategies. Front. Physiol. 2023, 14, 1183101. [Google Scholar] [CrossRef]
- Kerstens, T.P.; van Everdingen, W.M.; Habets, J.; van Dijk, A.P.; Helbing, W.A.; Thijssen, D.H.; Cate, F.E.U.T. Left ventricular deformation and myocardial fibrosis in pediatric patients with Duchenne muscular dystrophy. Int. J. Cardiol. 2023, 388, 131162. [Google Scholar] [CrossRef] [PubMed]
- Marchel, M.; Madej-Pilarczyk, A.; Tymińska, A.; Steckiewicz, R.; Ostrowska, E.; Wysińska, J.; Russo, V.; Grabowski, M.; Opolski, G. Cardiac Arrhythmias in Muscular Dystrophies Associated with Emerinopathy and Laminopathy: A Cohort Study. J. Clin. Med. 2021, 10, 732. [Google Scholar] [CrossRef]
- Sanna, T. Cardiac features of Emery–Dreifuss muscular dystrophy caused by lamin A/C gene mutations. Eur. Heart J. 2003, 24, 2227–2236. [Google Scholar] [CrossRef]
- Wang, S.; Peng, D. Cardiac Involvement in Emery-Dreifuss Muscular Dystrophy and Related Management Strategies. Int. Heart J. 2019, 60, 12–18. [Google Scholar] [CrossRef]
- Heller, S.A.; Shih, R.; Kalra, R.; Kang, P.B. Emery-Dreifuss muscular dystrophy. Muscle Nerve 2020, 61, 436–448. [Google Scholar] [CrossRef]
- Sanchez, F.; Weitz, C.; Gutierrez, J.M.; Mestroni, L.; Hanneman, K.; Vargas, D. Cardiac MR Imaging of Muscular Dystrophies. Curr. Probl. Diagn. Radiol. 2022, 51, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Iavarone, M.; Covino, S.; Petillo, R.; Russo, V. Interatrial block as a first clinical presentation of atrial cardiomyopathy related to a novel LMNA variant: A case report. Eur. Heart J.-Case Rep. 2023, 7, ytad532. [Google Scholar] [CrossRef] [PubMed]
- Thornton, C.A. Myotonic dystrophy. Neurol. Clin. 2014, 32, 705–719. [Google Scholar] [CrossRef]
- Russo, V.; Rago, A.; Ciardiello, C.; Russo, M.G.; Calabrò, P.; Politano, L.; Nigro, G. The Role of the Atrial Electromechanical Delay in Predicting Atrial Fibrillation in Myotonic Dystrophy Type 1 Patients. J. Cardiovasc. Electrophysiol. 2016, 27, 65–72. [Google Scholar] [CrossRef]
- Russo, V.; Rago, A.; Papa, A.A.; Politano, L.; Golino, P.; Russo, M.G.; Calabro, R.; Nigro, G. Does a high percentage of right ventricular pacing influence the incidence of paroxysmal atrial fibrillation in myotonic dystrophy type 1 patients? Pol. Heart J. 2013, 71, 1147–1153. [Google Scholar] [CrossRef]
- Russo, V.; Nigro, G.; Di Meo, F.; Papa, A.A.; Della Cioppa, N.; Proietti, R.; Russo, M.G.; Calabrò, R.; Politano, L. The effect of atrial preference pacing on atrial fibrillation electrophysiological substrate in Myotonic Dystrophy type 1 population. Acta Myol. 2014, 33, 127–135. [Google Scholar]
- Russo, V.; Di Meo, F.; Rago, A.; A Papa, A.; Molino, A.; Mosella, M.; Politano, L.; Russo, M.G.; Nigro, G. Paroxysmal atrial fibrillation in myotonic dystrophy type 1 patients: P wave duration and dispersion analysis. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 1241–1248. [Google Scholar]
- Russo, V.; Antonini, G.; Massa, R.; Casali, C.; Mauriello, A.; Martino, A.M.; Marconi, R.; Garibaldi, M.; Franciosa, P.; Zecchin, M.; et al. Comprehensive Cardiovascular Management of Myotonic Dystrophy Type 1 Patients: A Report from the Italian Neuro-Cardiology Network. J. Cardiovasc. Dev. Dis. 2024, 11, 63. [Google Scholar] [CrossRef]
- Russo, V.; Papa, A.A.; Nigro, G. The Controversial Epidemiology of Left Ventricular Dysfunction in Patients with Myotonic Dystrophy Type 1. JAMA Cardiol. 2017, 2, 1044. [Google Scholar] [CrossRef]
- Groh, W.J.; Bhakta, D.; Tomaselli, G.F.; Aleong, R.G.; Teixeira, R.A.; Amato, A.; Asirvatham, S.J.; Cha, Y.-M.; Corrado, D.; Duboc, D.; et al. 2022 HRS expert consensus statement on evaluation and management of arrhythmic risk in neuromuscular disorders. Heart Rhythm. 2022, 19, e61–e120. [Google Scholar] [CrossRef]
- Russo, V.; Sperlongano, S.; Gallinoro, E.; Rago, A.; Papa, A.A.; Golino, P.; Politano, L.; Nazarian, S.; Nigro, G. Prevalence of Left Ventricular Systolic Dysfunction in Myotonic Dystrophy Type 1: A Systematic Review. J. Card. Fail. 2020, 26, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Russo, V.; Papa, A.A.; Rago, A.; Nigro, G. Which Is the True Epidemiology of Atrial Fibrillation in Myotonic Dystrophy Type 1 Patients? Pacing Clin. Electrophysiol. 2016, 39, 1418–1419. [Google Scholar] [CrossRef] [PubMed]
- Petri, H.; Vissing, J.; Witting, N.; Bundgaard, H.; Køber, L. Cardiac manifestations of myotonic dystrophy type 1. Int. J. Cardiol. 2012, 160, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Russo, V.; Capolongo, A.; Bottino, R.; Carbone, A.; Palladino, A.; Liccardo, B.; Nigro, G.; Marchel, M.; Golino, P.; D’andrea, A. Echocardiographic Features of Cardiac Involvement in Myotonic Dystrophy 1: Prevalence and Prognostic Value. J. Clin. Med. 2023, 12, 1947. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, N.M.; Oliveira, R.B.; Dantas, R.O.; Iazigi, N. Effect of isosorbide dinitrate on gastroesophageal reflux in healthy volunteers and patients with Chagas’ disease. Dig. Dis. Sci. 1995, 40, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Marin-Neto, J.A.; Cunha-Neto, E.; Maciel, B.C.; Simões, M.V. Pathogenesis of Chronic Chagas Heart Disease. Circulation 2007, 115, 1109–1123. [Google Scholar] [CrossRef] [PubMed]
- Rassi, A., Jr.; Rassi, A.; Marin-Neto, J.A. Chagas heart disease: Pathophysiologic mechanisms, prognostic factors and risk stratification. Mem. Inst. Oswaldo Cruz 2009, 104 (Suppl. S1), 152–158. [Google Scholar] [CrossRef]
- Rochitte, C.E.; Oliveira, P.F.; Andrade, J.M.; Ianni, B.M.; Parga, J.R.; Ávila, L.F.; Kalil-Filho, R.; Mady, C.; Meneghetti, J.C.; Lima, J.A.; et al. Myocardial Delayed Enhancement by Magnetic Resonance Imaging in Patients with Chagas’ Disease. J. Am. Coll. Cardiol. 2005, 46, 1553–1558. [Google Scholar] [CrossRef]
- Miranda, C.H.; Figueiredo, A.B.; Maciel, B.C.; Marin-Neto, J.A.; Simões, M.V. Sustained Ventricular Tachycardia Is Associated with Regional Myocardial Sympathetic Denervation Assessed with 123I-Metaiodobenzylguanidine in Chronic Chagas Cardiomyopathy. J. Nucl. Med. 2011, 52, 504–510. [Google Scholar] [CrossRef]
- Køber, L.; Thune, J.J.; Nielsen, J.C.; Haarbo, J.; Videbæk, L.; Korup, E.; Jensen, G.; Hildebrandt, P.; Steffensen, F.H.; Bruun, N.E.; et al. Defibrillator Implantation in Patients with Nonischemic Systolic Heart Failure. N. Engl. J. Med. 2016, 375, 1221–1230. [Google Scholar] [CrossRef]
- Beggs, S.A.S.; Jhund, P.S.; E Jackson, C.; McMurray, J.J.V.; Gardner, R.S. Non-ischaemic cardiomyopathy, sudden death and implantable defibrillators: A review and meta-analysis. Heart 2018, 104, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Leoni, L.; Link, M.S.; Della Bella, P.; Gaita, F.; Curnis, A.; Salerno, J.U.; Igidbashian, D.; Raviele, A.; Disertori, M.; et al. Implantable Cardioverter-Defibrillator Therapy for Prevention of Sudden Death in Patients with Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia. Circulation 2003, 108, 3084–3091. [Google Scholar] [CrossRef] [PubMed]
- Hulot, J.S.; Jouven, X.; Empana, J.P.; Frank, R.; Fontaine, G. Natural History and Risk Stratification of Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. Circulation 2004, 110, 1879–1884. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Baldinger, S.H.; Gandjbakhch, E.; Maury, P.; Sellal, J.-M.; Androulakis, A.F.; Waintraub, X.; Charron, P.; Rollin, A.; Richard, P.; et al. Long-Term Arrhythmic and Nonarrhythmic Outcomes of Lamin A/C Mutation Carriers. J. Am. Coll. Cardiol. 2016, 68, 2299–2307. [Google Scholar] [CrossRef] [PubMed]
- Gigli, M.; Stolfo, D.; Graw, S.L.; Merlo, M.; Gregorio, C.; Nee Chen, S.; Dal Ferro, M.; PaldinoMD, A.; De Angelis, G.; Brun, F.; et al. Phenotypic Expression, Natural History, and Risk Stratification of Cardiomyopathy Caused by Filamin C Truncating Variants. Circulation 2021, 144, 1600–1611. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, M.M.; Lorenzini, M.; Pavlou, M.; Ochoa, J.P.; O’mahony, C.; Restrepo-Cordoba, M.A.; Segura-Rodriguez, D.; Bermúdez-Jiménez, F.; Molina, P.; Cuenca, S.; et al. Association of Left Ventricular Systolic Dysfunction among Carriers of Truncating Variants in Filamin C With Frequent Ventricular Arrhythmia and End-stage Heart Failure. JAMA Cardiol. 2021, 6, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Hodgkinson, K.; Connors, S.; Merner, N.; Haywood, A.; Young, T.; McKenna, W.; Gallagher, B.; Curtis, F.; Bassett, A.; Parfrey, P. The natural history of a genetic subtype of arrhythmogenic right ventricular cardiomyopathy caused by a p.S358L mutation in TMEM43. Clin. Genet. 2013, 83, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Hey, T.M.; Rasmussen, T.B.; Madsen, T.; Aagaard, M.M.; Harbo, M.; Mølgaard, H.; Møller, J.E.; Eiskjær, H.; Mogensen, J. Pathogenic RBM20-Variants Are Associated with a Severe Disease Expression in Male Patients with Dilated Cardiomyopathy. Circ. Heart Fail. 2019, 12, e005700. [Google Scholar] [CrossRef]
- Wahbi, K.; BEN Yaou, R.; Gandjbakhch, E.; Anselme, F.; Gossios, T.; Lakdawala, N.K.; Stalens, C.; Sacher, F.; Babuty, D.; Trochu, J.-N.; et al. Development and Validation of a New Risk Prediction Score for Life-Threatening Ventricular Tachyarrhythmias in Laminopathies. Circulation 2019, 140, 293–302. [Google Scholar] [CrossRef]
- Hodgkinson, K.A.; Howes, A.; Boland, P.; Shen, X.S.; Stuckless, S.; Young, T.-L.; Curtis, F.; Collier, A.; Parfrey, P.S.; Connors, S.P.; et al. Long-Term Clinical Outcome of Arrhythmogenic Right Ventricular Cardiomyopathy in Individuals with a p.S358L Mutation in TMEM43 Following Implantable Cardioverter Defibrillator Therapy. Circ. Arrhythm. Electrophysiol. 2016, 9, e003589. [Google Scholar] [CrossRef]
- Gigli, M.; Merlo, M.; Graw, S.L.; Barbati, G.; Rowland, T.J.; Slavov, D.B.; Stolfo, D.; Haywood, M.E.; Ferro, M.D.; Altinier, A.; et al. Genetic Risk of Arrhythmic Phenotypes in Patients with Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2019, 74, 1480–1490. [Google Scholar] [CrossRef]
- A McDonagh, T.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. Corrigendum to: 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) With the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2021, 42, 4901. [Google Scholar] [CrossRef]
- Gilljam, T.; Haugaa, K.H.; Jensen, H.K.; Svensson, A.; Bundgaard, H.; Hansen, J.; Dellgren, G.; Gustafsson, F.; Eiskjær, H.; Andreassen, A.K.; et al. Heart transplantation in arrhythmogenic right ventricular cardiomyopathy—Experience from the Nordic ARVC Registry. Int. J. Cardiol. 2018, 250, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Russo, V.; Ciabatti, M.; Brunacci, M.; Dendramis, G.; Santobuono, V.; Tola, G.; Picciolo, G.; Teresa, L.M.; D’andrea, A.; Nesti, M. Opportunities and drawbacks of the subcutaneous defibrillator across different clinical settings. Expert Rev. Cardiovasc. Ther. 2023, 21, 151–164. [Google Scholar] [CrossRef]
- AlTurki, A.; Proietti, R.; Russo, V.; Dhanjal, T.; Banerjee, P.; Essebag, V. Anti-arrhythmic drug therapy in implantable cardioverter-defibrillator recipients. Pharmacol. Res. 2019, 143, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Oloriz, T.; Silberbauer, J.; Maccabelli, G.; Mizuno, H.; Baratto, F.; Kirubakaran, S.; Vergara, P.; Bisceglia, C.; Santagostino, G.; Marzi, A.; et al. Catheter Ablation of Ventricular Arrhythmia in Nonischemic Cardiomyopathy. Circ. Arrhythm. Electrophysiol. 2014, 7, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Proietti, R.; Russo, V.; AlTurki, A. Anti-arrhythmic therapy in patients with non-ischemic cardiomyopathy. Pharmacol. Res. 2019, 143, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Sethi, Y.; Patel, N.; Kaka, N.; Kaiwan, O.; Kar, J.; Moinuddin, A.; Goel, A.; Chopra, H.; Cavalu, S. Precision Medicine and the future of Cardiovascular Diseases: A Clinically Oriented Comprehensive Review. J. Clin. Med. 2023, 12, 1799. [Google Scholar] [CrossRef] [PubMed]
- Leopold, J.A.; Loscalzo, J. Emerging Role of Precision Medicine in Cardiovascular Disease. Circ. Res. 2018, 122, 1302–1315. [Google Scholar] [CrossRef] [PubMed]
- Musunuru, K. Genome Editing: The Recent History and Perspective in Cardiovascular Diseases. J. Am. Coll. Cardiol. 2017, 70, 2808–2821. [Google Scholar] [CrossRef]
- Sethi, Y.; Mahtani, A.U.; Khehra, N.; Padda, I.; Patel, N.; Sebastian, S.A.; Malhi, G.; Kaiwan, O.; Saith, S.; Johal, G. Gene Editing as the Future of Cardiac Amyloidosis Therapeutics. Curr. Probl. Cardiol. 2023, 48, 101741. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Marti-Gutierrez, N.; Park, S.-W.; Wu, J.; Lee, Y.; Suzuki, K.; Koski, A.; Ji, D.; Hayama, T.; Ahmed, R.; et al. Correction of a pathogenic gene mutation in human embryos. Nature 2017, 548, 413–419. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Desmosomal |
| PKP2—Plakophilin C (OMIM: 602861) DSP—Desmoplakin (OMIM: 125647) DSG2—Desmoglein 2 (OMIM: 125671) DSC2—Desmocollin (OMIM: 125645) |
| Non-desmosomal |
| PLN—Phospholamban (OMIM: 172405) FLNC—Filamin C (OMIM: 1029565) DES—Desmin (OMIM: 125660) TTN—Titin (OMIM: 188840) LMNA—Lamin A/C (OMIM: 150330) TMEM 43—Transmembrane protein 43 (OMIM: 612048) TGFB3—Transforming growth factor-3 (OMIM: 190230) |
| ALVC | DCM | Myocarditis | CS | |
|---|---|---|---|---|
| LV function |
|
|
|
|
| LV dilation |
|
|
|
|
| LGE at CMR |
|
|
|
|
| Peculiar features |
|
|
|
|
| Tachy/Brady-arrhythmias |
|
|
|
|
| Further investigations |
|
|
|
|
| Genotype | Additional Risk Factors (If Present ICD Implantation Is Recommended) | Class of Recommendation—Level of Evidence |
|---|---|---|
| LMNA | ≥7% five-year risk of SCD estimated with LMNA-risk VTA calculator | IIa-C |
| FLNC-truncating variants | LGE on CMR LVEF < 45% | IIa-C |
| TMEM43 | Male Female and any of the following: LVEF < 45%, NSVT, LGE on CMR, >200 VE on 24 h Holter ECG | IIa-C |
| PLN p.Arg14del variant | ≥7% five-year risk of SCD estimated with PLN variant-specific risk calculator. | IIa-C |
| DSP | LGE on CMR LVEF < 45% | IIa-C |
| DES | No adjunctive risk factors | IIb-C |
| RBM20 | LGE on CMR LVEF < 45% | IIa-C |
| ALVC with any known causative gene variant and LVEF > 35% | No adjunctive risk factors | IIb-C |
| ALVC without known causative gene variant and LVEF > 35% | Syncope LGE presence on CMR | IIb-C |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mauriello, A.; Roma, A.S.; Ascrizzi, A.; Molinari, R.; Loffredo, F.S.; D’Andrea, A.; Russo, V. Arrhythmogenic Left Ventricular Cardiomyopathy: From Diagnosis to Risk Management. J. Clin. Med. 2024, 13, 1835. https://doi.org/10.3390/jcm13071835
Mauriello A, Roma AS, Ascrizzi A, Molinari R, Loffredo FS, D’Andrea A, Russo V. Arrhythmogenic Left Ventricular Cardiomyopathy: From Diagnosis to Risk Management. Journal of Clinical Medicine. 2024; 13(7):1835. https://doi.org/10.3390/jcm13071835
Chicago/Turabian StyleMauriello, Alfredo, Anna Selvaggia Roma, Antonia Ascrizzi, Riccardo Molinari, Francesco S. Loffredo, Antonello D’Andrea, and Vincenzo Russo. 2024. "Arrhythmogenic Left Ventricular Cardiomyopathy: From Diagnosis to Risk Management" Journal of Clinical Medicine 13, no. 7: 1835. https://doi.org/10.3390/jcm13071835