
The Specificities of Thrombotic Thrombocytopenic Purpura at Extreme Ages: A Narrative Review
 ,
,
Abstract
:1. Introduction
2. Immune-Mediated Thrombotic Thrombocytopenic Purpura in Children
2.1. Clinical Presentation and Diagnosis
2.2. Treatment
2.3. Prognosis
3. Immune Mediated Thrombotic Thrombocytopenic Purpura in Older Patients
3.1. Clinical Presentation and Diagnosis
3.2. Treatment
3.3. Prognosis
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Furlan, M.; Robles, R.; Galbusera, M.; Remuzzi, G.; Kyrle, P.A.; Brenner, B.; Krause, M.; Scharrer, I.; Aumann, V.; Mittler, U.; et al. Von Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura and the hemolytic-uremic syndrome. N. Engl. J. Med. 1998, 339, 1578–1584. [Google Scholar] [CrossRef] [PubMed]
- Joly, B.S.; Coppo, P.; Veyradier, A. Thrombotic thrombocytopenic purpura. Blood 2017, 129, 2836–2846. [Google Scholar] [CrossRef] [PubMed]
- Quintero, O.L.; Amador-Patarroyo, M.J.; Montoya-Ortiz, G.; Rojas-Villarraga, A.; Anaya, J.M. Autoimmune disease and gender: Plausible mechanisms for the female predominance of autoimmunity. J. Autoimmun. 2012, 38, J109–J119. [Google Scholar] [CrossRef]
- Mariotte, E.; Azoulay, E.; Galicier, L.; Rondeau, E.; Zouiti, F.; Boisseau, P.; Poullin, P.; de Maistre, E.; Provôt, F.; Delmas, Y.; et al. Epidemiology and pathophysiology of adulthood-onset thrombotic microangiopathy with severe ADAMTS13 deficiency (thrombotic thrombocytopenic purpura): A cross-sectional analysis of the French national registry for thrombotic microangiopathy. Lancet Haematol. 2016, 3, e237–e245. [Google Scholar] [CrossRef] [PubMed]
- Coppo, P.; Schwarzinger, M.; Buffet, M.; Wynckel, A.; Clabault, K.; Presne, C.; Poullin, P.; Malot, S.; Vanhille, P.; Azoulay, E.; et al. Predictive features of severe acquired ADAMTS13 deficiency in idiopathic thrombotic microangiopathies: The French TMA reference center experience. PLoS ONE 2010, 5, e10208. [Google Scholar] [CrossRef]
- Bendapudi, P.K.; Hurwitz, S.; Fry, A.; Marques, M.B.; Waldo, S.W.; Li, A.; Sun, L.; Upadhyay, V.; Hamdan, A.; Brunner, A.M.; et al. Derivation and external validation of the PLASMIC score for rapid assessment of adults with thrombotic microangiopathies: A cohort study. Lancet Haematol. 2017, 4, e157–e164. [Google Scholar] [CrossRef]
- Zheng, X.L.; Vesely, S.K.; Cataland, S.R.; Coppo, P.; Geldziler, B.; Iorio, A.; Matsumoto, M.; Mustafa, R.A.; Pai, M.; Rock, G.; et al. ISTH guidelines for the diagnosis of thrombotic thrombocytopenic purpura. J. Thromb. Haemost. 2020, 18, 2486–2495. [Google Scholar] [CrossRef]
- Tsai, H.M.; Lian, E.C. Antibodies to von Willebrand factor-cleaving protease in acute thrombotic thrombocytopenic purpura. N. Engl. J. Med. 1998, 339, 1585–1594. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.P.; Kaye, J.A.; Shea, K.; Ziyadeh, N.; Cali, C.; Black, C.; Walker, A.M. Incidence of thrombotic thrombocytopenic purpura/hemolytic uremic syndrome. Epidemiology 2004, 15, 208–215. [Google Scholar] [CrossRef]
- Terrell, D.R.; Williams, L.A.; Vesely, S.K.; Lämmle, B.; Hovinga, J.A.K.; George, J.N. The incidence of thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: All patients, idiopathic patients, and patients with severe ADAMTS-13 deficiency. J. Thromb. Haemost. 2005, 3, 1432–1436. [Google Scholar] [CrossRef]
- Amorosi, E.L.; Ultmann, J.E. Thrombotic thrombocytopenic purpura: Report of 16 cases and review of the literature. Medicine 1966, 45, 139–160. [Google Scholar] [CrossRef]
- Moschcowitz, E.L.I. Hyaline thrombosis of the terminal arterioles and capillaries: A hitherto undescribed disease. Proc. N. Y. Pathol. Soc. 1924, 24, 21–24. [Google Scholar]
- Joly, B.S.; Stepanian, A.; Leblanc, T.; Hajage, D.; Chambost, H.; Harambat, J.; Fouyssac, F.; Guigonis, V.; Leverger, G.; Ulinski, T.; et al. Child-onset and adolescent-onset acquired thrombotic thrombocytopenic purpura with severe ADAMTS13 deficiency: A cohort study of the French national registry for thrombotic microangiopathy. Lancet Haematol. 2016, 3, e537–e546. [Google Scholar] [CrossRef] [PubMed]
- Saleem, R.; Rogers, Z.R.; Neunert, C.; George, J.N. Maintenance rituximab for relapsing thrombotic thrombocytopenic purpura: A case report. Transfusion 2019, 59, 921–926. [Google Scholar] [CrossRef]
- Bhoopalan, S.V.; Hankins, J.; George, J.; Ryder, A.; Onder, A.M.; Puri, L. Use of caplacizumab in a child with refractory thrombotic thrombocytopenic purpura. Pediatr. Blood Cancer 2019, 66, e27737. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Kuwana, M.; Tanaka, H.; Hosomichi, K.; Hasegawa, A.; Uyama, H.; Nishio, K.; Omae, T.; Hishizawa, M.; Matsui, M.; et al. HLA loci predisposing to immune TTP in Japanese: Potential role of the shared ADAMTS13 peptide bound to different HLA-DR. Blood 2020, 135, 2413–2419. [Google Scholar] [CrossRef]
- Nagel, M.B.; Ryder, A.; Lobbins, M.; Bhatt, N. Refractory acquired thrombotic thrombocytopenic purpura treated with caplacizumab in a pediatric patient with systemic lupus erythematosus. Pediatr. Blood Cancer 2021, 68, e28534. [Google Scholar] [CrossRef]
- Tripiciano, C.; Zangari, P.; Montanari, M.; Leone, G.; Massella, L.; Garaboldi, L.; Massoud, M.; Lancellotti, S.; Strocchio, L.; Manno, E.C.; et al. Case Report: Two Cases of Pediatric Thrombotic Thrombocytopenic Purpura Treated with Combined Therapy. Front. Pediatr. 2021, 9, 743206. [Google Scholar] [CrossRef]
- Azapağasi, E.; Yazici, M.U.; Eroğlu, N.; Albayrak, M.; Kucur, Ö.; Fettah, A. Successful Treatment with Bortezomib for Refractory and Complicated Acquired Thrombotic Thrombocytopenic Purpura in an Adolescent Girl. J. Pediatr. Hematol. Oncol. 2021, 43, e587–e591. [Google Scholar] [CrossRef]
- Dutt, T.; Shaw, R.J.; Stubbs, M.; Yong, J.; Bailiff, B.; Cranfield, T.; Crowley, M.P.; Desborough, M.; Eyre, T.A.; Gooding, R.; et al. Real-world experience with caplacizumab in the management of acute TTP. Blood 2021, 137, 1731–1740. [Google Scholar] [CrossRef]
- Siddiqui, A.; Journeycake, J.M.; Borogovac, A.; George, J.N. Recognizing and managing hereditary and acquired thrombotic thrombocytopenic purpura in infants and children. Pediatr. Blood Cancer 2021, 68, e28949. [Google Scholar] [CrossRef] [PubMed]
- Boudali, J.; Hallak, B.; Haeck, M.; Sellier-Leclerc, A.L.; Ulrich, M.; Coppo, P.; Tellier, S.; Provôt, F. Immune-mediated thrombotic thrombocytopenic purpura in childhood treated by caplacizumab, about 3 cases. J. Nephrol. 2022, 35, 653–656. [Google Scholar] [CrossRef] [PubMed]
- Kirpalani, A.; Garabon, J.; Amos, K.; Patel, S.; Sharma, A.P.; Ganesan, S.L.; Barton, M.; Cacciotti, C.; Leppington, S.; Bakovic, L.; et al. Thrombotic thrombocytopenic purpura temporally associated with BNT162b2 vaccination in an adolescent successfully treated with caplacizumab. Br. J. Haematol. 2022, 196, e11–e14. [Google Scholar] [CrossRef]
- Veltroni, M.; Pegoraro, F.; Scappini, B.; Brugnolo, F.; Allegro, E.; Ermini, S.; Tondo, A.; Fotzi, I.; Bambi, F.; Favre, C. Off-label caplacizumab as add-on therapy in a 9-year-old boy with refractory aTTP. Ann. Hematol. 2022, 101, 1369–1371. [Google Scholar] [CrossRef]
- Reese, J.A.; Muthurajah, D.S.; Hovinga, J.A.K.; Vesely, S.K.; Terrell, D.R.; George, J.N. Children and adults with thrombotic thrombocytopenic purpura associated with severe, acquired Adamts13 deficiency: Comparison of incidence, demographic and clinical features. Pediatr. Blood Cancer 2013, 60, 1676–1682. [Google Scholar] [CrossRef] [PubMed]
- Joly, B.S.; Vanhoorelbeke, K.; Veyradier, A. Understanding therapeutic targets in thrombotic thrombocytopenic purpura. Intensive Care Med. 2017, 43, 1398–1400. [Google Scholar] [CrossRef]
- Zheng, X.L.; Vesely, S.K.; Cataland, S.R.; Coppo, P.; Geldziler, B.; Iorio, A.; Matsumoto, M.; Mustafa, R.A.; Pai, M.; Rock, G.; et al. ISTH guidelines for treatment of thrombotic thrombocytopenic purpura. J. Thromb. Haemost. 2020, 18, 2496–2502. [Google Scholar] [CrossRef]
- Scully, M.; Cataland, S.R.; Peyvandi, F.; Coppo, P.; Knöbl, P.; Hovinga, J.A.K.; Metjian, A.; de la Rubia, G.; Pavenski, K.; Callewaert, F.; et al. Caplacizumab Treatment for Acquired Thrombotic Thrombocytopenic Purpura. N. Engl. J. Med. 2019, 380, 335–346. [Google Scholar] [CrossRef]
- Bergstrand, M.; Hansson, E.; Delaey, B.; Callewaert, F.; De Passos Sousa, R.; Sargentini-Maier, M.L. Caplacizumab Model-Based Dosing Recommendations in Pediatric Patients with Acquired Thrombotic Thrombocytopenic Purpura. J. Clin. Pharmacol. 2022, 62, 409–421. [Google Scholar] [CrossRef]
- Amin Asnafi, A.; Jalali, M.T.; Pezeshki, S.M.S.; Jaseb, K.; Saki, N. The Association Between Human Leukocyte Antigens and ITP, TTP, and HIT. J. Pediatr. Hematol. Oncol. 2019, 41, 81–86. [Google Scholar] [CrossRef]
- Joly, B.S.; Loiseau, P.; Darmon, M.; Leblanc, T.; Chambost, H.; Fouyssac, F.; Guigonis, V.; Harambat, G.; Stepanian, A.; Coppo, P.; et al. HLA-DRB1*11 is a strong risk factor for acquired thrombotic thrombocytopenic purpura in children. Haematologica 2020, 105, e531. [Google Scholar] [CrossRef]
- Hassan, A.; Iqbal, M.; George, J.N. Additional autoimmune disorders in patients with acquired autoimmune thrombotic thrombocytopenic purpura. Am. J. Hematol. 2019, 94, E172–E174. [Google Scholar] [CrossRef] [PubMed]
- Prevel, R.; Roubaud-Baudron, C.; Gourlain, S.; Jamme, M.; Peres, K.; Benhamou, Y.; Galicier, L.; Azoulay, E.; Poullin, P.; Provôt, F.; et al. Immune thrombotic thrombocytopenic purpura in older patients: Prognosis and long-term survival. Blood 2019, 134, 2209–2217. [Google Scholar] [CrossRef]
- Agosti, P.; Mancini, I.; Gianniello, F.; Bucciarelli, P.; Artoni, A.; Ferrari, B.; Pontiggia, S.; Trisolini, S.M.; Facchini, L.; Carbone, C.; et al. Prevalence of the age-related diseases in older patients with acquired thrombotic thrombocytopenic purpura. Eur. J. Intern. Med. 2020, 75, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.; Zafrani, L.; Lemiale, V.; Stepanian, A.; Joly, B.; Azoulay, E.; Mariotte, E. The clinical picture of thrombotic microangiopathy in patients older than 60 years of age. Br. J. Haematol. 2021, 192, e25–e28. [Google Scholar] [CrossRef]
- Liu, A.; Dhaliwal, N.; Upreti, H.; Kasmani, J.; Dane, K.; Moliterno, A.; Braunstein, E.; Brodsky, R.; Chaturvedi, S. Reduced sensitivity of PLASMIC and French scores for the diagnosis of thrombotic thrombocytopenic purpura in older individuals. Transfusion 2021, 61, 266–273. [Google Scholar] [CrossRef]
- Joseph, A.; Eloit, M.; Azoulay, E.; Kaplanski, G.; Provot, F.; Presne, C.; Wynckel, A.; Grangé, S.; Rondeau; Pène, F.; et al. Immune-mediated thrombotic thrombocytopenic purpura prognosis is affected by blood pressure. Res. Pract. Thromb. Haemost. 2022, 6, e12702. [Google Scholar] [CrossRef]
- Fage, N.; Orvain, C.; Henry, N.; Mellaza, C.; Beloncle, F.; Tuffigo, M.; Geneviève, F.; Coppo, P.; Augusto, J.F.; Brilland, B.; et al. Proteinuria Increases the PLASMIC and French Scores Performance to Predict Thrombotic Thrombocytopenic Purpura in Patients with Thrombotic Microangiopathy Syndrome. Kidney Int. Rep. 2022, 7, 221–231. [Google Scholar] [CrossRef]
- Burguet, L.; Taton, B.; Prezelin-Reydit, M.; Rubin, S.; Picard, W.; Gruson, D.; Ryman, A.; Contin-Bordes, C.; Coppo, P.; Combe, C.; et al. Urine Protein/Creatinine Ratio in Thrombotic Microangiopathies: A Simple Test to Facilitate Thrombotic Thrombocytopenic Purpura and Hemolytic and Uremic Syndrome Diagnosis. J. Clin. Med. 2022, 11, 648. [Google Scholar] [CrossRef]
- Joseph, A.; Brilland, B.; Burguet, L.; Eloit, M.; Fage, N.; Augusto, J.F.; Delmas, Y.; Veyradier, A.; Halimi, J.-M.; Coppo, P. Predictive Scores for Early Identification of Immune-Mediated Thrombotic Thrombocytopenic Purpura: Room for Improvement? Kidney Int. Rep. 2022, 7, 2541–2542. [Google Scholar] [CrossRef]
- Coppo, P.; Bubenheim, M.; Azoulay, E.; Galicier, L.; Malot, S.; Bigé, N.; Poullin, P.; Provôt, F.; Martis, N.; Presne, C.; et al. A regimen with caplacizumab, immunosuppression, and plasma exchange prevents unfavorable outcomes in immune-mediated TTP. Blood 2021, 137, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Agosti, P.; Mancini, I.; Artoni, A.; Ferrari, B.; Pontiggia, S.; Trisolini, S.M.; Facchini, L.; Peyvandi, F.; Italian Group of TTP Investigators. The features of acquired thrombotic thrombocytopenic purpura occurring at advanced age. Thromb. Res. 2020, 187, 197–201. [Google Scholar] [CrossRef]
- De Louw, A.V.; Mariotte, E.; Darmon, M.; Cohrs, A.; Leslie, D.; Azoulay, E. Outcomes in 1096 patients with severe thrombotic thrombocytopenic purpura before the Caplacizumab era. PLoS ONE 2021, 16, e0256024. [Google Scholar] [CrossRef]
- Matsumoto, M.; Bennett, C.L.; Isonishi, A.; Qureshi, Z.; Hori, Y.; Hayakawa, M.; Yoshida, Y.; Yagi, H.; Fujimura, Y. Acquired idiopathic ADAMTS13 activity deficient thrombotic thrombocytopenic purpura in a population from Japan. PLoS ONE 2012, 7, e33029. [Google Scholar] [CrossRef] [PubMed]
- Mariotte, E.; Blet, A.; Galicier, L.; Darmon, M.; Parquet, N.; Lengline, E.; Boutboul, D.; Canet, E.; Traineau, R.; Schlemmer, B.; et al. Unresponsive thrombotic thrombocytopenic purpura in critically ill adults. Intensive Care Med. 2013, 39, 1272–1281. [Google Scholar] [CrossRef]
- Gui, R.; Huang, Q.; Cai, X.; Wu, J.; Liu, H.; Liu, Y.; Yang, L.; Zhang, J.; Cheng, Y.; Jiang, M.; et al. Development and validation of a prediction model (AHC) for early identification of refractory thrombotic thrombocytopenic purpura using nationally representative data. Br. J. Haematol. 2020, 191, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Adeyemi, A.; Razakariasa, F.; Chiorean, A.; de Passos Sousa, R. Epidemiology, treatment patterns, clinical outcomes, and disease burden among patients with immune-mediated thrombotic thrombocytopenic purpura in the United States. Res. Pract. Thromb. Haemost. 2022, 6, e12802. [Google Scholar] [CrossRef] [PubMed]
- Schofield, J.; Shaw, R.J.; Lester, W.; Thomas, W.; Toh, C.H.; Dutt, T. Intracranial hemorrhage in immune thrombotic thrombocytopenic purpura treated with caplacizumab. J. Thromb. Haemost. 2021, 19, 1922–1925. [Google Scholar] [CrossRef] [PubMed]
- Deford, C.C.; Reese, J.A.; Schwartz, L.H.; Perdue, J.J.; Hovinga, J.A.K.; Lämmle, B.; Terrell, D.; Vesely, S.; George, J.N. Multiple major morbidities and increased mortality during long-term follow-up after recovery from thrombotic thrombocytopenic purpura. Blood 2013, 122, 2023–2029. [Google Scholar] [CrossRef] [PubMed]
- Sukumar, S.; Brodsky, M.A.; Hussain, S.; Yanek, L.R.; Moliterno, A.R.; Brodsky, R.A.; Cataland, S.R.; Chaturvedi, S. Cardiovascular disease is a leading cause of mortality among TTP survivors in clinical remission. Blood Adv. 2022, 6, 1264–1270. [Google Scholar] [CrossRef]
- Upreti, H.; Kasmani, J.; Dane, K.; Braunstein, E.M.; Streiff, M.B.; Shanbhag, S.; Moliterno, A.R.; Sperati, C.J.; Gottesman, R.F.; Brodsky, R.A.; et al. Reduced ADAMTS13 activity during TTP remission is associated with stroke in TTP survivors. Blood 2019, 134, 1037–1045. [Google Scholar] [CrossRef]
- Chaturvedi, S.; Abbas, H.; McCrae, K.R. Increased morbidity during long-term follow-up of survivors of thrombotic thrombocytopenic purpura. Am. J. Hematol. 2015, 90, E208. [Google Scholar] [CrossRef]
- Brodsky, M.A.; Sukumar, S.; Selvakumar, S.; Yanek, L.; Hussain, S.; Mazepa, M.A.; Braunstein, E.M.; Moliterno, A.R.; Kickler, T.S.; Brodsky, R.A.; et al. Major adverse cardiovascular events in survivors of immune-mediated thrombotic thrombocytopenic purpura. Am. J. Hematol. 2021, 96, 1587–1594. [Google Scholar] [CrossRef]
- Chaturvedi, S.; Oluwole, O.; Cataland, S.; McCrae, K.R. Post-traumatic stress disorder and depression in survivors of thrombotic thrombocytopenic purpura. Thromb. Res. 2017, 151, 51–56. [Google Scholar]
- Falter, T.; Schmitt, V.; Herold, S.; Weyer, V.; von Auer, C.; Wagner, S.; Hefner, G.; Beutel, M.; Lackner, K.; Lämmle, B.; et al. Depression and cognitive deficits as long-term consequences of thrombotic thrombocytopenic purpura. Transfusion 2017, 57, 1152–1162. [Google Scholar] [CrossRef]
- Han, B.; Page, E.E.; Stewart, L.M.; Deford, C.C.; Scott, J.G.; Schwartz, L.H.; Perdue, J.J.; Terrell, D.R.; Vesely, S.K.; George, J.N. Depression and cognitive impairment following recovery from thrombotic thrombocytopenic purpura. Am. J. Hematol. 2015, 90, 709–714. [Google Scholar] [CrossRef]
- Goldberg, T.E.; Chen, C.; Wang, Y.; Jung, E.; Swanson, A.; Ing, C.; Garcia, P.S.; Whittington, R.A.; Moitra, V. Association of Delirium with Long-term Cognitive Decline: A Meta-analysis. JAMA Neurol. 2020, 77, 1373–1381. [Google Scholar] [CrossRef] [PubMed]
- Wolters, F.J.; Boender, J.; de Vries, P.S.; Sonneveld, M.A.; Koudstaal, P.J.; de Maat, M.P.; Franco, O.H.; Ikram, M.K.; Leebeek, F.W. Von Willebrand factor and ADAMTS13 activity in relation to risk of dementia: A population-based study. Sci. Rep. 2018, 8, 5474. [Google Scholar] [CrossRef] [PubMed]
- Stabler, S.; Giovannelli, J.; Launay, D.; Cotteau-Leroy, A.; Heusele, M.; Lefèvre, G.; Terriou, L.; Lambert, M.; Dubucquoi, S.; Hachulla, E.; et al. Serious Infectious Events and Immunoglobulin Replacement Therapy in Patients with Autoimmune Disease Receiving Rituximab: A Retrospective Cohort Study. Clin. Infect. Dis. 2021, 72, 727–737. [Google Scholar] [PubMed]
- Roriz, M.; Landais, M.; Desprez, J.; Barbet, C.; Azoulay, E.; Galicier, L.; Wynckel, A.; Baudel, J.-L.; Provôt, F.; Pène, F.; et al. Risk Factors for Autoimmune Diseases Development After Thrombotic Thrombocytopenic Purpura. Medicine 2015, 94, e1598. [Google Scholar] [CrossRef]
- Pereira, L.C.V.; Ercig, B.; Kangro, K.; Jamme, M.; Malot, S.; Galicier, L.; Poullin, P.; Provôt, F.; Presne, C.; Kanouni, T.; et al. Understanding the Health Literacy in Patients with Thrombotic Thrombocytopenic Purpura. Hemasphere 2020, 4, e462. [Google Scholar] [CrossRef] [PubMed]




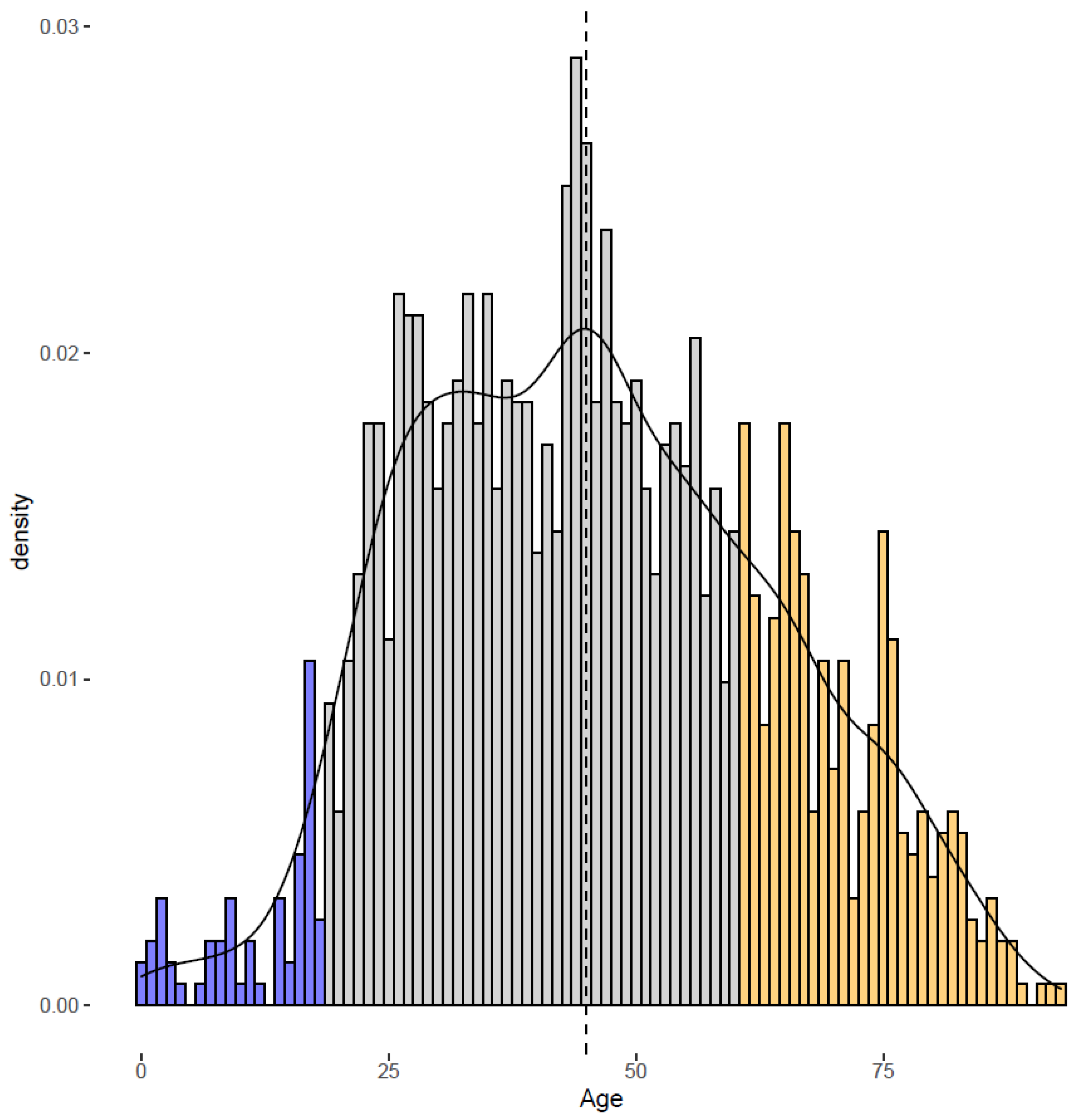{kind=link}
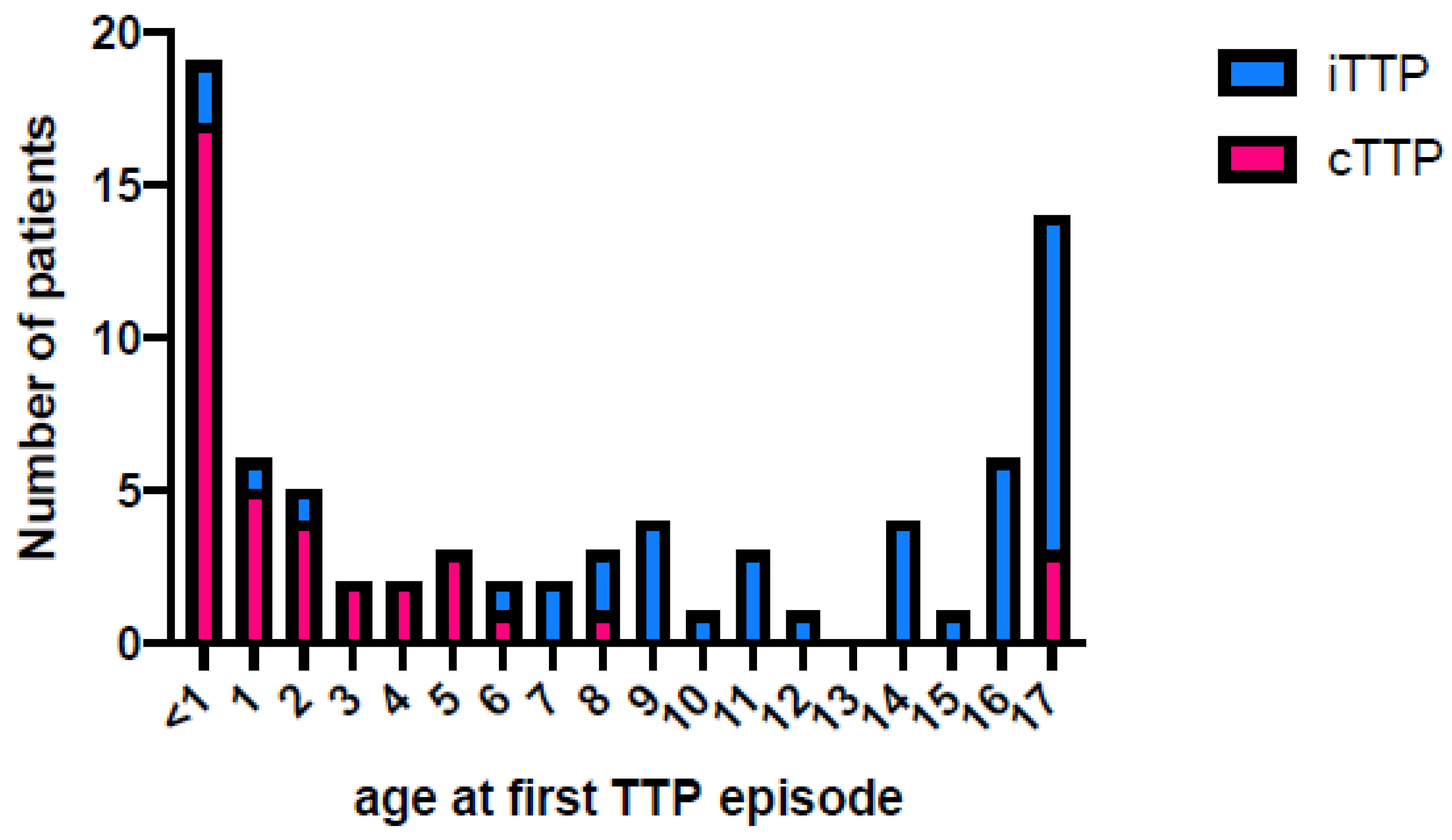{kind=link}
{kind=link}
| Recommendations | |
|---|---|
| Diagnosis | Physician awareness of atypical neurological presentations |
| Adaptation of clinical scores for older patients specificities | |
| Treatment | Rapid identification and intensification in unresponsive patients Medication optimization to avoid drug interactions Reassessment of prognostic factors in the caplacizumab era |
| Long-term follow-up | Comprehensive geriatric assessment and management of associated (cardiovascular) risk factors Careful ADAMTS13 monitoring ± preemptive rituximab |
| Characterization of cognitive impairment in older patients following iTTP episode |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joseph, A.; Joly, B.S.; Picod, A.; Veyradier, A.; Coppo, P. The Specificities of Thrombotic Thrombocytopenic Purpura at Extreme Ages: A Narrative Review. J. Clin. Med. 2023, 12, 3068. https://doi.org/10.3390/jcm12093068
Joseph A, Joly BS, Picod A, Veyradier A, Coppo P. The Specificities of Thrombotic Thrombocytopenic Purpura at Extreme Ages: A Narrative Review. Journal of Clinical Medicine. 2023; 12(9):3068. https://doi.org/10.3390/jcm12093068
Chicago/Turabian StyleJoseph, Adrien, Bérangère S. Joly, Adrien Picod, Agnès Veyradier, and Paul Coppo. 2023. "The Specificities of Thrombotic Thrombocytopenic Purpura at Extreme Ages: A Narrative Review" Journal of Clinical Medicine 12, no. 9: 3068. https://doi.org/10.3390/jcm12093068