
The Risk of Malignant Degeneration of Müllerian Derivatives in PMDS: A Review of the Literature
 , , , , ,
, , , , ,
Abstract
:1. Introduction
1.1. Sex Determination and Differentiation: An Overview
1.2. Anti-Müllerian Hormone
2. PMDS
2.1. Genetics and Molecular Aspects
- (1)
- PMDS1: the anti-Müllerian hormone gene is located on the short arm of chromosome 19 (19p13.3), has a length of 2.8 kb and is formed by five exons coding for a 535 amino acid protein, it is a member of the transforming growth factor beta (TGF-beta) family [9]. Several different AMH gene mutations have been identified: deletions, insertions, missense, nonsense and splicing mutations play a pathogenetic role in the determinism of the disease. The effect of mutations on the molecule function is heterogeneous: some prevent an adequate post-translational folding of the C-terminal region and this determines a rapid degradation of the molecule, even before its secretion; others result in the biosynthesis of a truncated protein in which the C-terminal portion, the one with biological activity, is missing; moreover others are associated with the synthesis and secretion of a hormone that has lost the ability to bind to its receptor [10]. As a consequence of this spectrum of mutations affecting the AMH gene, the AMH serum levels are characteristically low or undetectable in prepuberal patients with PMDS type I. The diagnostic value of AMH serum concentration is lost in adults in whom the hormone is physiologically undetectable.
- (2)
- PMDS2: the action of AMH on target tissues is made possible by the expression on them of a heterodimeric receptor consisting of AMH type 1 (AMHR1) and type 2 receptor (AMHR2). Only the type 2 receptor is AMH-specific and it is mutated in PMDS type 2 patients. It is a transmembrane receptor of 573 amino acid consisting of an N-terminal extracellular domain that binds AMH, a single transmembrane domain, and a C-terminal intracellular domain exhibiting serine/threonine kinase activity [10]. The AMHR2 is an 8 kb gene located on the long arm of chromosome 12 (12q13), consisting of 11 exons. The first three exons code for the extracellular domain that binds the hormone, exon four codes for the transmembrane domain, while the remaining seven exons code for the intracellular domain associated with the catalytic activity of the receptor. All exons can be affected by mutations; as for the AMH gene, it can be of various natures: missense, nonsense, deletions and splicing mutations have been detected [10]. Specifically, the mutation most frequently found in patients with PMDS2 is a 27-base pair deletion affecting exon 10 [1]. Mutations in the AMHR2 gene prevent the AMH from carrying out its action on target tissues. The insensitivity of the Müllerian ducts to AMH therefore means that in the fetus, they do not undergo the physiological process of involution but give rise to the development of the fallopian tubes, uterus and proximal third of the vagina. In contrast to type 1 PMDS, where the syndrome develops as a consequence of the receptor mutation, the serum concentration of AMH is normal in prepuberal males.
2.2. Clinical Features
- (1)
- Bilateral Cryptorchidism: the testes are both located in the pelvis assuming a position similar to the one normally presented by the ovaries; they are included in the broad ligament of the uterus, close to the fimbriae of the fallopian tubes. This is estimated to occur in 60–70% of patients [11].
- (2)
- Unilateral Cryptorchidism: one of the two testicles are located in the pelvis while the contralateral one is in the scrotum. The descended testicle is associated with the presence of an ipsilateral inguinal or inguinal-scrotal hernia whose content is represented by the fallopian tube of the same side and the uterus, so that this clinical presentation is often referred as a “hernia uteri inguinalis”. This is estimated to occur in 20–30% of patients [12].
- (3)
- Transverse Testicular Ectopia: this is the most specific but least common clinical presentation of PMDS and is characterized by the presence, in a single hemiscrotum, of a hernia sac containing both testes, both tubes and the uterus [12].
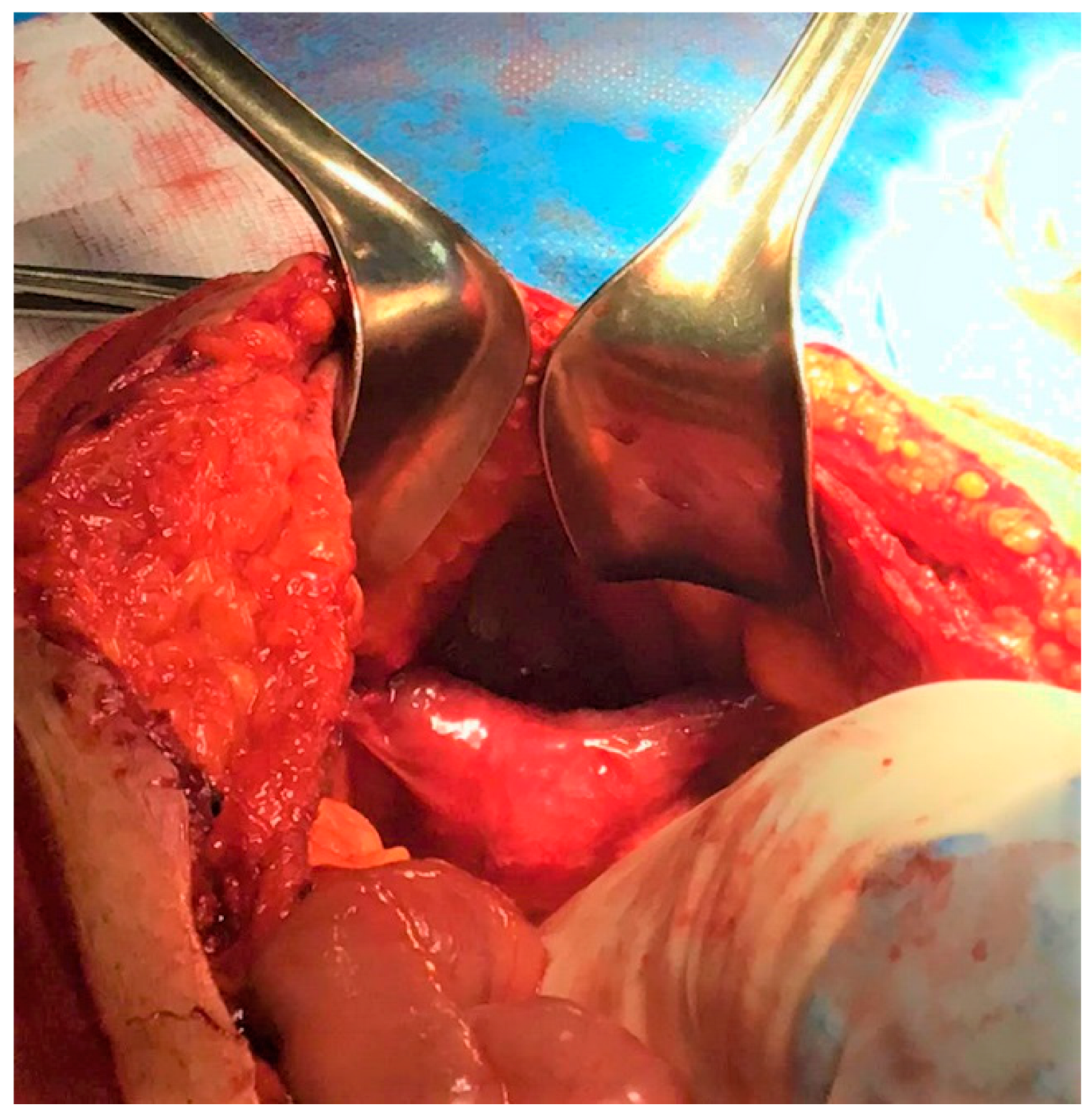
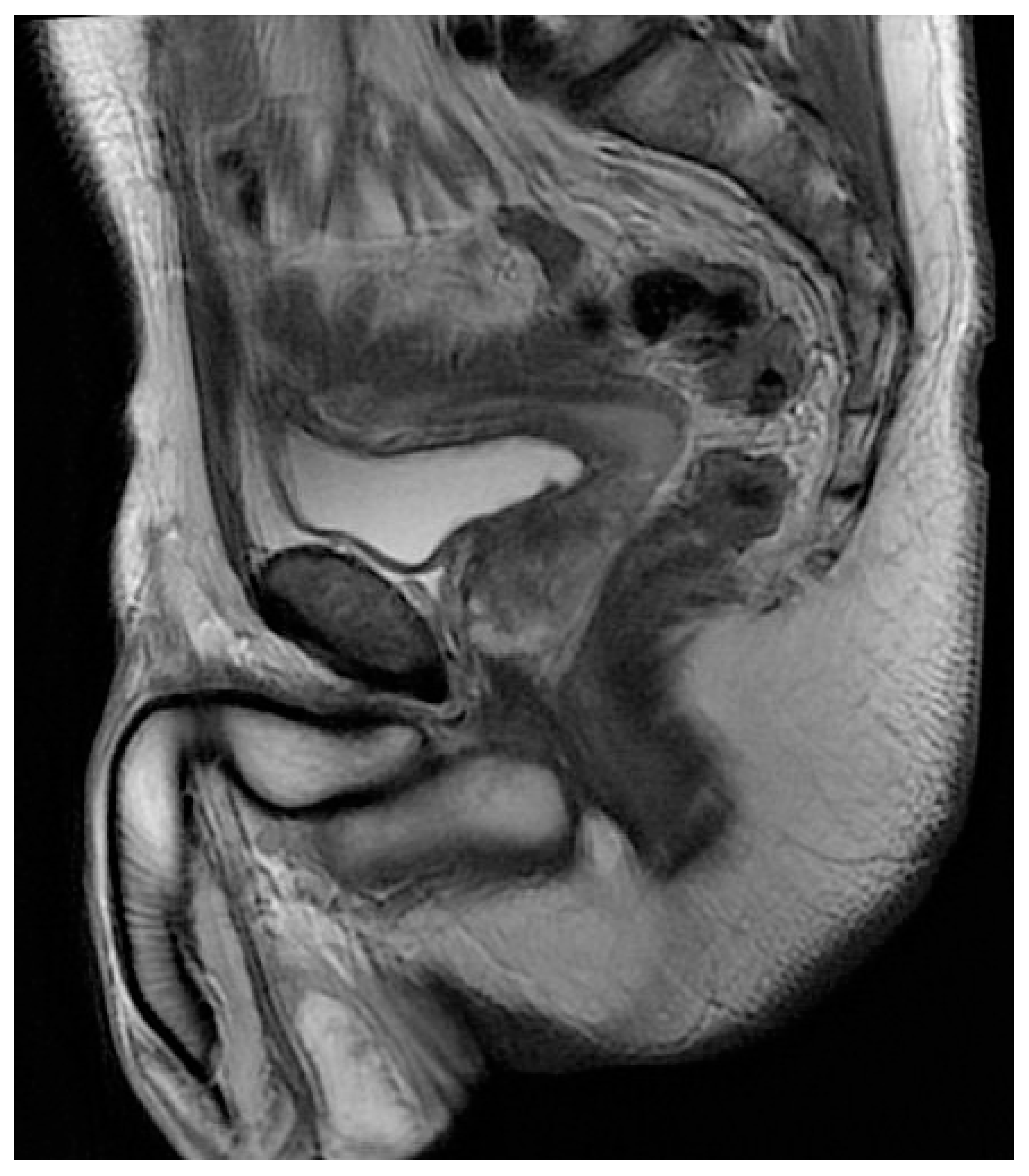
3. Case Report
4. Literature Review and Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Josso, N.; Belville, C.; di Clemente, N.; Picard, J.Y. AMH and AMH receptor defects in persistent Müllerian duct syndrome. Hum. Reprod Update 2005, 11, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Kucinskas, L.; Just, W. Human male sex determination and sexual differentiation: Pathways, molecular interactions and genetic disorders. Medicina 2005, 41, 633–640. [Google Scholar] [PubMed]
- Okashita, N.; Tachibana, M. Transcriptional Regulation of the Y-Linked Mammalian Testis-Determining Gene SRY. Sex Dev. 2021, 15, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Josso, N.; Rey, R.; Picard, J.Y. Testicular anti-Müllerian hormone: Clinical applications in DSD. Semin. Reprod Med. 2012, 30, 364–373. [Google Scholar] [CrossRef]
- Sajjad, Y. Development of the genital ducts and external genitalia in the early human embryo. J. Obs. Gynaecol. Res. 2010, 36, 929–937. [Google Scholar] [CrossRef]
- Josso, N.; Rey, R.A. What Does AMH Tell Us in Pediatric Disorders of Sex Development? Front. Endocrinol. 2020, 11, 619. [Google Scholar] [CrossRef]
- Rey, R. Anti-Müllerian hormone in disorders of sex determination and differentiation. Arq. Bras. Endocrinol. Metabol. 2005, 49, 26–36. [Google Scholar] [CrossRef]
- Edelsztein, N.Y.; Grinspon, R.P.; Schteingart, H.F.; Rey, R.A. Anti-Müllerian hormone as a marker of steroid and gonadotropin action in the testis of children and ado-lescents with disorders of the gonadal axis. Int. J. Pediatr. Endocrinol. 2016, 2016, 20. [Google Scholar] [CrossRef]
- Nishi, M.Y.; Domenice, S.; Maciel-Guerra, A.T.; Zaba Neto, A.; Silva, M.A.; Costa, E.M.; Guerra-Junior, G.; Mendonca, B.B. Analysis of anti-Müllerian hormone (AMH) and its receptor (AMHR2) genes in patients with persistent Müllerian duct syndrome. Arq. Bras. Endocrinol. Metabol. 2012, 56, 473–478. [Google Scholar] [CrossRef]
- Picard, J.Y.; Cate, R.L.; Racine, C.; Josso, N. The Persistent Müllerian Duct Syndrome: An Update Based Upon a Personal Experience of 157 Cases. Sex Dev. 2017, 11, 109–125. [Google Scholar] [CrossRef]
- Clarnette, T.D.; Sugita, Y.; Hutson, J.M. Genital anomalies in human and animal models reveal the mechanisms and hormones governing testicular descent. Br. J. Urol. 1997, 79, 99–112. [Google Scholar] [CrossRef]
- Ren, X.; Wu, D.; Gong, C. Persistent Müllerian duct syndrome: A case report and review. Exp. Ther. Med. 2017, 14, 5779–5784. [Google Scholar] [CrossRef] [PubMed]
- Farikullah, J.; Ehtisham, S.; Nappo, S.; Patel, L.; Hennayake, S. Persistent Müllerian duct syndrome: Lessons learned from managing a series of eight patients over a 10-year period and review of literature regarding malignant risk from the Müllerian remnants. BJU Int. 2012, 110, E1084–E1089. [Google Scholar] [CrossRef]
- Elmas, N.Z.; Esmat, H.A.; Osmani, G.M.; Ozcan, B.; Kızılay, F. Female form of persistent Müllerian duct syndrome: A rare case report and review of literature. Int. J. Surg. Case Rep. 2020, 77, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Beatty, J.S.; Bhalla, V.K.; Hatley, R.M.; Pipkin, W.L.; Howell, C.G. Neglected cryptorchidism: Delayed recognition of persistent müllerian duct syndrome and subsequent malignant degeneration. Urology 2013, 82, 511–514. [Google Scholar] [CrossRef] [PubMed]
- Bowen, D.K.; Matulewicz, R.S.; Gong, E.M. Preservation of müllerian structures with laparoscopic management of intra-abdominal testes in persistent müllerian duct syndrome. Pediatr. Urol. 2016, 12, 65–66. [Google Scholar] [CrossRef]
- Szemes, G.C.; Rubin, D.J. Squamous cell carcinoma in a Müllerian duct cyst. J. Urol. 1968, 100, 40–43. [Google Scholar] [CrossRef]
- Hodgson, N.B. Long-term survival from Müllerian duct carcinoma. J. Urol. 1976, 116, 313–315. [Google Scholar] [CrossRef]
- Novak, R.W.; Raines, R.B.; Sollee, A.N. Clear cell carcinoma in a Müllerian duct cyst. Am. J. Clin. Pathol. 1981, 76, 339–341. [Google Scholar] [CrossRef]
- Youngson, G.G. Squamous metaplasia complicating a müllerian duct remnant. Br. J. Urol. 1990, 65, 211–212. [Google Scholar] [CrossRef]
- Gilbert, R.F.; Ibarra, J.; Tansey, L.A.; Shanberg, A.M. Adenocarcinoma in a müllerian duct cyst. J. Urol. 1992, 148, 1262–1264. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Ito, H.; Kobayashi, K. Squamous cell carcinoma in a Müllerian duct cyst: Report of a case. Surg Today. 1996, 26, 645–648. [Google Scholar] [CrossRef]
- Shinmura, Y.; Yokoi, T.; Tsutsui, Y. A case of clear cell adenocarcinoma of the müllerian duct in persistent müllerian duct syndrome: The first reported case. Am. J. Surg. Pathol. 2002, 26, 1231–1234. [Google Scholar] [CrossRef] [PubMed]
- Thiel, D.D.; Erhard, M.J. Uterine adenosarcoma in a boy with persistent müllerian duct syndrome: First reported case. J. Pediatr. Surg. 2005, 40, e29–e31. [Google Scholar] [CrossRef] [PubMed]
- Romero, F.R.; Fucs, M.; Castro, M.G.; Garcia, C.R.; Fernandes, R.d.e.C.; Perez, M.D. Adenocarcinoma of persistent müllerian duct remnants: Case report and differential diagnosis. Urology 2005, 66, 194–195. [Google Scholar] [CrossRef]
- Xing, J.P.; Dang, J.G.; Wu, D.P.; Long, Q.Z.; Chen, X.F.; Nan, X.Y. Papillary cystadenocarcinoma in a Müllerian duct cyst: Report of a case with literature. Zhonghua Nan Ke Xue 2006, 12, 218–221. [Google Scholar]
- Warmann, S.W.; Vogel, M.; Wehrmann, M.; Scheel-Walter, H.G.; Artlich, A.; Pereira, P.L.; Fuchs, J. Giant mullerian duct cyst with malignant transformation in 15-year-old boy. Urology 2006, 67, 424.e3–424.e6. [Google Scholar] [CrossRef]
- Dimasis, N.; Koukourikis, P.; Klampatsas, A.; Xirou, P.; Sountoulides, P. A unique case of aggressive uterine cancer in a 45-year-old man with persistent Müllerian duct syndrome. Arch. Esp. Urol. 2019, 72, 435–438. [Google Scholar]
- Ovidiu, B.; Marcu, D.R.; Mischianu, D.L.D.; Poiana, C.; Diaconu, C.C.; Bungau, S.G.; Tit, D.M.; Cumpanas, A.; Bohiltea, R. The challenges of androgen insensitivity syndrome. Arch. Med. Sci. 2021, 18, 881–889. [Google Scholar] [CrossRef]
- Kosti, K.; Athanasiadis, L.; Goulis, D.G. Long-term consequences of androgen insensitivity syndrome. Maturitas 2019, 127, 51–54. [Google Scholar] [CrossRef]
- Smith-Harrison, L.I.; Patel, M.S.; Smith, R.P.; Schenkman, N.S. Persistent Müllerian duct structures presenting as hematuria in an adult: Case report of robotic surgical removal and review of the literature. Urol. Ann. 2015, 7, 544–546. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.H.; Wang, N.L.; Ting, W.H.; Du, Y.C.; Fu, Y.W. Excision of Mullerian duct remnant for persistent Mullerian duct syndrome provides favorable short- and mid-term outcomes. J. Pediatr. Urol. 2014, 10, 929–933. [Google Scholar] [CrossRef]
- Shalaby, M.M.; Kurkar, A.; Zarzour, M.A.; Faddan, A.A.; Khalil, M.; Abdelhafez, M.F. The management of the persistent Müllerian duct syndrome. Arab. J. Urol. 2014, 12, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Kolon, T.F.; Herndon, C.D.; Baker, L.A.; Baskin, L.S.; Baxter, C.G.; Cheng, E.Y.; Diaz, M.; Lee, P.A.; Seashore, C.J.; Tasian, G.E.; et al. Evaluation and treatment of cryptorchidism: AUA guideline. J. Urol. 2014, 192, 337–345. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Reference | Case n. | Year | Age at Diagnosis (Years) | Previous Orchidopexy or Other Relevant Procedures Before Diagnosis | Presentation | Type of Müllerian Malignancy | Outcome |
|---|---|---|---|---|---|---|---|
| [17] | 1 | 1968 | 44 | At age 18 hypospadias repair, bilateral inguinal exploration for cryptorchidism and left orchidopexy. | Recurrent UTIs, Back pain, Urethral discharge | Squamous cell carcinoma |
|
| [18] | 2 | 1976 | 68 | none | Lower abdominal pain, Irritative bladder symptoms | Papillary cystadenocarcinoma |
|
| [19] | 3 | 1981 | 33 | Several years of hematuria: Cystoscopy, retrograde studies. | Hematuria, Right flank pain | Clear cell carcinoma |
|
| [20] | 4 | 1990 | 4 | none | UTI, Hematuria | Squamous cell carcinoma |
|
| [21] | 5 | 1992 | 50 | none | Hematuria | Adenocarcinoma |
|
| [22] | 6 | 1996 | 36 | Radical surgery for hypospadias. | Lower abdominal pain, Fever | Squamous cell carcinoma |
|
| [23] | 7 | 2002 | 67 | none | Autopsy | Clear cell adenocarcinoma |
|
| [24] | 8 | 2005 | 14 | Left nephrectomy at 2 months for multicystic kidney. Right orchiectomy at age 8, simultaneous left side inguinal orchidopexy for inguinal testicle. | Hematuria, Increasing lower abdominal protuberance | Adenosarcoma |
|
| [25] | 9 | 2005 | 39 | none | Abdominal pain, Hematuria, Urinary retention, Bilateral cryptorchidism | Endocervical adenocarcinoma |
|
| [26] | 10 | 2006 | 44 | none | Hemospermia, Hematuria, Infertility for 15 years | Papillary cystadenocarcinoma | Not known |
| [27] | 11 | 2006 | 15 | none | Lower abdominal protrusion, Urinary retention | Clear cell adenocarcinoma |
|
| [28] | 12 | 2017 | 45 | Right inguinal hernia repair at 8 years | Painless hematuria, lower abdominal protuberance | Clear cell adenocarcinoma |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gagliardi, F.; Lauro, A.; De Anna, L.; Tripodi, D.; Esposito, A.; Forte, F.; Pironi, D.; Lori, E.; Gentile, P.A.; Marino, I.R.; et al. The Risk of Malignant Degeneration of Müllerian Derivatives in PMDS: A Review of the Literature. J. Clin. Med. 2023, 12, 3115. https://doi.org/10.3390/jcm12093115
Gagliardi F, Lauro A, De Anna L, Tripodi D, Esposito A, Forte F, Pironi D, Lori E, Gentile PA, Marino IR, et al. The Risk of Malignant Degeneration of Müllerian Derivatives in PMDS: A Review of the Literature. Journal of Clinical Medicine. 2023; 12(9):3115. https://doi.org/10.3390/jcm12093115
Chicago/Turabian StyleGagliardi, Federica, Augusto Lauro, Livia De Anna, Domenico Tripodi, Anna Esposito, Flavio Forte, Daniele Pironi, Eleonora Lori, Patrizia Alba Gentile, Ignazio R. Marino, and et al. 2023. "The Risk of Malignant Degeneration of Müllerian Derivatives in PMDS: A Review of the Literature" Journal of Clinical Medicine 12, no. 9: 3115. https://doi.org/10.3390/jcm12093115