
Myelofibrosis and Survival Prognostic Models: A Journey between Past and Future

 , , , , ,
, , , , ,
Abstract
:1. Introduction
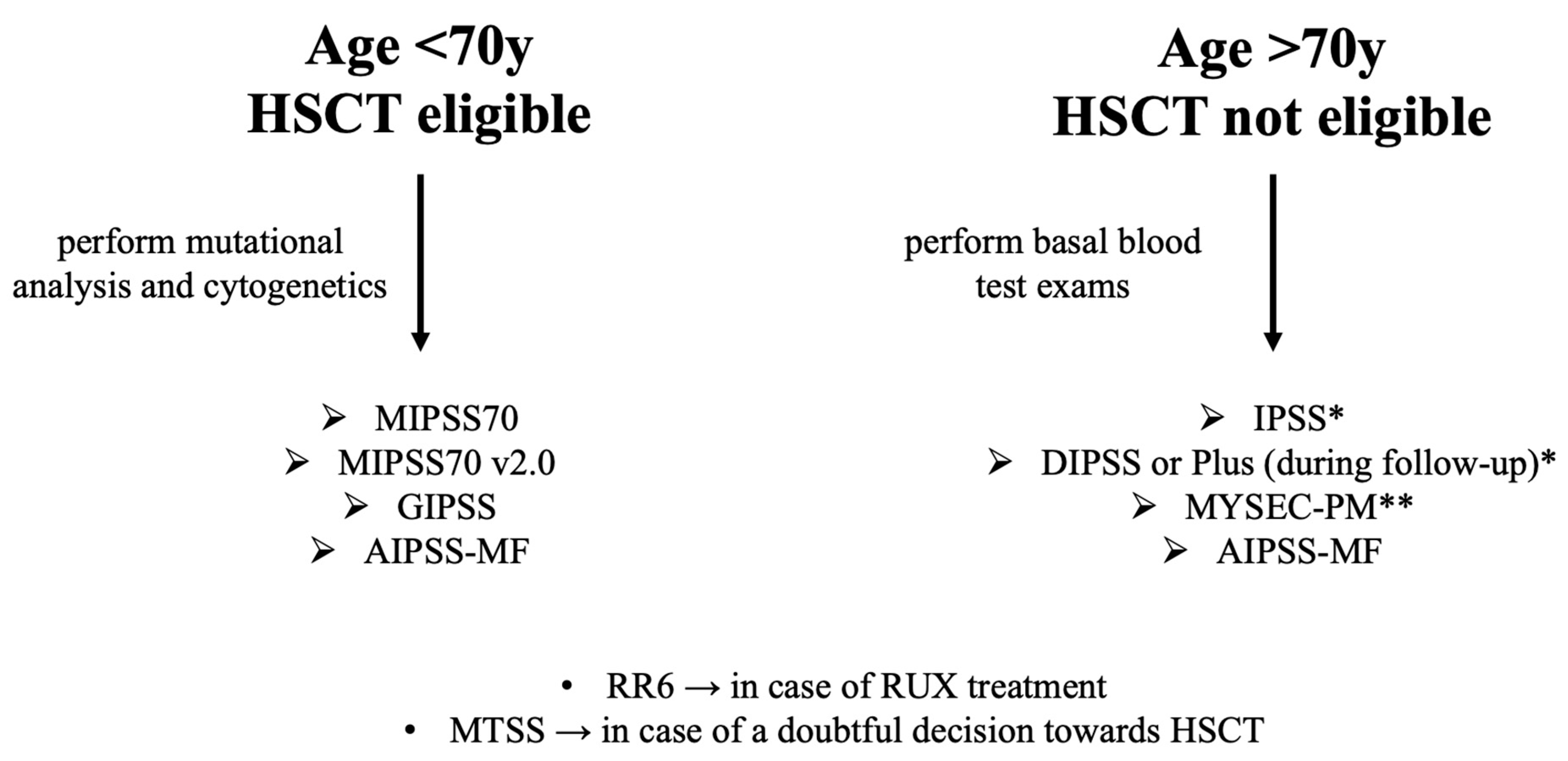
2. Prognostic Model Journey
2.1. IPSS (International Prognostic Scoring System)
2.2. DIPSS (Dynamic IPSS) and DIPSS Plus
2.3. MIPSS70 (Mutation-Enhanced International Prognostic Score System) and MIPSS70+
2.4. GIPSS (Genetically Inspired Prognostic Scoring System)
2.5. MYSEC-PM (Myelofibrosis Secondary to PV and ET-Prognostic Model)
2.6. MTSS (Myelofibrosis Transplant Scoring System)
2.7. RR6 (Response to Ruxolitinib after 6 Months)
2.8. AIPSS-MF (Artificial Intelligence Prognostic Scoring System for Myelofibrosis)
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hussein, K.; van Dyke, D.L.; Tefferi, A. Conventional cytogenetics in myelofibrosis: Literature review and discussion. Eur. J. Haematol. 2009, 82, 329–338. [Google Scholar] [CrossRef]
- Iurlo, A.; Cattaneo, D.; Gianelli, U. Molecular sciences blast transformation in myeloproliferative neoplasms: Risk factors, biological findings, and targeted therapeutic options. Int. J. Mol. Sci. 2019, 20, 1839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palumbo, G.A.; Stella, S.; Pennisi, M.S.; Pirosa, C.; Fermo, E.; Fabris, S.; Cattaneo, D.; Iurlo, A. The role of new technologies in myeloproliferative neoplasms. Front. Oncol. 2019, 9, 321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bose, P.; Verstovsek, S. JAK inhibition for the treatment of myelofibrosis: Limitations and future perspectives. Hemasphere 2020, 4, e424. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Jimma, T.; Finke, C.M.; Gangat, N.; Vaidya, R.; Begna, K.H.; Al-Kali, A.; Ketterling, R.P.; Hanson, C.A.; et al. One thousand patients with primary myelofibrosis: The mayo clinic experience. Mayo. Clin. Proc. 2012, 87, 25–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, M.A.; Verstovsek, S.; Dingli, D.; Schwager, S.M.; Mesa, R.A.; Li, C.Y.; Tefferi, A. Monocytosis Is an adverse prognostic factor for survival in younger patients with primary myelofibrosis. Leuk. Res. 2007, 31, 1503–1509. [Google Scholar] [CrossRef] [PubMed]
- Cervantes, F.; Pereira, A.; Esteve, J.; Rafel, M.; Cobo, F.; Rozman, C.; Montserrat, E. Identification of ‘short-lived’ and ‘long-lived’ patients at presentation of idiopathic myelofibrosis. Br. J. Haematol. 1997, 97, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Dupriez, B.; Morel, P.; Demon, J.L.; Lai, J.L.; Simon, M.; Plantier, I.; Bauters, F. Prognostic factors in agnogenic myeloid metaplasia: A report on 195 cases with a new scoring system. Blood 1996, 88, 1013–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dingli, D.; Schwager, S.M.; Mesa, R.A.; Li, C.Y.; Tefferi, A. Prognosis in transplant-eligible patients with agnogenic myeloid metaplasia. Cancer 2006, 106, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Mosquera-Orgueira, A.; Pérez-Encinas, M.; Hernández-Sánchez, A.; González-Martínez, T.; Arellano-Rodrigo, E.; Martínez-Elicegui, J.; Villaverde-Ramiro, Á.; Raya, J.M.; Ayala, R.; Ferrer-Marín, F.; et al. Machine learning improves risk stratification in myelofibrosis: An analysis of the spanish registry of myelofibrosis. Hemasphere 2023, 7, E818. [Google Scholar] [CrossRef]
- Cervantes, F.; Dupriez, B.; Pereira, A.; Passamonti, F.; Reilly, J.T.; Morra, E.; Vannucchi, A.M.; Mesa, R.A.; Demory, J.L.; Barosi, G.; et al. New prognostic scoring system for primary myelofibrosis based on a study of the international working group for myelofibrosis research and treatment. Blood 2009, 113, 2895–2901. [Google Scholar] [CrossRef]
- Passamonti, F.; Cervantes, F.; Vannucchi, A.M.; Morra, E.; Rumi, E.; Pereira, A.; Guglielmelli, P.; Pungolino, E.; Caramella, M.; Maffioli, M.; et al. A dynamic prognostic model to predict survival in primary myelofibrosis: A study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood 2010, 115, 1703–1708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangat, N.; Caramazza, D.; Vaidya, R.; George, G.; Begna, K.; Schwager, S.; van Dyke, D.; Hanson, C.; Wu, W.; Pardanani, A.; et al. DIPSS plus: A refined dynamic international prognostic scoring system for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J. Clin. Oncol. 2011, 29, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Luque Paz, D.; Riou, J.; Verger, E.; Cassinat, B.; Chauveau, A.; Ianotto, J.C.; Dupriez, B.; Boyer, F.; Renard, M.; Ugo, V.; et al. Genomic analysis of primary and secondary myelofibrosis redefines the prognostic impact of ASXL1 mutations: A fim study. Blood Adv. 2021, 5, 1442–1451. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Mudireddy, M.; Mannarelli, C.; Nicolosi, M.; Pacilli, A.; Pardanani, A.; Rumi, E.; Rosti, V.; et al. MIPSS70: Mutation-enhanced international prognostic score system for transplantation-age patients with primary myelofibrosis. J. Clin. Oncol. 2018, 36, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Caramazza, D.; Begna, K.H.; Gangat, N.; Vaidya, R.; Siragusa, S.; van Dyke, D.L.; Hanson, C.; Pardanani, A.; Tefferi, A. Refined cytogenetic-risk categorization for overall and leukemia-free survival in primary myelofibrosis: A single center study of 433 patients. Leukemia 2011, 25, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Guglielmelli, P.; Nicolosi, M.; Mannelli, F.; Mudireddy, M.; Bartalucci, N.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; et al. GIPSS: Genetically inspired prognostic scoring system for primary myelofibrosis. Leukemia 2018, 32, 1631–1642. [Google Scholar] [CrossRef] [Green Version]
- Gowin, K.; Coakley, M.; Kosiorek, H.; Mesa, R. Discrepancies of applying primary myelofibrosis prognostic scores for patients with post polycythemia vera/essential thrombocytosis myelofibrosis. Haematologica 2016, 101, e405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passamonti, F.; Giorgino, T.; Mora, B.; Guglielmelli, P.; Rumi, E.; Maffioli, M.; Rambaldi, A.; Caramella, M.; Komrokji, R.; Gotlib, J.; et al. A clinical-molecular prognostic model to predict survival in patients with post polycythemia vera and post essential thrombocythemia myelofibrosis. Leukemia 2017 31:12 2017, 31, 2726–2731. [Google Scholar] [CrossRef]
- Palandri, F.; Palumbo, G.A.; Iurlo, A.; Polverelli, N.; Benevolo, G.; Breccia, M.; Abruzzese, E.; Tiribelli, M.; Bonifacio, M.; Tieghi, A.; et al. Differences in presenting features, outcome and prognostic models in patients with primary myelofibrosis and post-polycythemia vera and/or post-essential thrombocythemia myelofibrosis treated with ruxolitinib. New perspective of the mysec-pm in a large multicenter study⁎. Semin. Hematol. 2018, 55, 248–255. [Google Scholar] [CrossRef]
- Breccia, M.; Baratè, C.; Benevolo, G.; Bonifacio, M.; Elli, E.M.; Guglielmelli, P.; Maffioli, M.; Malato, A.; Mendicino, F.; Palumbo, G.A.; et al. Tracing the decision-making process for myelofibrosis: Diagnosis, stratification, and management of ruxolitinib therapy in real-word practice. Ann. Hematol. 2020, 99, 65–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breccia, M.; Palandri, F.; Guglielmelli, P.; Palumbo, G.A.; Malato, A.; Mendicino, F.; Ricco, A.; Sant’Antonio, E.; Tiribelli, M.; Iurlo, A. Management of myelofibrosis during treatment with ruxolitinib: A real-world perspective in case of resistance and/or intolerance. Curr. Oncol. 2022, 29, 4970–4980. [Google Scholar] [CrossRef]
- Young, J.A.H.; Logan, B.R.; Wu, J.; Wingard, J.R.; Weisdorf, D.J.; Mudrick, C.; Knust, K.; Horowitz, M.M.; Confer, D.L.; Dubberke, E.R.; et al. Infections following transplantation of bone marrow or peripheral-blood stem cells from unrelated donors. Biol. Blood Marrow. Transpl. 2016, 22, 359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, V.; Kolli, S.S.; Strowd, L.C. Review of graft-versus-host disease. Derm. Clin. 2019, 37, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Milone, G.; Bellofiore, C.; Leotta, S.; Milone, G.A.; Cupri, A.; Duminuco, A.; Garibaldi, B.; Palumbo, G. Endothelial dysfunction after hematopoietic stem cell transplantation: A review based on physiopathology. J. Clin. Med. 2022, 11, 623. [Google Scholar] [CrossRef]
- Gagelmann, N.; Ditschkowski, M.; Bogdanov, R.; Bredin, S.; Robin, M.; Cassinat, B.; Shahswar, R.; Thol, F.; Heuser, M.; Socié, G.; et al. Comprehensive clinical-molecular transplant scoring system for myelofibrosis undergoing stem cell transplantation. Blood 2019, 133, 2233–2242. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T.; et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. New Engl. J. Med. 2012, 366, 799–807. [Google Scholar] [CrossRef] [Green Version]
- Harrison, C.N.; Vannucchi, A.M.; Kiladjian, J.J.; Al-Ali, H.K.; Gisslinger, H.; Knoops, L.; Cervantes, F.; Jones, M.M.; Sun, K.; McQuitty, M.; et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs. best available therapy for myelofibrosis. Leukemia 2016, 30, 1701. [Google Scholar] [CrossRef] [Green Version]
- Coltro, G.; Sant’Antonio, E.; Palumbo, G.A.; Mannelli, F.; De Stefano, V.; Ruggeri, M.; Elli, E.M.; Zanotti, R.; Borsani, O.; Bertozzi, I.; et al. Assessment of the efficacy and tolerability of ruxolitinib for the treatment of myelofibrosis patients in a real-life setting: An Italian MYNERVA project. Cancer Med. 2023, 00, 1. [Google Scholar] [CrossRef]
- Palandri, F.; Palumbo, G.A.; Abruzzese, E.; Iurlo, A.; Polverelli, N.; Elli, E.; Bonifacio, M.; Bergamaschi, M.; Martino, B.; Tiribelli, M.; et al. Impact of 2016 WHO diagnosis of early and overt primary myelofibrosis on presentation and outcome of 232 patients treated with ruxolitinib. Hematol. Oncol. 2019, 37, 418–423. [Google Scholar] [CrossRef]
- Palandri, F.; Palumbo, G.A.; Bonifacio, M.; Breccia, M.; Latagliata, R.; Martino, B.; Polverelli, N.; Abruzzese, E.; Tiribelli, M.; Nicolosi, M.; et al. Durability of spleen response affects the outcome of ruxolitinib-treated patients with myelofibrosis: Results from a multicentre study on 284 patients. Leuk Res. 2018, 74, 86–88. [Google Scholar] [CrossRef]
- Palandri, F.; Catani, L.; Bonifacio, M.; Benevolo, G.; Heidel, F.; Palumbo, G.A.; Crugnola, M.; Abruzzese, E.; Bartoletti, D.; Polverelli, N.; et al. Ruxolitinib in elderly patients with myelofibrosis: Impact of age and genotype. A multicentre study on 291 elderly patients. Br. J. Haematol. 2018, 183, 35–46. [Google Scholar] [CrossRef]
- Palandri, F.; Breccia, M.; Bonifacio, M.; Polverelli, N.; Elli, E.M.; Benevolo, G.; Tiribelli, M.; Abruzzese, E.; Iurlo, A.; Heidel, F.H.; et al. Life after ruxolitinib: Reasons for discontinuation, impact of disease phase, and outcomes in 218 patients with myelofibrosis. Cancer 2020, 126, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Pardanani, A. Serious adverse events during ruxolitinib treatment discontinuation in patients with myelofibrosis. Mayo. Clin. Proc. 2011, 86, 1188. [Google Scholar] [CrossRef]
- Lussana, F.; Cattaneo, M.; Rambaldi, A.; Squizzato, A. Ruxolitinib-associated infections: A systematic review and meta-analysis. Am. J. Hematol. 2018, 93, 339–347. [Google Scholar] [CrossRef] [Green Version]
- Duminuco, A.; Scarso, S.; Cupri, A.; Parrinello, N.L.; Villari, L.; Scuderi, G.; Giunta, G.; Leotta, S.; Milone, G.A.; Giuffrida, G.; et al. Leishmania infection during ruxolitinib treatment: The cytokines-based immune response in the setting of immunocompromised patients. J. Clin. Med. 2023, 12, 578. [Google Scholar] [CrossRef]
- Maffioli, M.; Mora, B.; Ball, S.; Iurlo, A.; Elli, E.M.; Finazzi, M.C.; Polverelli, N.; Rumi, E.; Caramella, M.; Carraro, M.C.; et al. A prognostic model to predict survival after 6 months of ruxolitinib in patients with myelofibrosis. Blood Adv. 2022, 6, 1855–1864. [Google Scholar] [CrossRef] [PubMed]
- Duminuco, A.; Nardo, A.; Garibaldi, B.; Vetro, C.; Longo, A.; Giallongo, C.; di Raimondo, F.; Palumbo, G.A. Prediction of survival and prognosis migration from gold-standard scores in myelofibrosis patients treated with ruxolitinib applying the rr6 prognostic model in a monocentric real-life setting. J. Clin. Med. 2022, 11, 7418. [Google Scholar] [CrossRef]
- Scalzulli, E.; Ielo, C.; Luise, C.; Musiu, P.; Bisegna, M.L.; Carmosino, I.; Assanto, G.M.; Martelli, M.; Breccia, M. RR6 prognostic model provides information about survival for myelofibrosis treated with ruxolitinib: Validation in a real-life cohort. Blood Adv. 2022, 6, 4424–4426. [Google Scholar] [CrossRef] [PubMed]
- Vannucchi, A.M.; Guglielmelli, P. Molecular prognostication in ph-negative mpns in 2022. Hematology 2022, 2022, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Rozovski, U.; Verstovsek, S.; Manshouri, T.; Dembitz, V.; Bozinovic, K.; Newberry, K.; Zhang, Y.; Bove, J.E.; Pierce, S.; Kantarjian, H.; et al. An accurate, simple prognostic model consisting of age, JAK2, CALR, and mpl mutation status for patients with primary myelofibrosis. Haematologica 2017, 102, 79–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallapureddy, R.R.; Mudireddy, M.; Penna, D.; Lasho, T.L.; Finke, C.M.; Hanson, C.A.; Ketterling, R.P.; Begna, K.H.; Gangat, N.; Pardanani, A.; et al. Leukemic transformation among 1306 patients with primary myelofibrosis: Risk factors and development of a predictive model. Blood Cancer J. 2019, 9, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skov, V. Next Generation Sequencing in MPNs. Lessons from the past and prospects for use as predictors of prognosis and treatment responses. Cancers 2020, 12, 2194. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.; Bose, P.; Dunbar, A.; Elshoury, A.; George, T.I.; Gundabolu, K.; Buffett Cancer Center, P.; Hexner, E.; Hobbs, G.S.; Jamieson, C.; et al. NCCN Guidelines Version 3.2022 Myeloproliferative Neoplasms. J. Natl. Compr. Cancer Netw. 2022, 20, 1033–1062. [Google Scholar]
- Mora, B.; Passamonti, F. Towards a personalized definition of prognosis in philadelphia-negative myeloproliferative neoplasms. Curr. Hematol. Malig. Rep. 2022, 17, 127–139. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Prognostic Model | Variable | Points | Risk Category | Score | Median Survival (Years) |
|---|---|---|---|---|---|
| IPSS | Constitutional symptoms | 1 | LOW | 0 | 11.3 |
| Hemoglobin < 10 g/dL | 1 | INTERMEDIATE-1 | 1 | 7.9 | |
| WBC > 25 × 109/L | 1 | INTERMEDIATE-2 | 2 | 4 | |
| Blast ≥ 1% | 1 | HIGH | ≥3 | 2.3 | |
| Age > 65 | 1 | / | / | / | |
| DIPSS | Constitutional symptoms | 1 | LOW | 0 | NR |
| Hemoglobin < 10 g/dL | 2 | INTERMEDIATE-1 | 1–2 | 14.2 | |
| WBC > 25 × 109/L | 1 | INTERMEDIATE-2 | 3–4 | 4 | |
| Blast ≥ 1% | 1 | HIGH | ≥5 | 1.5 | |
| Age > 65 | 1 | / | / | / | |
| DIPSS-Plus | DIPSS added to | LOW | 0 | 15 | |
| transfusion requirement | 1 | INTERMEDIATE-1 | 1 | 6.7 | |
| Platelet count < 100 × 109/L | 1 | INTERMEDIATE-2 | 2–3 | 2.9 | |
| Unfavorable karyotype | 1 | HIGH | ≥4 | 1.3 | |
| MYSEC-PM | Age | 0.15 per year | LOW | <11 | NR |
| Hemoglobin < 11 g/d | 2 | INTERMEDIATE 1 | ≥11, <14 | 9.3 | |
| Platelet count < 150 × 109/L | 1 | INTERMEDIATE 2 | ≥14, <16 | 4.4 | |
| Blasts > 3% | 2 | HIGH | ≥16 | 2 | |
| CALR-unmutated | 2 | / | / | / | |
| Constitutional symptoms | 1 | / | / | / | |
| MIPSS70 | Hemoglobin < 10 g/dL | 1 | LOW | 0–1 | 27.7 |
| WBC > 25 × 109/L | 2 | INTERMEDIATE | 2–4 | 7.1 | |
| Platelet count < 100 × 109/L | 2 | HIGH | ≥5 | 2.3 | |
| Blasts ≥ 2% | 1 | / | / | / | |
| Constitutional symptoms | 1 | / | / | / | |
| BM fibrosis grade ≥ 2 | 1 | / | / | / | |
| Single HMR mutation | 1 | / | / | / | |
| HMR mutations ≥ 2 | 2 | / | / | / | |
| Non-CALR mutation type 1 | 1 | / | / | / | |
| MIPSS70+ v2.0 | Severe anemia | 2 | VERY LOW | 0 | NR |
| Moderate anemia | 1 | LOW | 1–2 | 16.4 | |
| VHR karyotype | 4 | INTERMEDIATE | 3–4 | 7.7 | |
| Blasts ≥ 2% | 1 | HIGH | 5–8 | 4.1 | |
| Constitutional symptoms | 2 | VERY HIGH | ≥9 | 1.8 | |
| Unfavorable karyotype | 3 | / | / | / | |
| Single HMR mutation | 2 | / | / | / | |
| HMR mutations ≥ 2 | 3 | / | / | / | |
| Non-CALR mutation type 1 | 2 | / | / | / | |
| GIPSS | Non-CALR mutation type 1 | 1 | LOW | 0 | 26.4 |
| VHR karyotype | 2 | INTERMEDIATE 1 | 1 | 8 | |
| Unfavorable karyotype | 1 | INTERMEDIATE 2 | 2 | 4.2 | |
| ASXL1 mutation | 1 | HIGH | ≥3 | 2 | |
| SRSF2 mutation | 1 | / | / | / | |
| U2AF1 Q157 mutation | 1 | / | / | / | |
| MTSS | HLA-mismatched donor | 2 | LOW | 0–2 | 5 years-OS 83% |
| Non CALR/MPL mutation | 2 | INTERMEDIATE 1 | 3–4 | 5 years-OS 64% | |
| Age > 57 | 1 | INTERMEDIATE 2 | 5 | 5 years-OS 37% | |
| WBC > 25 × 109/L | 1 | HIGH | >5 | 5 years-OS 22% | |
| Platelet count < 150 × 109/L | 1 | / | / | / | |
| Karnofsky score < 90% | 1 | / | / | / | |
| ASXL1 mutation | 1 | / | / | / | |
| RR6 | Spleen length reduction ≤ 30% with respect to baseline at 3 and 6 months | 1.5 | LOW | 0 | NR |
| RBC transfusions requirement at baseline/3 months/6 months | 1 or 1.5 | INTERMEDIATE | 1–2 | 5 | |
| RUX treatment at dose < 20 mg twice daily at baseline, 3, and 6 months | 1 | HIGH | >2 | 2.7 | |
| AIPSS-MF | Sex, age (years), blood blasts (%), hemoglobin (g/L), leukocytes (×109/L), platelet count (×109/L), constitutional symptoms, leukoerythroblastosis | The model was designed to provide personalized predictions of overall survival and leukemia-free survival. Outcome predictions are from disease diagnosis. | |||
| Prognostic Model | Threshold | |
|---|---|---|
| Lower Risk | MIPSS-70 | ≤3 |
| MIPSS-70+ Version 2.0 | ≤3 | |
| DIPSS-Plus | ≤1 | |
| DIPSS | ≤2 | |
| MYSEC-PM * | <14 | |
| Higher Risk | MIPSS-70 | >3 |
| MIPSS-70+ Version 2.0 | >3 | |
| DIPSS-Plus | >1 | |
| DIPSS | >2 | |
| MYSEC-PM * | >14 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duminuco, A.; Nardo, A.; Giuffrida, G.; Leotta, S.; Markovic, U.; Giallongo, C.; Tibullo, D.; Romano, A.; Di Raimondo, F.; Palumbo, G.A. Myelofibrosis and Survival Prognostic Models: A Journey between Past and Future. J. Clin. Med. 2023, 12, 2188. https://doi.org/10.3390/jcm12062188
Duminuco A, Nardo A, Giuffrida G, Leotta S, Markovic U, Giallongo C, Tibullo D, Romano A, Di Raimondo F, Palumbo GA. Myelofibrosis and Survival Prognostic Models: A Journey between Past and Future. Journal of Clinical Medicine. 2023; 12(6):2188. https://doi.org/10.3390/jcm12062188
Chicago/Turabian StyleDuminuco, Andrea, Antonella Nardo, Gaetano Giuffrida, Salvatore Leotta, Uros Markovic, Cesarina Giallongo, Daniele Tibullo, Alessandra Romano, Francesco Di Raimondo, and Giuseppe A. Palumbo. 2023. "Myelofibrosis and Survival Prognostic Models: A Journey between Past and Future" Journal of Clinical Medicine 12, no. 6: 2188. https://doi.org/10.3390/jcm12062188