
Autosomal Dominant Retinitis Pigmentosa Secondary to TOPORS Mutations: A Report of a Novel Mutation and Clinical Findings

Abstract
:1. Introduction
2. Methods
2.1. Patient Population
2.2. Genetic Analysis
3. Results
3.1. Identification of TOPORS Mutations
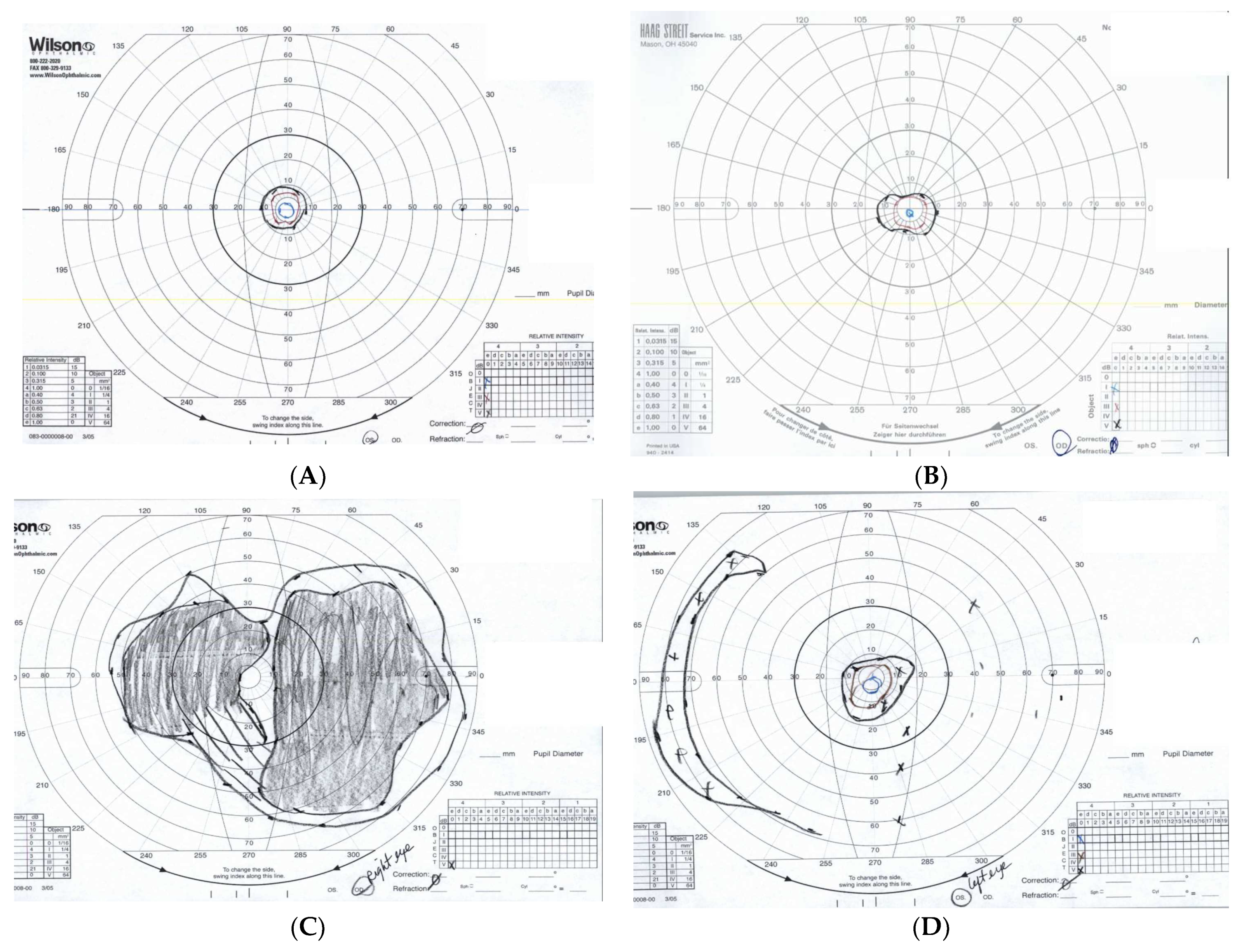
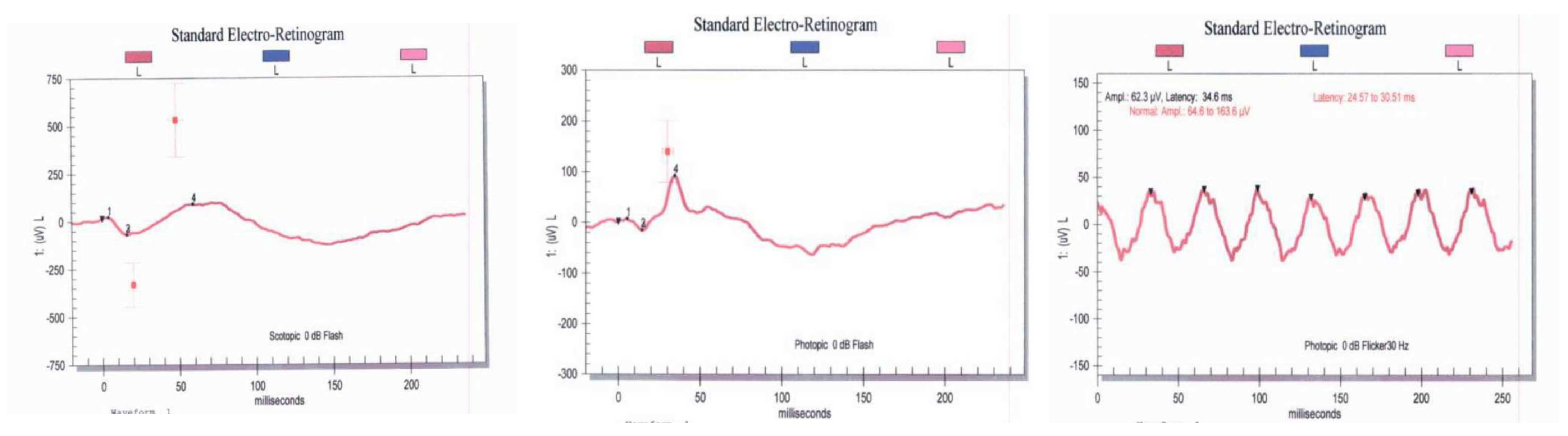
3.2. Clinical Features of Patients with TOPORS Mutations
3.3. An Isolated Case of Full Thickness Macular Hole Leading to Rapid Vision Loss with Successful Repair
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bowne, S.J.; Sullivan, L.S.; Gire, A.I.; Birch, D.G.; Hughbanks-Wheaton, D.; Heckenlively, J.R.; Daiger, S.P. Mutations in the TOPORS gene cause 1% of autosomal dominant retinitis pigmentosa. Mol. Vis. 2008, 14, 922–927. [Google Scholar] [PubMed]
- Schob, C.; Orth, U.; Gal, A.; Kindler, S.; Chakarova, C.F.; Bhattacharya, S.S.; Rüther, K. Mutations in TOPORS: A Rare Cause of Autosomal Dominant Retinitis Pigmentosa in Continental Europe? Ophthalmic Genet. 2009, 30, 96–98. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Zhou, Y.; Li, N. Mutations of TOPORS identified in families with retinitis pigmentosa. Ophthalmic Genet. 2022, 43, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Chakarova, C.F.; Khanna, H.; Shah, A.Z.; Patil, S.B.; Sedmak, T.; Murga-Zamalloa, C.A.; Papaioannou, M.G.; Nagel-Wolfrum, K.; Lopez, I.; Munro, P.; et al. TOPORS, implicated in retinal degeneration, is a cilia-centrosomal protein. Hum. Mol. Genet. 2011, 20, 975. [Google Scholar] [CrossRef] [PubMed]
- Saleem, A.; Dutta, J.; Malegaonkar, D.; Rasheed, F.; Rasheed, Z.; Rajendra, R.; Marshall, H.; Luo, M.; Li, H.; Rubin, E.H. The topoisomerase I- and p53-binding protein topors is differentially expressed in normal and malignant human tissues and may function as a tumor suppressor. Oncogene 2004, 23, 5293–5300. [Google Scholar] [CrossRef] [PubMed]
- Rajendra, R.; Malegaonkar, D.; Pungaliya, P.; Marshall, H.; Rasheed, Z.; Brownell, J.; Liu, L.F.; Lutzker, S.; Saleem, A.; Rubin, E.H. Topors functions as an E3 ubiquitin ligase with specific E2 enzymes and ubiquitinates p53. J. Biol. Chem. 2004, 279, 36440–36444. [Google Scholar] [CrossRef] [PubMed]
- Chakarova, C.F.; Papaioannou, M.G.; Khanna, H.; Lopez, I.; Waseem, N.; Shah, A.; Theis, T.; Friedman, J.; Maubaret, C.; Bujakowska, K.; et al. Mutations in TOPORS Cause Autosomal Dominant Retinitis Pigmentosa with Perivascular Retinal Pigment Epithelium Atrophy. Am. J. Hum. Genet. 2007, 81, 1098–1103. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, Y.; Jiang, Y.; Li, X.; Xiao, X.; Li, S.; Jia, X.; Sun, W.; Wang, P.; Zhang, Q. Autosomal Dominant Retinitis Pigmentosa–Associated TOPORS Protein Truncating Variants Are Exclusively Located in the Region of Amino Acid Residues 807 to 867. Investig. Opthalmol. Vis. Sci. 2022, 63, 19. [Google Scholar] [CrossRef] [PubMed]
- VCV001510617.4—ClinVar—NCBI. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/1510617/ (accessed on 9 May 2023).
- Coussa, R.G.; Chakarova, C.; Ajlan, R.; Taha, M.; Kavalec, C.; Gomolin, J.; Khan, A.; Lopez, I.; Ren, H.; Waseem, N.; et al. Genotype and Phenotype Studies in Autosomal Dominant Retinitis Pigmentosa (adRP) of the French Canadian Founder Population. Investig. Ophthalmol. Vis. Sci. 2015, 56, 8297–8305. [Google Scholar] [CrossRef] [PubMed]
- Genead, M.A.; Fishman, G.A. Efficacy of sustained topical dorzolamide therapy for cystic macular lesions in patients with retinitis pigmentosa and usher syndrome. Arch. Ophthalmol. Ill 1960 2010, 128, 1146–1150. [Google Scholar] [CrossRef]
- Duker, J.S.; Kaiser, P.K.; Binder, S.; de Smet, M.D.; Gaudric, A.; Reichel, E.; Sadda, S.R.; Sebag, J.; Spaide, R.F.; Stalmans, P. The International Vitreomacular Traction Study Group classification of vitreomacular adhesion, traction, and macular hole. Ophthalmology 2013, 120, 2611–2619. [Google Scholar] [CrossRef]
- Giusti, C.; Forte, R.; Vingolo, E.M. Clinical pathogenesis of macular holes in patients affected by retinitis pigmentosa. Eur. Rev. Med. Pharmacol. Sci. 2002, 6, 45–48. [Google Scholar] [PubMed]
- Lee, C.Y.; Yang, C.M.; Yang, C.H.; Hu, F.R.; Chen, T.C. Flap technique-assisted surgeries for advanced retinitis pigmentosa complicated with macular hole: A case report and literature review. BMC Ophthalmol. 2021, 21, 322. [Google Scholar] [CrossRef] [PubMed]
- All Variants in the TOPORS Gene—Global Variome Shared LOVD. Available online: https://databases.lovd.nl/shared/variants/TOPORS?search_position_c_start=2556&search_position_c_start_intron=0&search_position_c_end=2557&search_position_c_end_intron=0&search_vot_clean_dna_change=%3D%22c.2556_2557del%22&search_transcriptid=00021645 (accessed on 15 May 2023).
- Unique Variants in the TOPORS Gene—Global Variome Shared LOVD. Available online: https://databases.lovd.nl/shared/variants/TOPORS/unique (accessed on 9 May 2023).
- Sullivan, L.S.; Bowne, S.J.; Reeves, M.J.; Blain, D.; Goetz, K.; NDifor, V.; Vitez, S.; Wang, X.; Tumminia, S.J.; Daiger, S.P. Prevalence of mutations in eyeGENE probands with a diagnosis of autosomal dominant retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2013, 54, 6255–6261. [Google Scholar] [CrossRef] [PubMed]
- TOPORS|gnomAD v2.1.1|gnomAD. Available online: https://gnomad.broadinstitute.org/gene/ENSG00000197579?dataset=gnomad_r2_1 (accessed on 4 June 2023).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Family | Sex | Age at Last Follow Up | Age of Onset | Follow Up Time | Past Medical History | Past Ocular History | TOPORS Mutational Variant | BCVA OD | BCVA OS | Visual Field Testing |
|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | F1 | F | 12 | NA | 2 Years | Autism, ADHD, Gait abnormality | None | c.2556_2557del | 20/30 − 1 | 20/25 + 2 | Mild Restriction OD. Smallest target seen is I3e. |
| P2 | F1 | F | 35 | 20 s | 2 Years | GAD, Smoker | Dry Eye | c.2556_2557del | 20/25 − 1 | 20/40 | NA |
| P3 | F1 | F | 32 | 18 | 13 Years | IBS, GAD, Endometriosis | None | c.2556_2557del | 20/25 − 1 | 20/25 − 1 | About 80 central vision OU. Smallest target seen is V4e OD and I4e OS. |
| P4 | F2 | F | 43 | 20 s | 4 Years | GERD, Hypothyroidism | PSC OU | c.2556_2557del | 20/25 − 1 | 20/25 − 1 | Ring scotoma OU. Smallest target seen is I4e. |
| P5 | F2 | F | 25 | 20 s | 2 Months | GAD | Photophobia | c.2556_2557del | 20/20 | 20/20 | NA |
| P6 | F3 | F | 51 | 20s | 17 Years | Nephrolithiasis, Smoker, GAD | PSC OD, CME OU, PCIOL OD | c.2431C>T | 20/60 − 1 | 20/60 + 2 | GVF OU limited to central 10 degrees. Smallest target is I4e. |
| P7 | F3 | F | 70 | 20 s | 16 Years | Nephrolithiasis, Hypertension, Hyperlipidemia | PCIOL OU, Hx of PSC OU, CME OU, ERM OU, FTMH OD, PTMH OS | c.2431C>T | 20/60 | 20/40 | GVF of less than 10 degrees OU. Smallest target seen is V4e. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eid, A.T.; Eid, K.T.; Odom, J.V.; Hinkle, D.; Leys, M. Autosomal Dominant Retinitis Pigmentosa Secondary to TOPORS Mutations: A Report of a Novel Mutation and Clinical Findings. J. Clin. Med. 2024, 13, 1498. https://doi.org/10.3390/jcm13051498
Eid AT, Eid KT, Odom JV, Hinkle D, Leys M. Autosomal Dominant Retinitis Pigmentosa Secondary to TOPORS Mutations: A Report of a Novel Mutation and Clinical Findings. Journal of Clinical Medicine. 2024; 13(5):1498. https://doi.org/10.3390/jcm13051498
Chicago/Turabian StyleEid, Alen T., Kevin Toni Eid, James Vernon Odom, David Hinkle, and Monique Leys. 2024. "Autosomal Dominant Retinitis Pigmentosa Secondary to TOPORS Mutations: A Report of a Novel Mutation and Clinical Findings" Journal of Clinical Medicine 13, no. 5: 1498. https://doi.org/10.3390/jcm13051498