
The Potential Roles of Exosomes Carrying APP and Tau Cleavage Products in Alzheimer’s Disease

Abstract
:1. Introduction
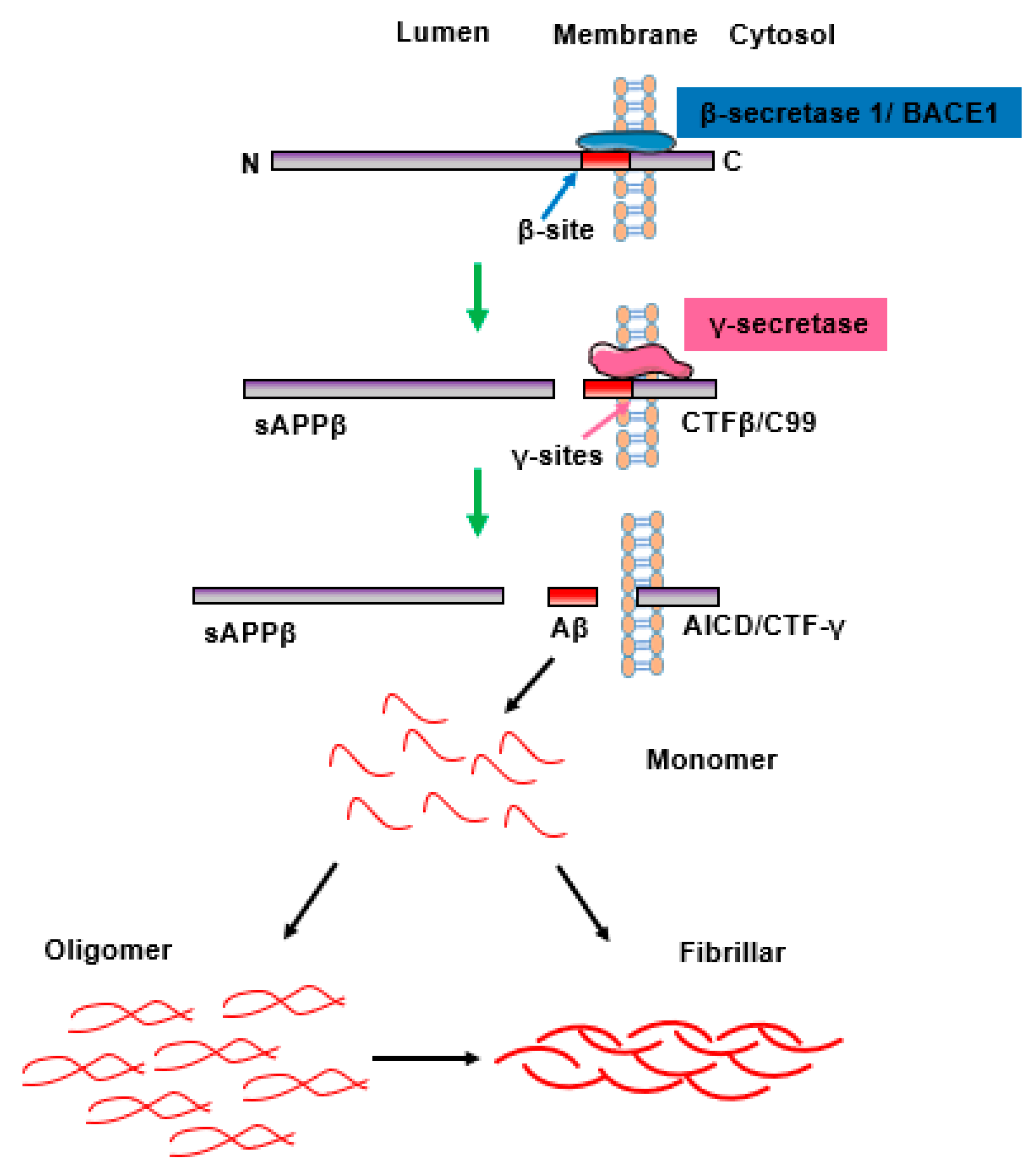
2. The Aggregation and Accumulation of Aβ Peptides and Senile Plaques Is Linked to AD Pathology
3. The Role of Hyperphosphorylated Tau and Neurofibrillary Tangles in the Progression of AD Pathology
4. Exosomes
5. The Propagation of Toxic Aβ Can Be Mediated by Exosomes in AD Pathology
5.1. Exosomes Acting as a Carrier to Transfer Neurotoxic Aβ between Neuronal Cells
5.2. The Interaction (Crosstalk) between Aβ and Glial-Cell-Derived Exosomes
5.3. The Formation Process of Exosomes Containing APP and Its Cleavage Products Is Associated with the Endosomal—Lysosomal Pathway
6. The Aggregation of Phosphorylated Tau Protein Can Be Regulated by Exosomes in AD Pathology
7. Exosomes including Aβ and Tau Acting as Biomarkers in the AD Pathological Process
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Jia, J.; Wei, C.; Chen, S.; Li, F.; Tang, Y.; Qin, W.; Zhao, L.; Jin, H.; Xu, H.; Wang, F.; et al. The cost of Alzheimer’s disease in China and re-estimation of costs worldwide. Alzheimers Dement. 2018, 14, 483–491. [Google Scholar] [CrossRef]
- Haukedal, H.; Freude, K.K. Implications of Glycosylation in Alzheimer’s Disease. Front. Neurosci. 2020, 14, 625348. [Google Scholar] [CrossRef]
- Wang, J.; Gu, B.J.; Masters, C.L.; Wang, Y.J. A systemic view of Alzheimer disease—Insights from amyloid-beta metabolism beyond the brain. Nat. Rev. Neurol. 2017, 13, 703. [Google Scholar] [CrossRef] [Green Version]
- Lakshmi, S.; Essa, M.M.; Hartman, R.E.; Guillemin, G.J.; Sivan, S.; Elumalai, P. Exosomes in Alzheimer’s Disease: Potential Role as Pathological Mediators, Biomarkers and Therapeutic Targets. Neurochem. Res. 2020, 45, 2553–2559. [Google Scholar] [CrossRef]
- Mohamed, T.; Shakeri, A.; Rao, P.P. Amyloid cascade in Alzheimer’s disease: Recent advances in medicinal chemistry. Eur. J. Med. Chem. 2016, 113, 258–272. [Google Scholar] [CrossRef]
- Suh, Y.H.; Checler, F. Amyloid precursor protein, presenilins, and alpha-synuclein: Molecular pathogenesis and pharmacological applications in Alzheimer’s disease. Pharmacol. Rev. 2002, 54, 469–525. [Google Scholar] [CrossRef] [Green Version]
- Lei, Y.; Renyuan, Z. Effects of Androgens on the Amyloid-β Protein in Alzheimer’s Disease. Endocrinology 2018, 159, 3885–3894. [Google Scholar] [CrossRef]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554. [Google Scholar] [CrossRef] [Green Version]
- Behl, T.; Kaur, I.; Fratila, O.; Brata, R.; Bungau, S. Exploring the Potential of Therapeutic Agents Targeted towards Mitigating the Events Associated with Amyloid-β Cascade in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 7443. [Google Scholar] [CrossRef]
- Barage, S.H.; Sonawane, K.D. Amyloid cascade hypothesis: Pathogenesis and therapeutic strategies in Alzheimer’s disease. Neuropeptides 2015, 52, 1–18. [Google Scholar] [CrossRef]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.E.; Lim, D.; Lee, J.Y.; Lim, S.M.; Pae, A.N. Development of tau-directed small molecule modulators for Alzheimer’s disease: A recent patent review (2014-2018). Pharm. Pat. Anal. 2019, 8, 15–39. [Google Scholar] [CrossRef]
- Naseri, N.N.; Wang, H.; Guo, J.; Sharma, M.; Luo, W. The complexity of tau in Alzheimer’s disease. Neurosci. Lett. 2019, 705, 183–194. [Google Scholar] [CrossRef]
- Hanger, D.P.; Seereeram, A.; Noble, W. Mediators of tau phosphorylation in the pathogenesis of Alzheimer’s disease. Expert Rev. Neurother. 2009, 9, 1647–1666. [Google Scholar] [CrossRef]
- Hanger, D.P.; Anderton, B.H.; Noble, W. Tau phosphorylation: The therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 2009, 15, 112–119. [Google Scholar] [CrossRef]
- Gratuze, M.; Joly-Amado, A.; Vieau, D.; Buée, L.; Blum, D. Mutual Relationship between Tau and Central Insulin Signalling: Consequences for AD and Tauopathies? Neuroendocrinology 2018, 107, 181–195. [Google Scholar] [CrossRef]
- Maccioni, R.B.; Farías, G.; Morales, I.; Navarrete, L. The revitalized tau hypothesis on Alzheimer’s disease. Arch. Med. Res. 2010, 41, 226–231. [Google Scholar] [CrossRef]
- Gao, Y.; Tan, L.; Yu, J.T.; Tan, L. Tau in Alzheimer’s Disease: Mechanisms and Therapeutic Strategies. Curr. Alzheimer Res. 2018, 15, 283–300. [Google Scholar] [CrossRef]
- Cooper, L.F.; Ravindran, S.; Huang, C.C.; Kang, M. A Role for Exosomes in Craniofacial Tissue Engineering and Regeneration. Front. Physiol. 2019, 10, 1569. [Google Scholar] [CrossRef]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef]
- Mashouri, L.; Yousefi, H.; Aref, A.R.; Ahadi, A.M.; Molaei, F.; Alahari, S.K. Exosomes: Composition, biogenesis, and mechanisms in cancer metastasis and drug resistance. Mol. Cancer 2019, 18, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, J.; Zhang, Y.; Zhu, Z.; Gu, C.; Waqas, A.; Chen, L. Emerging Exosomes and Exosomal MiRNAs in Spinal Cord Injury. Front. Cell Dev. Biol. 2021, 9, 703989. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Huang, W. New Developments in Exosomal lncRNAs in Cardiovascular Diseases. Front. Cardiovasc. Med. 2021, 8, 709169. [Google Scholar] [CrossRef] [PubMed]
- Heidarzadeh, M.; Gürsoy-Özdemir, Y.; Kaya, M.; Eslami Abriz, A.; Zarebkohan, A.; Rahbarghazi, R.; Sokullu, E. Exosomal delivery of therapeutic modulators through the blood-brain barrier; promise and pitfalls. Cell Biosci. 2021, 11, 142. [Google Scholar] [CrossRef]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Riau, A.K.; Ong, H.S.; Yam, G.H.F.; Mehta, J.S. Sustained Delivery System for Stem Cell-Derived Exosomes. Front. Pharmacol. 2019, 10, 1368. [Google Scholar] [CrossRef]
- Eitan, E.; Suire, C.; Zhang, S.; Mattson, M.P. Impact of lysosome status on extracellular vesicle content and release. Ageing Res. Rev. 2016, 32, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Lötvall, J.; Hill, A.F.; Hochberg, F.; Buzás, E.I.; Di Vizio, D.; Gardiner, C.; Gho, Y.S.; Kurochkin, I.V.; Mathivanan, S.; Quesenberry, P.; et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: A position statement from the International Society for Extracellular Vesicles. J. Extracell. Vesicles 2014, 3, 26913. [Google Scholar] [CrossRef]
- Poliakov, A.; Spilman, M.; Dokland, T.; Amling, C.L.; Mobley, J.A. Structural heterogeneity and protein composition of exosome-like vesicles (prostasomes) in human semen. Prostate 2009, 69, 159–167. [Google Scholar] [CrossRef]
- van der Pol, E.; Coumans, F.A.; Grootemaat, A.E.; Gardiner, C.; Sargent, I.L.; Harrison, P.; Sturk, A.; van Leeuwen, T.G.; Nieuwland, R. Particle size distribution of exosomes and microvesicles determined by transmission electron microscopy, flow cytometry, nanoparticle tracking analysis, and resistive pulse sensing. J. Thromb. Haemost. JTH 2014, 12, 1182–1192. [Google Scholar] [CrossRef]
- Thej, C.; Kishore, R. Unfathomed Nanomessages to the Heart: Translational Implications of Stem Cell-Derived, Progenitor Cell Exosomes in Cardiac Repair and Regeneration. Cells 2021, 10, 1811. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Wang, D.; Han, Y.; Huang, T.; He, X.; Wang, J.; Ou, C. Emerging Role of Cancer-Associated Fibroblasts-Derived Exosomes in Tumorigenesis. Front. Immunol. 2021, 12, 795372. [Google Scholar] [CrossRef]
- Polanco, J.C.; Li, C.; Durisic, N.; Sullivan, R.; Götz, J. Exosomes taken up by neurons hijack the endosomal pathway to spread to interconnected neurons. Acta Neuropathol. Commun. 2018, 6, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-González, R.; Kim, Y.; Miller, C.; Pacheco-Quinto, J.; Eckman, E.A.; Levy, E. Extracellular vesicles: Where the amyloid precursor protein carboxyl-terminal fragments accumulate and amyloid-β oligomerizes. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 12922–12931. [Google Scholar] [CrossRef]
- Choi, Y.; Kim, S.M.; Heo, Y.; Lee, G.; Kang, J.Y.; Yoon, D.S. Nanoelectrical characterization of individual exosomes secreted by Aβ(42)-ingested cells using electrostatic force microscopy. Nanotechnology 2021, 32, 025705. [Google Scholar] [CrossRef] [PubMed]
- Zheng, T.; Pu, J.; Chen, Y.; Mao, Y.; Guo, Z.; Pan, H.; Zhang, L.; Zhang, H.; Sun, B.; Zhang, B. Plasma Exosomes Spread and Cluster Around β-Amyloid Plaques in an Animal Model of Alzheimer’s Disease. Front. Aging Neurosci. 2017, 9, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, T.; Wu, X.; Wei, X.; Wang, M.; Zhang, B. The release and transmission of amyloid precursor protein via exosomes. Neurochem. Int. 2018, 114, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Sardar Sinha, M.; Ansell-Schultz, A.; Civitelli, L.; Hildesjö, C.; Larsson, M.; Lannfelt, L.; Ingelsson, M.; Hallbeck, M. Alzheimer’s disease pathology propagation by exosomes containing toxic amyloid-beta oligomers. Acta Neuropathol. 2018, 136, 41–56. [Google Scholar] [CrossRef] [Green Version]
- Laulagnier, K.; Javalet, C.; Hemming, F.J.; Chivet, M.; Lachenal, G.; Blot, B.; Chatellard, C.; Sadoul, R. Amyloid precursor protein products concentrate in a subset of exosomes specifically endocytosed by neurons. Cell Mol. Life Sci. 2018, 75, 757–773. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Cortez, L.; Kamali-Jamil, R.; Sim, V.; Wille, H.; Kar, S. Implications of exosomes derived from cholesterol-accumulated astrocytes in Alzheimer’s disease pathology. Dis. Model Mech. 2021, 14, dmm048929. [Google Scholar] [CrossRef]
- Dinkins, M.B.; Enasko, J.; Hernandez, C.; Wang, G.; Kong, J.; Helwa, I.; Liu, Y.; Terry, A.V., Jr.; Bieberich, E. Neutral Sphingomyelinase-2 Deficiency Ameliorates Alzheimer’s Disease Pathology and Improves Cognition in the 5XFAD Mouse. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 8653–8667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinkins, M.B.; Dasgupta, S.; Wang, G.; Zhu, G.; Bieberich, E. Exosome reduction in vivo is associated with lower amyloid plaque load in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 1792–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falker, C.; Hartmann, A.; Guett, I.; Dohler, F.; Altmeppen, H.; Betzel, C.; Schubert, R.; Thurm, D.; Wegwitz, F.; Joshi, P.; et al. Exosomal cellular prion protein drives fibrillization of amyloid beta and counteracts amyloid beta-mediated neurotoxicity. J. Neurochem. 2016, 137, 88–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, K.; Zhao, L.; Gregory, C.; Solanki, A.; Mastrianni, J.A. “Dual Disease” TgAD/GSS mice exhibit enhanced Alzheimer’s disease pathology and reveal PrP(C)-dependent secretion of Aβ. Sci. Rep. 2019, 9, 8524. [Google Scholar] [CrossRef] [Green Version]
- Gao, G.; Zhao, S.; Xia, X.; Li, C.; Li, C.; Ji, C.; Sheng, S.; Tang, Y.; Zhu, J.; Wang, Y.; et al. Glutaminase C Regulates Microglial Activation and Pro-inflammatory Exosome Release: Relevance to the Pathogenesis of Alzheimer’s Disease. Front. Cell. Neurosci. 2019, 13, 264. [Google Scholar] [CrossRef]
- Chiarini, A.; Armato, U.; Gardenal, E.; Gui, L.; Dal Prà, I. Amyloid β-Exposed Human Astrocytes Overproduce Phospho-Tau and Overrelease It within Exosomes, Effects Suppressed by Calcilytic NPS 2143-Further Implications for Alzheimer’s Therapy. Front. Neurosci. 2017, 11, 217. [Google Scholar] [CrossRef] [Green Version]
- Abdullah, M.; Takase, H.; Nunome, M.; Enomoto, H.; Ito, J.; Gong, J.S.; Michikawa, M. Amyloid-β Reduces Exosome Release from Astrocytes by Enhancing JNK Phosphorylation. J. Alzheimers Dis. 2016, 53, 1433–1441. [Google Scholar] [CrossRef]
- Nafar, F.; Williams, J.B.; Mearow, K.M. Astrocytes release HspB1 in response to amyloid-β exposure in vitro. J. Alzheimers Dis. 2016, 49, 251–263. [Google Scholar] [CrossRef]
- Elsherbini, A.; Kirov, A.S.; Dinkins, M.B.; Wang, G.; Qin, H.; Zhu, Z.; Tripathi, P.; Crivelli, S.M.; Bieberich, E. Association of Aβ with ceramide-enriched astrosomes mediates Aβ neurotoxicity. Acta Neuropathol. Commun. 2020, 8, 60. [Google Scholar] [CrossRef]
- Deng, Z.; Wang, J.; Xiao, Y.; Li, F.; Niu, L.; Liu, X.; Meng, L.; Zheng, H. Ultrasound-mediated augmented exosome release from astrocytes alleviates amyloid-β-induced neurotoxicity. Theranostics 2021, 11, 4351–4362. [Google Scholar] [CrossRef]
- Pitt, J.; Wilcox, K.C.; Tortelli, V.; Diniz, L.P.; Oliveira, M.S.; Dobbins, C.; Yu, X.W.; Nandamuri, S.; Gomes, F.C.A.; DiNunno, N.; et al. Neuroprotective astrocyte-derived insulin/insulin-like growth factor 1 stimulates endocytic processing and extracellular release of neuron-bound Aβ oligomers. Mol. Biol. Cell 2017, 28, 2623–2636. [Google Scholar] [CrossRef]
- Fernandes, A.; Ribeiro, A.R.; Monteiro, M.; Garcia, G.; Vaz, A.R.; Brites, D. Secretome from SH-SY5Y APP(Swe) cells trigger time-dependent CHME3 microglia activation phenotypes, ultimately leading to miR-21 exosome shuttling. Biochimie 2018, 155, 67–82. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Liao, X.; Wu, J.; Zhang, X.; Li, Y.; Xiang, D.; Luo, S. The Microglial membrane receptor TREM2 mediates exosome secretion to promote phagocytosis of amyloid-β by microglia. FEBS Lett. 2022, 596, 1059–1071. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Kollmer, M.; Markx, D.; Claus, S.; Walther, P.; Fändrich, M. Amyloid plaque structure and cell surface interactions of β-amyloid fibrils revealed by electron tomography. Sci. Rep. 2017, 7, 43577. [Google Scholar] [CrossRef] [Green Version]
- Williamson, R.L.; Laulagnier, K.; Miranda, A.M.; Fernandez, M.A.; Wolfe, M.S.; Sadoul, R.; Di Paolo, G. Disruption of amyloid precursor protein ubiquitination selectively increases amyloid β (Aβ) 40 levels via presenilin 2-mediated cleavage. J. Biol. Chem. 2017, 292, 19873–19889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauritzen, I.; Pardossi-Piquard, R.; Bourgeois, A.; Pagnotta, S.; Biferi, M.G.; Barkats, M.; Lacor, P.; Klein, W.; Bauer, C.; Checler, F. Intraneuronal aggregation of the β-CTF fragment of APP (C99) induces Aβ-independent lysosomal-autophagic pathology. Acta Neuropathol. 2016, 132, 257–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauritzen, I.; Bécot, A.; Bourgeois, A.; Pardossi-Piquard, R.; Biferi, M.G.; Barkats, M.; Checler, F. Targeting γ-secretase triggers the selective enrichment of oligomeric APP-CTFs in brain extracellular vesicles from Alzheimer cell and mouse models. Transl. Neurodegener. 2019, 8, 35. [Google Scholar] [CrossRef] [Green Version]
- Cone, A.S.; Hurwitz, S.N.; Lee, G.S.; Yuan, X.; Zhou, Y.; Li, Y.; Meckes, D.G., Jr. Alix and Syntenin-1 direct amyloid precursor protein trafficking into extracellular vesicles. BMC Mol. Cell Biol. 2020, 21, 58. [Google Scholar] [CrossRef]
- Miranda, A.M.; Lasiecka, Z.M.; Xu, Y.; Neufeld, J.; Shahriar, S.; Simoes, S.; Chan, R.B.; Oliveira, T.G.; Small, S.A.; Di Paolo, G. Neuronal lysosomal dysfunction releases exosomes harboring APP C-terminal fragments and unique lipid signatures. Nat. Commun. 2018, 9, 291. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.C.; Ma, X.Y.; Liu, X.H.; Yu, J.; Zhao, Y.C.; Tan, Y.; Liu, X.Y.; Zhao, Y.X. Hypoxia increases amyloid-β level in exosomes by enhancing the interaction between CD147 and Hook1. Am. J. Transl. Res. 2018, 10, 150–163. [Google Scholar]
- Pacheco-Quinto, J.; Clausen, D.; Pérez-González, R.; Peng, H.; Meszaros, A.; Eckman, C.B.; Levy, E.; Eckman, E.A. Intracellular metalloprotease activity controls intraneuronal Aβ aggregation and limits secretion of Aβ via exosomes. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 3758–3771. [Google Scholar] [CrossRef]
- Guix, F.X.; Sannerud, R.; Berditchevski, F.; Arranz, A.M.; Horré, K.; Snellinx, A.; Thathiah, A.; Saido, T.; Saito, T.; Rajesh, S.; et al. Tetraspanin 6: A pivotal protein of the multiple vesicular body determining exosome release and lysosomal degradation of amyloid precursor protein fragments. Mol. Neurodegener. 2017, 12, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winston, C.N.; Goetzl, E.J.; Akers, J.C.; Carter, B.S.; Rockenstein, E.M.; Galasko, D.; Masliah, E.; Rissman, R.A. Prediction of conversion from mild cognitive impairment to dementia with neuronally derived blood exosome protein profile. Alzheimer’s Dement. 2016, 3, 63–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polanco, J.C.; Scicluna, B.J.; Hill, A.F.; Götz, J. Extracellular Vesicles Isolated from the Brains of rTg4510 Mice Seed Tau Protein Aggregation in a Threshold-dependent Manner. J. Biol. Chem. 2016, 291, 12445–12466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, S.; Polanco, J.C.; Götz, J. Extracellular Vesicles Containing P301L Mutant Tau Accelerate Pathological Tau Phosphorylation and Oligomer Formation but Do Not Seed Mature Neurofibrillary Tangles in ALZ17 Mice. J. Alzheimers Dis. 2016, 54, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Balaji, V.; Kaniyappan, S.; Krüger, L.; Irsen, S.; Tepper, K.; Chandupatla, R.; Maetzler, W.; Schneider, A.; Mandelkow, E.; et al. The release and trans-synaptic transmission of Tau via exosomes. Mol. Neurodegener. 2017, 12, 5. [Google Scholar] [CrossRef] [Green Version]
- Guix, F.X.; Corbett, G.T.; Cha, D.J.; Mustapic, M.; Liu, W.; Mengel, D.; Chen, Z.; Aikawa, E.; Young-Pearse, T.; Kapogiannis, D.; et al. Detection of Aggregation-Competent Tau in Neuron-Derived Extracellular Vesicles. Int. J. Mol. Sci. 2018, 19, 663. [Google Scholar] [CrossRef] [Green Version]
- Winston, C.N.; Aulston, B.; Rockenstein, E.M.; Adame, A.; Prikhodko, O.; Dave, K.N.; Mishra, P.; Rissman, R.A.; Yuan, S.H. Neuronal Exosome-Derived Human Tau is Toxic to Recipient Mouse Neurons in vivo. J. Alzheimers Dis. 2019, 67, 541–553. [Google Scholar] [CrossRef]
- Podvin, S.; Jones, A.; Liu, Q.; Aulston, B.; Ransom, L.; Ames, J.; Shen, G.; Lietz, C.B.; Jiang, Z.; O’Donoghue, A.J.; et al. Dysregulation of Exosome Cargo by Mutant Tau Expressed in Human-induced Pluripotent Stem Cell (iPSC) Neurons Revealed by Proteomics Analyses. Mol. Cell. Proteom. MCP 2020, 19, 1017–1034. [Google Scholar] [CrossRef]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kügler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef] [Green Version]
- Polanco, J.C.; Hand, G.R.; Briner, A.; Li, C.; Götz, J. Exosomes induce endolysosomal permeabilization as a gateway by which exosomal tau seeds escape into the cytosol. Acta Neuropathol. 2021, 141, 235–256. [Google Scholar] [CrossRef] [PubMed]
- Marcus, C.; Mena, E.; Subramaniam, R.M. Brain PET in the diagnosis of Alzheimer’s disease. Clin. Nucl. Med. 2014, 39, e413–e422, quiz e423–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welge, V.; Fiege, O.; Lewczuk, P.; Mollenhauer, B.; Esselmann, H.; Klafki, H.W.; Wolf, S.; Trenkwalder, C.; Otto, M.; Kornhuber, J.; et al. Combined CSF tau, p-tau181 and amyloid-beta 38/40/42 for diagnosing Alzheimer’s disease. J. Neural Transm. 2009, 116, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Eitan, E.; Hutchison, E.R.; Marosi, K.; Comotto, J.; Mustapic, M.; Nigam, S.M.; Suire, C.; Maharana, C.; Jicha, G.A.; Liu, D.; et al. Extracellular Vesicle-Associated Aβ Mediates Trans-Neuronal Bioenergetic and Ca(2+)-Handling Deficits in Alzheimer’s Disease Models. NPJ Aging Mech. Dis. 2016, 2, 16019. [Google Scholar] [CrossRef] [Green Version]
- Gibbons, G.S.; Lee, V.M.Y.; Trojanowski, J.Q. Mechanisms of Cell-to-Cell Transmission of Pathological Tau: A Review. JAMA Neurol. 2019, 76, 101–108. [Google Scholar] [CrossRef]
- Perrotte, M.; Haddad, M.; Le Page, A.; Frost, E.H.; Fulöp, T.; Ramassamy, C. Profile of pathogenic proteins in total circulating extracellular vesicles in mild cognitive impairment and during the progression of Alzheimer’s disease. Neurobiol. Aging 2020, 86, 102–111. [Google Scholar] [CrossRef]
- Sun, R.; Wang, H.; Shi, Y.; Sun, Z.; Jiang, H.; Zhang, J. Changes in the Morphology, Number, and Pathological Protein Levels of Plasma Exosomes May Help Diagnose Alzheimer’s Disease. J. Alzheimers Dis. 2020, 73, 909–917. [Google Scholar] [CrossRef]
- Shi, M.; Kovac, A.; Korff, A.; Cook, T.J.; Ginghina, C.; Bullock, K.M.; Yang, L.; Stewart, T.; Zheng, D.; Aro, P.; et al. CNS tau efflux via exosomes is likely increased in Parkinson’s disease but not in Alzheimer’s disease. Alzheimers Dement. 2016, 12, 1125–1131. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Meng, S.; Di, W.; Xia, M.; Dong, L.; Zhao, Y.; Ling, S.; He, J.; Xue, X.; Chen, X.; et al. Amyloid-β protein and MicroRNA-384 in NCAM-Labeled exosomes from peripheral blood are potential diagnostic markers for Alzheimer’s disease. CNS Neurosci. Ther. 2022, 28, 1093–1107. [Google Scholar] [CrossRef]
- Eren, E.; Hunt, J.F.V.; Shardell, M.; Chawla, S.; Tran, J.; Gu, J.; Vogt, N.M.; Johnson, S.C.; Bendlin, B.B.; Kapogiannis, D. Extracellular vesicle biomarkers of Alzheimer’s disease associated with sub-clinical cognitive decline in late middle age. Alzheimers Dement. 2020, 16, 1293–1304. [Google Scholar] [CrossRef]
- Abner, E.L.; Jicha, G.A.; Shaw, L.M.; Trojanowski, J.Q.; Goetzl, E.J. Plasma neuronal exosomal levels of Alzheimer’s disease biomarkers in normal aging. Ann. Clin. Transl. Neurol. 2016, 3, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Qiu, Q.; Zhang, H.; Chu, L.; Du, Y.; Zhang, J.; Zhou, C.; Liang, F.; Shi, S.; Wang, S.; et al. Concordance between the assessment of Aβ42, T-tau, and P-T181-tau in peripheral blood neuronal-derived exosomes and cerebrospinal fluid. Alzheimers Dement. 2019, 15, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Zhao, A.; Li, Y.; Yan, Y.; Qiu, Y.; Li, B.; Xu, W.; Wang, Y.; Liu, J.; Deng, Y. Increased prediction value of biomarker combinations for the conversion of mild cognitive impairment to Alzheimer’s dementia. Transl. Neurodegener. 2020, 9, 30. [Google Scholar] [CrossRef] [PubMed]
- Porsteinsson, A.P.; Isaacson, R.S.; Knox, S.; Sabbagh, M.N.; Rubino, I. Diagnosis of Early Alzheimer’s Disease: Clinical Practice in 2021. J. Prev. Alzheimer’s Dis. 2021, 8, 371–386. [Google Scholar] [CrossRef]
- Soares Martins, T.; Marçalo, R.; da Cruz, E.S.C.B.; Trindade, D.; Catita, J.; Amado, F.; Melo, T.; Rosa, I.M.; Vogelgsang, J.; Wiltfang, J.; et al. Novel Exosome Biomarker Candidates for Alzheimer’s Disease Unravelled Through Mass Spectrometry Analysis. Mol. Neurobiol. 2022, 59, 2838–2854. [Google Scholar] [CrossRef] [PubMed]
- Soares Martins, T.; Marçalo, R.; Ferreira, M.; Vaz, M.; Silva, R.M.; Martins Rosa, I.; Vogelgsang, J.; Wiltfang, J.; da Cruz, E.S.O.A.B.; Henriques, A.G. Exosomal Aβ-Binding Proteins Identified by “In Silico” Analysis Represent Putative Blood-Derived Biomarker Candidates for Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 3933. [Google Scholar] [CrossRef]
- Wang, D.; Wang, P.; Bian, X.; Xu, S.; Zhou, Q.; Zhang, Y.; Ding, M.; Han, M.; Huang, L.; Bi, J.; et al. Elevated plasma levels of exosomal BACE1-AS combined with the volume and thickness of the right entorhinal cortex may serve as a biomarker for the detection of Alzheimer’s disease. Mol. Med. Rep. 2020, 22, 227–238. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Mustapic, M.; Kapogiannis, D.; Eitan, E.; Lobach, I.V.; Goetzl, L.; Schwartz, J.B.; Miller, B.L. Cargo proteins of plasma astrocyte-derived exosomes in Alzheimer’s disease. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2016, 30, 3853–3859. [Google Scholar] [CrossRef] [Green Version]
- Cohn, W.; Melnik, M.; Huang, C.; Teter, B.; Chandra, S.; Zhu, C.; McIntire, L.B.; John, V.; Gylys, K.H.; Bilousova, T. Multi-Omics Analysis of Microglial Extracellular Vesicles From Human Alzheimer’s Disease Brain Tissue Reveals Disease-Associated Signatures. Front. Pharmacol. 2021, 12, 766082. [Google Scholar] [CrossRef]
- Delgado-Peraza, F.; Nogueras-Ortiz, C.J.; Volpert, O.; Liu, D.; Goetzl, E.J.; Mattson, M.P.; Greig, N.H.; Eitan, E.; Kapogiannis, D. Neuronal and Astrocytic Extracellular Vesicle Biomarkers in Blood Reflect Brain Pathology in Mouse Models of Alzheimer’s Disease. Cells 2021, 10, 993. [Google Scholar] [CrossRef]
- Sun, R.; Wang, H.; Shi, Y.; Gao, D.; Sun, Z.; Chen, Z.; Jiang, H.; Zhang, J. A Pilot Study of Urinary Exosomes in Alzheimer’s Disease. Neuro-Degener. Dis. 2019, 19, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Rani, K.; Rastogi, S.; Vishwakarma, P.; Bharti, P.S.; Sharma, V.; Renu, K.; Modi, G.P.; Vishnu, V.Y.; Chatterjee, P.; Dey, A.B.; et al. A novel approach to correlate the salivary exosomes and their protein cargo in the progression of cognitive impairment into Alzheimer’s disease. J. Neurosci. Methods 2021, 347, 108980. [Google Scholar] [CrossRef]
- Khan, S.; Barve, K.H.; Kumar, M.S. Recent Advancements in Pathogenesis, Diagnostics and Treatment of Alzheimer’s Disease. Curr. Neuropharmacol. 2020, 18, 1106–1125. [Google Scholar] [CrossRef] [PubMed]
- Barile, L.; Vassalli, G. Exosomes: Therapy delivery tools and biomarkers of diseases. Pharmacol. Ther. 2017, 174, 63–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Promotes or Inhibits Exosome Release | Major Contents of Exosome | References |
|---|---|---|---|
| PI3K-dependent pathway | Promotes | APP and its cleaved products | [40] |
| nSMase2 | Inhibits | Aβ42 | [41,42] |
| PrPC | Balances | Aβ42 | [43,44] |
| GAC | Promotes | APP and Aβ | [45] |
| CaSR antagonist NPS2143 | Inhibits | Tau and p-Tau | [46] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.; Gu, Y.; Zhang, Q.; Liu, H.; Liu, Y. The Potential Roles of Exosomes Carrying APP and Tau Cleavage Products in Alzheimer’s Disease. J. Clin. Med. 2023, 12, 1883. https://doi.org/10.3390/jcm12051883
Zhao Y, Gu Y, Zhang Q, Liu H, Liu Y. The Potential Roles of Exosomes Carrying APP and Tau Cleavage Products in Alzheimer’s Disease. Journal of Clinical Medicine. 2023; 12(5):1883. https://doi.org/10.3390/jcm12051883
Chicago/Turabian StyleZhao, Yanfang, Yujin Gu, Qili Zhang, Hongliang Liu, and Yingying Liu. 2023. "The Potential Roles of Exosomes Carrying APP and Tau Cleavage Products in Alzheimer’s Disease" Journal of Clinical Medicine 12, no. 5: 1883. https://doi.org/10.3390/jcm12051883